

SUMMARY
Transient expression of the transcription factor neurogenin-3 marks progenitor cells in the pancreas as they differentiate into islet cells. We developed a transgenic mouse line in which the surrogate markers secreted alkaline phosphatase (SeAP) and enhanced green florescent protein (EGFP) can be used to monitor neurogenin-3 expression, and thus islet cell genesis. In transgenic embryos, cells expressing EGFP lined the pancreatic ducts. SeAP was readily detectable in embryos, in the media of cultured embryonic pancreases and in the serum of adult animals. Treatment with the γ-secretase inhibitor DAPT, which blocks Notch signaling, enhanced SeAP secretion rates and increased the number of EGFP-expressing cells as assayed by fluorescence-activated cell sorting (FACS) and immunohistochemistry in cultured pancreases from embryos at embryonic day 11.5, but not in pancreases harvested 1 day later. By contrast, treatment with growth differentiation factor 11 (GDF11) reduced SeAP secretion rates. In adult mice, partial pancreatectomy decreased, whereas duct ligation increased, circulating SeAP levels. This model will be useful for studying signals involved in islet cell genesis in vivo and developing therapies that induce this process.
INTRODUCTION
Therapeutic methods for generating new insulin-producing islet cells remain an unrealized goal of diabetes treatment. Currently, the normal developmental pathways by which islets form during pancreatic development and regeneration remain the only definitive method for generating truly normal islet cells. Therefore, models by which these processes can be tracked in vivo can provide the means for testing methods for manipulating islet cell generation.
During mammalian development, the pancreas first appears as clusters of apparently identical cells on the dorsal and ventral aspects of the gut tube at the foregut-midgut junction. The exocrine, endocrine and duct cells differentiate from these undifferentiated pancreatic progenitor cells (Slack, 1995; Wilson et al., 2003). Understanding and controlling this process of differentiation could ultimately provide us with the cells needed to treat diabetes mellitus.
A single transcription factor, the pro-endocrine basic helix-loop-helix (bHLH) factor neurogenin-3, is both necessary and sufficient to drive these progenitor cells to differentiate into the endocrine cells that form the islets of Langerhans. Mice homozygous for a targeted deletion of the neurogenin-3 gene fail to develop any endocrine cells in the pancreas (Gradwohl et al., 2000). Conversely, expression of neurogenin-3 in all of the epithelial cells of the early pancreatic bud drives all of those cells to differentiate into endocrine cells (Apelqvist et al., 1999; Schwitzgebel et al., 2000). Neurogenin-3 only appears transiently during pancreatic development, in cells along or adjacent to the developing ducts (Jensen et al., 2000a; Schwitzgebel et al., 2000). Although these cells do not express markers of mature endocrine cells such as insulin and glucagon, lineage-tracing experiments have demonstrated that the neurogenin-3-expressing cells are the progenitors of the mature endocrine cells in the islets of Langerhans (Gu et al., 2002). Because its expression rapidly wanes prior to final differentiation, neurogenin-3 must activate a gene expression program that then completes the differentiation of these cells. Consistent with this model, neurogenin-3 activates the expression of several key islet differentiation factors (Heremans et al., 2002; Gasa et al., 2004), including NeuroD1 (Huang et al., 2000), Pax4 (Smith et al., 2003), Nkx2.2 (Watada et al., 2003), Myt1 (Wang et al., 2008) and Insm1 (Mellitzer et al., 2006).
Given the decisive role of neurogenin-3 in islet development, the mechanisms that control its expression in the developing pancreas thereby control the generation of islet cells. Both positive and negative regulators of neurogenin-3 expression in the pancreas have been identified. Several transcription factors, including Sox9, FoxA2, HNF1β and HNF6, have been implicated as activators of neurogenin-3 expression (Jacquemin et al., 2000; Lee et al., 2001; Maestro et al., 2003; Lynn et al., 2007), whereas the inhibitory bHLH transcription factor Hes1 suppresses neurogenin-3 expression (Jensen et al., 2000b; Lee et al., 2001). Hes1 expression in turn is activated by the Notch signaling pathway, and Notch signaling in the developing pancreas limits the number of cells in which neurogenin-3 expression is activated (Apelqvist et al., 1999). In addition, loss-of-function studies suggest that the TGFβ family member GDF11 can also restrict neurogenin-3 expression (Dichmann et al., 2003; Harmon et al., 2004).
To further explore the mechanisms that regulate neurogenin-3 expression and thereby control islet cell genesis, we designed a transgene construct with the coding sequence for neurogenin-3 replaced by genes encoding the marker proteins secreted alkaline phosphatase (SeAP) and enhanced green florescent protein (EGFP) in a large human bacterial artificial chromosome (BAC NEUROG3-SeAP/EGFP). Transgenic mice produced with the BAC NEUROG3-SeAP/EGFP construct can be used to study neurogenin-3 gene expression and islet cell genesis in intact cells and living mice. Ultimately, this information can be used to guide the development of therapies for diabetes.
RESULTS
Generation of transgenic BAC NEUROG3-SeAP/EGFP mice
To produce a transgene that would allow for the assessment of neurogenin-3 gene expression in vivo, we started with a BAC containing the human genomic sequence 134-kb upstream and 30-kb downstream of the human neurogenin-3 (NEUROG3) gene, and replaced the coding sequence for neurogenin-3 with two marker genes, SeAP and EGFP, separated by a viral internal ribosomal entry site (IRES) (Fig. 1A). This construct was used to derive three independent transgenic mouse founders. One founder did not produce any progeny. Transgene expression patterns in the remaining two lines were indistinguishable.
Fig. 1.

The BAC NEUROG3-SeAP/EGFP transgenes are expressed in the developing pancreas. (A) Homologous recombination (Yang et al., 1997) was used to replace the human neurogenin-3 coding sequence in the NEUROG3 BAC (RP11-343J3T) with two reporter genes, SeAP and EGFP, flanked on the 5′ end by the human β-globulin intron and the 3′ end by the SV40 polyadenylation signal, and separated by a viral IRES. (B) Levels of neurogenin-3 mRNA in mouse pancreas were measured by real-time TaqMan RT-PCR at the embryonic dates shown and in adult islets, and are expressed relative to levels of histone H3.3a mRNA. (C) Levels of the SeAP/EGFP mRNA in mouse pancreas were measured by real-time SYBR Green RT-PCR at the embryonic dates shown and are expressed relative to levels of mouse β-actin mRNA. (D) Tissue SeAP activity was measured in pancreas homogenates at the embryonic dates shown and is expressed relative to total protein. All data represent mean + s.e.m. from at least three independent experiments.
The transgenes are expressed in the endocrine lineage during development
To characterize transgene expression, we compared it to the expression of native neurogenin-3 RNA and protein. The concentration of neurogenin-3 mRNA as measured by TaqMan real-time reverse transcriptase (RT)-PCR peaked in the mouse pancreas at embryonic day 13.5 (E13.5; Fig. 1B), when the pancreas enters the secondary transition, a period of rapid differentiation of duct, exocrine and endocrine cells. Timing of the peak in SeAP/EGFP mRNA in the pancreases from transgenic embryos was similar to that of neurogenin-3, whereas SeAP protein expression as measured by SeAP activity in the pancreas was somewhat delayed relative to the mRNA peak (Fig. 1C,D).
As expected, EGFP protein in the pancreases of BAC NEUROG3-SeAP/EGFP embryos was detected in cells along the ducts and peaked at E14.5-E15.5, shortly after the peak in neurogenin-3 mRNA (Fig. 2A and data not shown). Quantification by fluorescence-activated cell sorting (FACS) yielded 62,000 EGFP-expressing cells from 20 pancreases at E15.5, which is approximately 3000 cells per pancreas or ∼5% of the total cell population (data not shown). Although EGFP-positive cells were still present 1 day later, at E16.5, the density of EGFP-expressing cells had decreased (Fig. 2B).
Fig. 2.

The EGFP transgene is expressed in the endocrine lineage in the embryonic pancreas of BAC NEUROG3-SeAP/EGFP transgenic mice. (A,B) Native EGFP fluorescence from whole-mounted pancreas at E15.5 (A) and E16.5 (B). (C) Immunofluorescent staining of whole-mounted E15.5 pancreas for mucin-1 (blue) together with immunofluorescent staining for EGFP (green). (D–I) Immunofluorescent staining of frozen E15.5 pancreatic sections for EGFP (green) and neurogenin-3 (red) together with glucagon (blue; D and E) or insulin (blue; G and H). White arrowheads mark representative EGFP positive cells co-stained with neurogenin-3. Yellow arrowheads mark representative EGFP-positive cells co-stained with glucagon or insulin. Scale bars: 50 μm.
Co-staining for EGFP and the ductal marker mucin-1 demonstrated that all of the EGFP-expressing cells lay within a short distance of the duct lumen (Fig. 2C). Co-staining for EGFP and neurogenin-3 at E15.5 revealed an obvious but incomplete overlap between the two proteins (Fig. 2D–I; supplementary material Fig. S1A–F). This difference between the expression of the native protein and the transgenic protein has been observed before (Lee et al., 2001), is also seen in mice in which the EGFP coding sequence replaces the neurogenin-3 coding sequence directly in the Neurog3 locus (Neurog3EGFP/+ mice; supplementary material Fig. S1G–L), and most probably reflects differences in the turnover of the two proteins. Most cells that stained positive for insulin or glucagon at this stage co-stained weakly for EGFP, but not for the endogenous neurogenin-3 protein.
All EGFP-expressing cells coexpressed the endocrine marker chromogranin-A (Fig. 3A–C), demonstrating that EGFP expression is restricted to the endocrine lineage. The same pattern was also seen in EGFP knock-in mice (Neurog3EGFP/+ mice; Fig. 3D–F). These data again support the conclusion that EGFP is expressed in the endocrine cell progenitors in both models but, unlike the shorter half-life neurogenin-3 protein, EGFP protein is not completely cleared prior to terminal differentiation of the endocrine cells.
Fig. 3.

The EGFP transgene is expressed in the endocrine pancreas lineage in BAC NEUROG3-SeAP/EGFP transgenic mice and Neurog3EGFP/+ mice at E18.5. Panels show fluorescent staining of frozen pancreatic sections from BAC NEUROG3-SeAP/EGFP transgenic mice (line 29) (A–C) and Neurog3EGFP/+ mice (D–F) at E18.5 for Dolichos biflorus agglutinin (DBA; blue) along with immunofluorescent staining for EGFP (green; A,C,D,F) and chromogranin-A (red; B,C,E,F).
SeAP was readily detected not only by measurement of enzymatic activity in serum and pancreatic extracts from the transgenic fetuses, but also by direct histological detection of SeAP activity with chromogenic substrate NBT/BCIP in pancreatic sections at E14.5 and E18.5 (Fig. 4A,B). Interestingly, SeAP staining with NBT/BCIP colocalized with neurogenin-3 protein more closely than did EGFP (no cells lacking neurogenin-3 protein had robust SeAP activity).
Fig. 4.
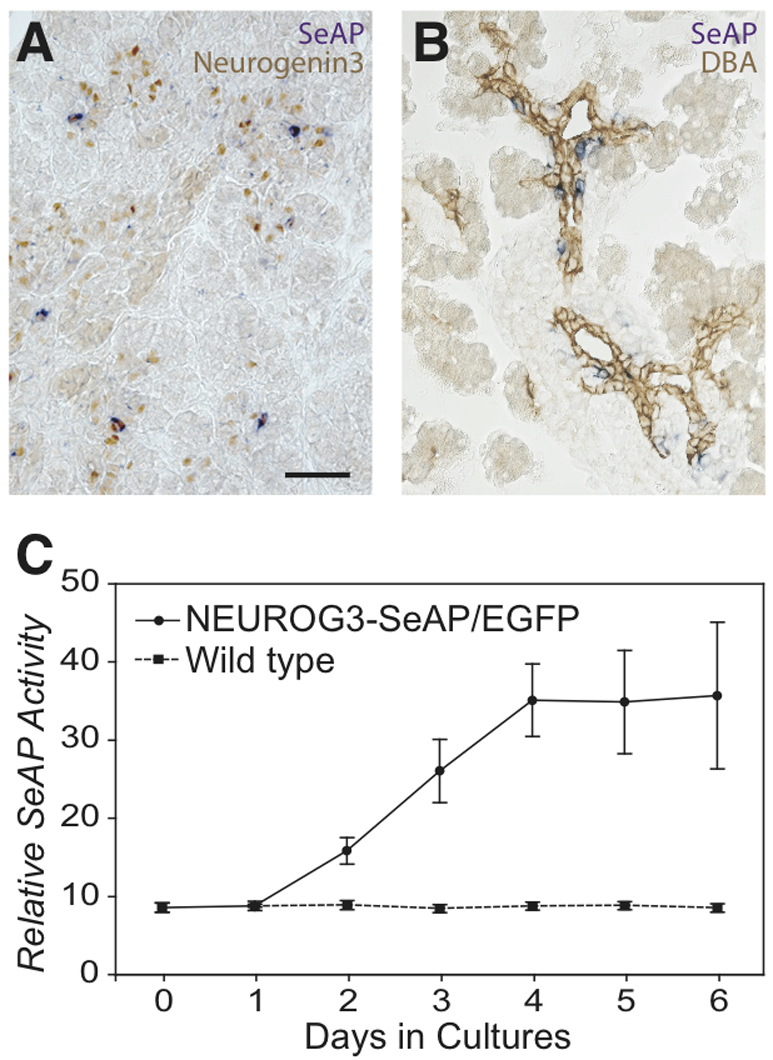
The SeAP transgene is expressed in the endocrine lineage the embryonic pancreas of BAC NEUROG3-SeAP/EGFP transgenic mice. (A,B) Enzymatic SeAP activity (purple) was colocalized with immunohistochemical staining of endogenous neurogenin-3 (brown, A) in E14.5 fetal pancreases or ductal marker DBA lectin (brown, B) in E18.5 fetal pancreases. Scale bar: 50 μm. (C) Pancreases were harvested from E12.5 embryos and placed in culture at day 1. At 24-hour intervals, SeAP activity in the media was assayed [reported as relative light units (RLUs)], and media was replaced. The solid line indicates media cultured with transgenic pancreases; the dashed line indicates media cultured with non-transgenic pancreases. Data represent mean ± s.e.m. from six pancreases in each group.
When placed in organ culture, E12.5 fetal pancreases secreted SeAP into the media, reaching a maximum in 4 days (Fig. 4C), whereas no SeAP activity was detected in media from cultured pancreases from non-transgenic littermates.
The transgenes are expressed in the adult pancreas
Transgene expression persisted at low levels in the adult, as demonstrated by the presence of detectable levels of SeAP activity in the serum (Fig. 5). It is not clear whether this activity originated from expression in endocrine progenitors in the gut, or persistent expression in the pancreas, because adult islets express low but consistently detectable levels of neurogenin-3 mRNA (Fig. 1B) (Wang et al., 2009). After 50% pancreatectomy, serum SeAP levels initially fell at 24 hours, but recovered within 2 weeks without evidence of a spike in neurogenin-3 gene expression (Fig. 5A). To explore the possibility that the surgery reduced the contribution from the gut to serum SeAP levels, we also measured the tissue SeAP levels in gut. Transgenic mice that underwent a 50% pancreatectomy had no difference in gut SeAP content compared with mice subjected to a sham operation (26672±3207 light units/μg protein in sham-operated animals vs 28674±3055 in pancreatectomized animals). By contrast, when the pancreatic duct was ligated, SeAP levels rose relative to sham-operated animals and peaked at 2 weeks after the placement of the ligation (Fig. 5B).
Fig. 5.

SeAP activity falls in the serum of adult BAC NEUROG3-SeAP/EGFP transgenic mice after partial pancreatectomy but increases after pancreatic duct ligation. Day 0 is prior to surgery. (A) The white bar at day 0 shows the background signal for SeAP activity in the serum of non-transgenic mice. Gray bars show the activity of SeAP in the serum of transgenic animals that underwent a sham operation. Black bars show the activity of SeAP in the serum of transgenic animals that underwent partial pancreatectomy. Data represent mean + s.e.m. from three mice in each treated group, and four mice in the untreated, non-transgenic group. ***P<0.001 compared with serum from sham-operated mice by Student’s t-test. (B) The white bars show the background signal for SeAP activity in the serum of control transgenic mice that did not have surgery. Gray bars show the activity of SeAP in the serum of animals that underwent a sham operation. Black bars show the activity of SeAP in the serum of animals that underwent duct ligation. Data represent mean + s.e.m. from six mice in the untreated group, and seven mice in each treated group. *P<0.03, **P<0.01, ***P<0.001 by Student’s t-test for the comparisons indicated.
GDF11 and Notch signaling regulate the SeAP/EGFP transgene in fetal pancreas explants
To test whether potential regulators of neurogenin-3 could influence expression of the transgene in vitro, we first measured SeAP activity in the media of fetal pancreatic bud harvested at E12.5 and cultured with TGFβ family member GDF11. Treatment with GDF11 for 4–6 days strongly inhibited SeAP secretion (Fig. 6A).
Fig. 6.

GDF11 and Notch signaling regulate the SeAP transgene in fetal pancreas explants. Regulation of SeAP secretion from cultured fetal pancreases from NEUROG3-SeAP/EGFP transgenic mice. (A) Pancreases were harvested from E12.5 embryos and treated with 250 ng/ml GDF11 (dashed lines) or control media (solid lines) for 6 days. SeAP activity was assayed on the days of culture shown. Data represent mean ± s.e.m. from three pancreases in each group. (B,C) Pancreases were harvested from E11.5 embryos (B) or E12.5 embryos (C), placed in culture and treated with the γ-secretase inhibitor DAPT at 50 mM in 0.1% DMSO (black bars) or 0.1% DMSO alone (white bars). SeAP activity was assayed after 48 hours of treatment. Data represent mean + s.e.m. for the number of cultured pancreases shown.
Next, we tested the regulation of the transgene by Notch signaling by blocking signaling through Notch receptors with the γ-secretase inhibitor DAPT (Dovey et al., 2001). Pancreatic buds were harvested from E11.5 transgenic embryos and placed in organ culture. DAPT was added after 24 hours in culture (equivalent to E12.5). An initial dose-response curve was performed, and a near maximal effect was seen between 50 and 100 nM DAPT (data not shown). Subsequent experiments used 50 nM DAPT. Treatment of transgenic pancreatic buds harvested at E11.5 with DAPT caused a fourfold increase in SeAP in the culture media after 48 hours (Fig. 6B). Interestingly, pancreatic buds harvested 24 hours later, at E12.5, gave very little response to DAPT (Fig. 6C).
After SeAP measurement, the number of EGFP-expressing cells from the cultured buds harvested at E11.5 was assayed by FACS (Fig. 7). From 42 pancreases cultured without DAPT, 10,000 of 253,000 cells were EGFP positive (4.0%). From 47 pancreases cultured with DAPT, 17,000 of 155,000 cells were EGFP positive (11.0%). These results were confirmed by direct examination of the buds: treated buds at 48 hours contained an increased number of neurogenin-3-expressing cells and endocrine cells relative to untreated buds, as detected by immunohistochemistry (data not shown).
Fig. 7.

Blocking Notch signaling increases the number of EGFP-expressing cells in fetal pancreas explants. Pancreases were harvested from E11.5 embryos, placed in culture and treated with 50 mM DAPT in 0.1% DMSO (A) or 0.1% DMSO alone (B). After 48 hours of treatment, cultured pancreases were dispersed into single cells, pooled and sorted by FACS to quantify the number of EGFP-expressing cells. Cells are separated on the y-axis by side scatter (SSC) and on the x-axis by EGFP fluorescence.
DISCUSSION
Neurogenin-3 is both necessary and sufficient to drive the early pancreatic epithelial cells to differentiate into islet cells, and uniquely marks the transient endocrine progenitor cells (Apelqvist et al., 1999; Gradwohl et al., 2000; Schwitzgebel et al., 2000). We have developed a BAC transgenic mouse model in which two gene products, SeAP and EGFP, function as reporters for neurogenin-3 gene expression, and have used it to assess the expression of neurogenin-3 in intact cells, in living mice and in pancreatic organ cultures. In this model, transgene expression recapitulates normal neurogenin-3 expression, albeit with a slight delay.
The lack of complete overlap between the expression of the EGFP and SeAP transgene products and native neurogenin-3 protein has several possible explanations; however, because this phenomenon has been observed with several different mouse lines using different promoters (Lee et al., 2001; Gu et al., 2002), different transgenes (Lee et al., 2001; Gu et al., 2002; Mellitzer et al., 2004) and even insertion into the native gene (Lee et al., 2002; Mellitzer et al., 2004), this divergence most probably results from differences among the reporter proteins and neurogenin-3 in their rates of accumulation and removal, and in their detection sensitivity. EGFP has a prolonged half-life in eukaryotic cells, and we found that immunofluorescent detection further increased the ability to detect low levels of remaining EGFP in cells (data not shown). By contrast, the rapid secretion of SeAP shortened the period of detectable expression of this reporter in cells and a tissue half-life more closely approximating that of neurogenin-3. These data show that neurogenin-3 protein accumulates very rapidly, but briefly, and is rapidly degraded. In fact, neurogenin-3 shuts off its own expression (Smith et al., 2004). Once neurogenin-3 activates the endocrine differentiation program, it is quickly removed.
Although dramatically reduced, neurogenin-3 expression is not completely extinguished in the adult pancreas, because we can still detect the mRNA in adult islets (Fig. 2), as seen before (Dror et al., 2007; Wilson et al., 2005; Wang et al., 2009). SeAP activity in the serum of adult transgenic animals also confirms neurogenin-3 gene activity in the adult, but some of this activity might derive from the neurogenin-3-expressing cells of the gut (Jenny et al., 2002). Interestingly, within 24 hours after 50% pancreatectomy in adult mice, neurogenin-3 gene expression, as indicated by serum SeAP activity, was reduced by half, suggesting that the pancreas contributes a significant fraction of the circulating SeAP activity in the adult transgenic mice. Although partial pancreatectomy induces a compensatory increase in the remaining islet cell population (Lee et al., 1989), we did not detect any transient increase post-surgery that would suggest that neurogenin-3 gene expression and islet cell neogenesis is reactivated during recovery from pancreatic damage, consistent with a previous study that also did not observe any increase in neurogenin-3 expression post-pancreatectomy (Lee et al., 2006). However, we cannot rule out the possibility that the decrease in background SeAP activity caused by the partial pancreatectomy could obscure a post-pancreatectomy peak in SeAP secretion. We did, however, detect a peak in circulating SeAP activity after duct ligation in adult mice. In this model, we did not see a decrease in SeAP after surgery as we did with partial pancreatectomy, perhaps because islet mass is never reduced in this model. The peak in SeAP at 7 and 14 days post-ligation is consistent with the previous demonstration that new islet cells differentiate from transiently induced neurogenin-3-expressing progenitors after duct ligation (Xu et al., 2008), and that these new islet cells derive from duct cells (Inada et al., 2008).
It must be kept in mind, of course, that the expression of any surrogate marker gene can vary relative to the true gene for any of a variety of reasons. In the case of SeAP, its expression might decrease in the setting of endoplasmic reticulum (ER) stress (Hiramatsu et al., 2006), and β-cells with their high rates of insulin synthesis are particularly susceptible to ER stress (Laybutt et al., 2007).
Several groups have reported that the Notch signaling pathway restricts the expression of neurogenin-3 (Apelqvist et al., 1999; Jensen et al., 2000b; Lee et al., 2001; Hald et al., 2003; Murtaugh et al., 2003). We also found that inhibition of Notch signaling with DAPT caused an increase in neurogenin-3 expression, but only within an early and brief developmental window. Stimulation of neurogenin-3 expression by DAPT was markedly attenuated by E13.5, just at the start of the secondary transition and the normal peak of neurogenin-3 expression. The data in Fig. 6 suggests three possible interpretations: first, there might simply be fewer DAPT-sensitive progenitor cells present at E13.5 relative to E12.5. Such differences might reflect distinct windows during which neurogenin-3 expression is normally activated (Villasenor et al., 2008) and possibly during which distinct endocrine cell types are generated (Johansson et al., 2007). Second, most of the DAPT-sensitive progenitor cells might already be in the process of activating neurogenin-3 expression at E13.5. Finally, the high levels of SeAP secreted by the E13.5 pancreases might obscure the additional promoter activation and SeAP secretion induced by DAPT treatment. To distinguish these possibilities, it would be interesting to test more mature fetal pancreases in which endogenous neurogenin-3 expression has waned, but intact older pancreases are difficult to maintain in culture. Alternatively, one could test purified populations of fetal or adult pancreatic cells for the ability to activate neurogenin-3 expression in response to DAPT or other stimulators of neurogenin-3 expression.
Even if blockade of Notch signaling alone is insufficient to activate neurogenin-3 expression and initiate islet cell neogenesis in mature pancreatic cells, activation might be possible by modulating other signaling pathways, including GDF11-TGFβ (Yamaoka et al., 1998; Hald et al., 2003; Kodama et al., 2005; Baeyens et al., 2006), either alone or in conjunction with Notch blockade. The present study demonstrates the utility of a model that permits the facile measurement of neurogenin-3 expression in live animals and intact cells. This model can be used to dissect the mechanisms that control the expression of neurogenin-3 in vivo, and to screen for molecules that control neurogenin-3 expression and thereby control the formation of islet cells. Therefore this model provides a tool both for understanding how new islet cells differentiate, and for applying that knowledge to the testing of molecules that could be used to generate new islet cells for patients with diabetes.
METHODS
BAC transgene construct
The human β-globin gene first intron and SeAP gene (Clontech, Mountain View, CA) were inserted upstream of the Encephalomyocarditis virus (ECMV) IRES in pIRES2-EGFP (Clontech). 380 bp of 5′-flanking sequence from the NEUROG3 gene were inserted upstream of the β-globin gene intron, and 1.3 kb of 3′-flanking sequence were inserted downstream of the simian virus 40 (SV40) late gene polyadenylation signal (polyA) at the 3′ end of the EGFP gene. This construct was then used for homologous recombination (Yang et al., 1997) into the NEUROG3 BAC (RP11-343J3; Sanger Institute, UK) such that the intron-SeAP-IRES-EGFP-polyA replaced the coding sequence for neurogenin-3 (Fig. 1). Recombined BAC clones were selected by chloramphenicol and kanamycin treatment, and confirmed by restriction fragment length analysis on pulse field gel electrophoresis and by sequencing.
Mice
Mice were housed on a 12-hour light-dark cycle with controlled climate, and all studies involving mice followed the guidelines of the University of California San Francisco Institutional Animal Care and Use Committee (see http://www.research.ucsf.edu/aw/Policies/awPolicies.asp). Noon of the day on which the vaginal plug was discovered was considered E0.5 in the timing of embryo collection. CD1 mice were obtained from Charles River Laboratories and used for all studies on normal mice.
BAC NEUROG3-SeAP/EGFP DNA was purified with the QIAGEN Large-Construct kit (QIAGEN, Valencia, CA) and dialysis. Transgenic mice were generated by pronuclear injection of intact circular BAC DNA (∼0.5–2 ng/μl) into oocytes from B6/SJL F1 parents. Genotypes were determined by PCR analysis of genomic DNA from tail biopsies. Three transgenic founders were obtained, and two of these were successfully bred and used in the studies described here. Line 27 was used for Fig. 1C,D, Fig. 2D–L, Fig. 4A,B, Fig. 5B and Fig. 6A. All other data were generated with line 29.
For adult mouse survival surgeries, mice were anesthetized by intraperitoneal injection of sodium pentobarbital. The abdomen was shaved and a mid-line incision was made. Intestine and pancreas was detached from loose connective tissue with cotton sponge and Q-tips.
For partial pancreatectomy, after ligation of small arteries and vessels, the spleen and the distal 50% of the pancreas were carefully removed. The surgical field was checked for bleeding and the abdominal incision was sutured. The animals were monitored to ensure recovery from surgery without signs of distress. For sham surgery, the same procedure was followed, except that after mobilization of the intestine and pancreas, the abdominal incision was closed and sutured without removal of any portion of the pancreas.
For duct ligation, 8-week-old male BAC NEUROG3-SeAP/EGFP homozygous transgenic mice were treated as described previously (Wang et al., 1995). The duct from the splenic lobe (tail) of pancreas was isolated and ligated with wax-coated and braided silk (Sofsilk; Fisher Scientific, Pittsburgh, PA). The same procedure without ligation was performed in sham-operated animals. The proper ligation was confirmed by necropsy at the end of experiments.
Fetal pancreatic bud culture
Pancreatic buds from BAC NEUROG3-SeAP/EGFP transgenic mouse embryos were harvested at E11.5 or E12.5. Pancreatic primordia were manually micro-dissected from stomach, duodenal loop and spleen, and placed on Matrigel matrix without phenol red (BD Biosciences, San Jose, CA) contained within Millicell culture plate inserts (Millipore, Billerica, MA). The explants were incubated in 50% DME H-16 supplemented with non-essential amino acids, 1.4 g/l glucose, 0.11 g/l sodium pyruvate, 0.365 g/l linoleic acid, 1.2 g/l NaHCO3, 10% fetal bovine serum (FBS), penicillin/streptomycin, fungizone and Insulin/Transferrin/Selenium supplement (Sigma-Aldrich, St Louis, MO) in 12-well plates at 37°C in 5% CO2.
GDF11 (R&D Systems, Minneapolis, MN) was reconstituted in phosphate buffered saline (PBS) containing 0.1% bovine serum albumin (BSA) and 4 mM HCl, and added to the media to yield a final concentration of 250 ng/ml. The γ-secretase inhibitor DAPT (7{N-[N-(3,5-difluorophenyl)-L-alanyl]-S-phenyl-glycine t-butyl ester}) (Calbiochem, San Diego, CA) was diluted from stock solutions in dimethylsulfoxide (DMSO) to yield a final concentration equal to 0.1% DMSO in media.
SeAP assay
SeAP activity was measured with the Great EscApe SeAP chemiluminescence detection kit (Clontech). SeAP is heat stable, so endogenous alkaline phosphatase activity was destroyed by heat treatment of the samples at 65°C for 30 minutes. Embryonic pancreas or adult tissues were homogenized in RIPA buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate and 0.1% SDS). 25 μl of homogenate, culture medium or serum was used for each assay, and all assays were performed in duplicate. Blood samples were obtained from adult mouse tail veins.
Immunohistochemical staining
For direct detection of native EGFP fluorescence in Fig. 2A,B, E15.5 and E16.5 pancreases were harvested in PBS and mounted with VECTASHIELD (Vector Labs, Burlingame, CA).
For the whole-mount staining in Fig. 2C, pancreases were fixed for 2 hours at room temperature with 4.0% paraformaldehyde (PFA) in PEM [0.1 M PIPES (1,4-piperazinediethanesulfonic acid), 1.0 mM MgSO4, 2 mM EGTA, pH to 7 with NaOH] and washed in PBS before incubating overnight at 4°C in primary antibody diluted with PBSTS (0.3% Triton X-100 and 4% goat serum in PBS). After removal of the primary antibody, the tissue was washed in PBS containing 0.3% Triton X-100, and secondary antibodies diluted in PBSTS were added and incubated for 4 hours at 4°C prior to washing in PBS and mounting on slides with VECTASHIELD (Vector Labs). Primary antibodies were used for whole-mount staining at the following dilutions: hamster anti-mucin-1 Ab-5 (Lab Vision, Fremont, CA), 1:200; rabbit anti-GFP (MBL International, Woburn, MA), 1:500. Secondary antibodies were used for whole-mount staining at the following dilutions: fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA), 1:200; aminomethylcoumarin acetate (AMCA)-conjugated anti-hamster IgG (Jackson ImmunoResearch), 1:50. All slides were visualized with a Zeiss Axioskop II microscope and imaging with a Hamamatsu ORCA100 digital camera and Open Lab software.
For frozen sections in Fig. 2D–I, Fig. 3 and supplementary material Fig. S1, pancreases were fixed in 4% PFA-PBS at room temperature for 2 hours. The tissues were rinsed in PBS and transferred into 30% sucrose-PBS overnight at 4°C. They were embedded in O.C.T. compound (Sakura Finetek USA, Torrance, CA), frozen at −80°C and cut in 10-μm sections. Sections were washed and treated with 1% donkey serum for 30 minutes and incubated overnight at 4°C with primary antibodies. After removal of the primary antibody, sections were washed in PBS, and secondary antibodies were added and incubated for 1 hour at room temperature. Primary antibodies and Dolichos biflorus agglutinin (DBA) lectin were used for staining frozen sections at the following dilutions: mouse anti-insulin (Sigma-Aldrich), 1:2000; mouse anti-glucagon antibody (Sigma-Aldrich), 1:2000; rabbit anti-chromogranin-A antibody (Lab Vision), 1:200; rabbit anti-GFP (MBL International), 1:500; guinea-pig anti-neurogenin-3 (Schwitzgebel et al., 2000), 1:2000; FITC-conjugated DBA lectin (Vector Labs), 1:200. Secondary antibodies (Jackson ImmunoResearch) were used for staining frozen sections at the following dilutions: Cy3-conjugated anti-guinea-pig IgG, 1:400; AMCA-conjugated anti-mouse IgG, 1:50; FITC-conjugated anti-rabbit IgG, 1:200. Slides were washed in PBS and mounted with VECTASHIELD prior to visualization with a Zeiss Axioskop II microscope.
For chromogenic detection of SeAP activity, frozen sections were first incubated at 65°C in PBS for 30 minutes to inactivate endogenous alkaline phosphatase activity and were subsequently incubated with NBT/BCIP (nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl-phosphate; Roche Diagnostic, Indianapolis, IN) in 0.1 M Tris-HCl, pH 9.5, 0.1 M NaCl and 50 mM MgCl2 overnight at room temperature. After washing with PBS, the primary anti-neurogenin-3 antibody and biotinylated DBA lectin was used as described above, and peroxidase staining was performed by using the avidin biotin complex (ABC) Elite Kit (Vector Labs).
RNA isolation and RT-PCR
Adult mouse islets were handpicked after collagenase digestion of the isolated pancreas and purification on a Ficoll gradient (Giddings et al., 1985). Total RNA was isolated with the RNAqueous kit (Ambion, Austin, TX) from tissues at the indicated dates and from adult islets. First-strand cDNA was prepared from 1 μg of total RNA with the Invitrogen SuperScript II first strand cDNA synthesis kit (Invitrogen, Carlsbad, CA) and oligo-dT primers. Quantification of neurogenin-3 cDNA was performed by TaqMan PCR on an Applied Biosystems Prism 7700HT sequence detection system as previously described (Wilson et al., 2005) and reported relative to levels of the cDNA encoding mouse histone H3.3a. The following probe and primer sets were used: 5′-ATCCATCACT-TTTTCCAGGGTG-3′ (neurogenin-3 forward primer), 5′-TCATCTATGGGCCAAGAGCTG-3′ (neurogenin-3 reverse primer), 5′-AATCCAGTGTTGCGTCTTACCTCACTGGC-3′ (neurogenin-3 TaqMan probe), CGCTTCCAGAGTGCAGCTATT-3′(Histone3.3A forward primer), 5′-TCTTCAAAAAGGCCAACCAGA-3′ (Histone3.3A reverse primer), 5′-GCCTCACTTGCCTCCTGCAAAGCA-3′ (Histone3.3A TaqMan probe).
Quantification of SeAP/EGFP cDNA was performed with SYBR Green using Fast SYBR Master Mix (Applied Biosystems) and reported relative to levels of the cDNA encoding mouse β-actin. The following primer sets were used: 5′-TGTGCTGCTCACCGAGGCC-3′ (beta-actin forward primer), 5′-CGGAGTCCATCACAATGCCTG-3′ (beta-actin reverse primer), 5′-CCTACGGCGTGCAGTGCTTCAGC-3′ (EGFP forward primer), 5′-GGGTCTTGTAGTTGCCGTCGTCC-3′ (EGFP reverse primer).
FACS
Freshly or cultured fetal pancreases were digested in Earle’s balanced salt solution (EBSS) (HyClone, South Logan, UT), containing 2.5 mg/ml collagenase (Roche Diagnostics, Indianapolis, IN), 0.1 mg/ml DNase I (Sigma-Aldrich) and 1% fetal calf serum (FCS). Dissociated cells were collected and treated with 0.5% trypsin-EDTA for 10 minutes at 37°. Cells were washed with complete Dulbucco’s modified Eagle’s medium (DMEM) with 10% FCS, and the trypsin-EDTA digestion was repeated. Dissociated cells were washed two more times before resuspension in Ca/Mg-free PBS containing 0.1% BSA and 0.5 mM EDTA. Cells were subjected to cell sorting in PBS on a MoFlo high performance cell sorter (DakoCytomation, Carpinteria, CA) based on side scatter (SSC) and EGFP florescence, and resuspended immediately in DMEM containing 20% BSA.
Acknowledgments
We thank Gerold Grodsky and members of the German laboratory for helpful advice and criticism. This work was supported by Larry L. Hillblom Foundation grants no. 2002/1(E) and no. 2007/1B, a grant from the Nora Eccles Treadwell Foundation, NIH grant DK21344, cores from NIH grant P30 DK063720, Juvenile Diabetes Research Foundation fellowship awards 3-2003-213 (Y.S.) and 3-2004-276 (F.C.L.), and an American Diabetes Association Mentor-Based Postdoctoral Fellowship grant (Y.K.).
Footnotes
COMPETING INTERESTS
M.S.G. is an the inventor on one issued US patent for human neurogenin-3 and two issued US patents for the use of neurogenin-3 for the production of insulin-producing cells. All of these patents are held by the University of California.
AUTHOR CONTRIBUTIONS
Y.S., Y.K. and M.S.G. conceived and designed the experiments; Y.S., Y.K., D.W.S., F.C.L., N.K., J.W. and S.Z. performed the experiments; Y.S., Y.K. and M.S.G. analyzed the data; and Y.S., Y.K. and M.S.G. wrote the paper.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.002998/-/DC1
REFERENCES
- Apelqvist A., Li H., Sommer L., Beatus P., Anderson D. J., Honjo T., Hrabe de Angelis M., Lendahl U., Edlund H. (1999). Notch signaling controls pancreatic cell differentiation. Nature 400, 877–881 [DOI] [PubMed] [Google Scholar]
- Baeyens L., Bonne S., German M. S., Ravassard P., Heimberg H., Bouwens L. (2006). Ngn3 expression during postnatal in vitro beta cell neogenesis induced by the JAK/STAT pathway. Cell Death Differ. 13, 1892–1899 [DOI] [PubMed] [Google Scholar]
- Dichmann D. S., Miller C. P., Jensen J., Scott Heller R., Serup P. (2003). Expression and misexpression of members of the FGF and TGFbeta families of growth factors in the developing mouse pancreas. Dev. Dyn. 226, 663–674 [DOI] [PubMed] [Google Scholar]
- Dovey H. F., John V., Anderson J. P., Chen L. Z., de Saint Andrieu P., Fang L. Y., Freedman S. B., Folmer B., Goldbach E., Holsztynska E. J., et al. (2001). Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J. Neurochem. 76, 173–181 [DOI] [PubMed] [Google Scholar]
- Dror V., Nguyen V., Walia P., Kalynyak T. B., Hill J. A., Johnson J. D. (2007). Notch signalling suppresses apoptosis in adult human and mouse pancreatic islet cells. Diabetologia 50, 2504–2515 [DOI] [PubMed] [Google Scholar]
- Gasa R., Mrejen C., Leachman N., Otten M., Barnes M., Wang J., Chakrabarti S., Mirmira R., German M. (2004). Proendocrine genes coordinate the pancreatic islet differentiation program in vitro. Proc. Natl. Acad. Sci. USA 101, 13245–13250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giddings S. J., Chirgwin J., Permutt M. A. (1985). Evaluation of rat insulin messenger RNA in pancreatic and extra pancreatic tissues. Diabetologia 28, 343–347 [DOI] [PubMed] [Google Scholar]
- Gradwohl G., Dierich A., LeMeur M., Guillemot F. (2000). neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl. Acad. Sci. USA 97, 1607–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G., Dubauskaite J., Melton D. A. (2002). Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129, 2447–2457 [DOI] [PubMed] [Google Scholar]
- Hald J., Hjorth J. P., German M. S., Madsen O. D., Serup P., Jensen J. (2003). Activated Notch1 prevents differentiation of pancreatic acinar cells and attenuate endocrine development. Dev. Biol. 260, 426–437 [DOI] [PubMed] [Google Scholar]
- Harmon E. B., Apelqvist A. A., Smart N. G., Gu X., Osborne D. H., Kim S. K. (2004). GDF11 modulates NGN3+ islet progenitor cell number and promotes beta-cell differentiation in pancreas development. Development 131, 6163–6174 [DOI] [PubMed] [Google Scholar]
- Heremans Y., Van De Casteele M., in’t Veld P., Gradwohl G., Serup P., Madsen O., Pipeleers D., Heimberg H. (2002). Recapitulation of embryonic neuroendocrine differentiation in adult human pancreatic duct cells expressing neurogenin 3. J. Cell Biol. 159, 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiramatsu N., Kasai A., Hayakawa K., Nagai K., Kubota T., Yao J., Kitamura M. (2006). Secreted protein-based reporter systems for monitoring inflammatory events: critical interference by endoplasmic reticulum stress. J. Immunol. Methods 315, 202–207 [DOI] [PubMed] [Google Scholar]
- Huang H. P., Liu M., El-Hodiri H. M., Chu K., Jamrich M., Tsai M. J. (2000). Regulation of the pancreatic islet-specific gene BETA2 (neuroD) by neurogenin 3. Mol. Cell. Biol. 20, 3292–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada A., Nienaber C., Katsuta H., Fujitani Y., Levine J., Morita R., Sharma A., Bonner-Weir S. (2008). Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc. Natl. Acad. Sci. USA 105, 19915–19919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemin P., Durviaux S. M., Jensen J., Godfraind C., Gradwohl G., Guillemot F., Madsen O. D., Carmeliet P., Dewerchin M., Collen D., et al. (2000). Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol. Cell. Biol. 20, 4445–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenny M., Uhl C., Roche C., Duluc I., Guillermin V., Guillemot F., Jensen J., Kedinger M., Gradwohl G. (2002). Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. EMBO J. 21, 6338–6347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen J., Heller R. S., Funder-Nielsen T., Pedersen E. E., Lindsell C., Weinmaster G., Madsen O. D., Serup P. (2000a). Independent development of pancreatic alpha- and beta-cells from neurogenin3-expressing precursors: a role for the notch pathway in repression of premature differentiation. Diabetes 49, 163–176 [DOI] [PubMed] [Google Scholar]
- Jensen J., Pedersen E. E., Galante P., Hald J., Heller R. S., Ishibashi M., Kageyama R., Guillemot F., Serup P., Madsen O. D. (2000b). Control of endodermal endocrine development by Hes-1. Nat. Genet. 24, 36–44 [DOI] [PubMed] [Google Scholar]
- Johansson K. A., Dursun U., Jordan N., Gu G., Beermann F., Gradwohl G., Grapin-Botton A. (2007). Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev. Cell 12, 457–465 [DOI] [PubMed] [Google Scholar]
- Kodama S., Toyonaga T., Kondo T., Matsumoto K., Tsuruzoe K., Kawashima J., Goto H., Kume K., Kume S., Sakakida M., et al. (2005). Enhanced expression of PDX-1 and Ngn3 by exendin-4 during beta cell regeneration in STZ-treated mice. Biochem. Biophys. Res. Commun. 327, 1170–1178 [DOI] [PubMed] [Google Scholar]
- Laybutt D. R., Preston A. M., Akerfeldt M. C., Kench J. G., Busch A. K., Biankin A. V., Biden T. J. (2007). Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 50, 752–763 [DOI] [PubMed] [Google Scholar]
- Lee C. S., Perreault N., Brestelli J. E., Kaestner K. H. (2002). Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes Dev. 16, 1488–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. S., De Leon D. D., Kaestner K. H., Stoffers D. A. (2006). Regeneration of pancreatic islets after partial pancreatectomy in mice does not involve the reactivation of neurogenin-3. Diabetes 55, 269–272 [PubMed] [Google Scholar]
- Lee H. C., Bonner-Weir S., Weir G. C., Leahy J. L. (1989). Compensatory adaption to partial pancreatectomy in the rat. Endocrinology 124, 1571–1575 [DOI] [PubMed] [Google Scholar]
- Lee J. C., Smith S. B., Watada H., Lin J., Scheel D., Wang J., Mirmira R. G., German M. S. (2001). Regulation of the pancreatic pro-endocrine gene neurogenin3. Diabetes 50, 928–936 [DOI] [PubMed] [Google Scholar]
- Lynn F. C., Smith S. B., Wilson M. E., Yang K. Y., Nekrep N., German M. S. (2007). Sox9 coordinates a transcriptional network in pancreatic progenitor cells. Proc. Natl. Acad. Sci. USA 104, 10500–10505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestro M. A., Boj S. F., Luco R. F., Pierreux C. E., Cabedo J., Servitja J. M., German M. S., Rousseau G. G., Lemaigre F. P., Ferrer J. (2003). Hnf6 and Tcf2 (MODY5) are linked in a gene network operating in a precursor cell domain of the embryonic pancreas. Hum. Mol. Genet. 12, 3307–3314 [DOI] [PubMed] [Google Scholar]
- Mellitzer G., Martin M., Sidhoum-Jenny M., Orvain C., Barths J., Seymour P. A., Sander M., Gradwohl G. (2004). Pancreatic islet progenitor cells in neurogenin 3-yellow fluorescent protein knock-add-on mice. Mol. Endocrinol. 18, 2765–2776 [DOI] [PubMed] [Google Scholar]
- Mellitzer G., Bonne S., Luco R. F., Van De Casteele M., Lenne-Samuel N., Collombat P., Mansouri A., Lee J., Lan M., Pipeleers D., et al. (2006). IA1 is NGN3-dependent and essential for differentiation of the endocrine pancreas. EMBO J. 25, 1344–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh L. C., Stanger B. Z., Kwan K. M., Melton D. A. (2003). Notch signaling controls multiple steps of pancreatic differentiation. Proc. Natl. Acad. Sci. USA 100, 14920–14925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwitzgebel V. M., Scheel D. W., Conners J. R., Kalamaras J., Lee J. E., Anderson D. J., Sussel L., Johnson J. D., German M. S. (2000). Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development 127, 3533–3542 [DOI] [PubMed] [Google Scholar]
- Slack J. M. (1995). Developmental biology of the pancreas. Development 121, 1569–1580 [DOI] [PubMed] [Google Scholar]
- Smith S. B., Gasa R., Watada H., Wang J., Griffen S. C., German M. S. (2003). Neurogenin3 and hepatic nuclear factor 1 cooperate in activating pancreatic expression of Pax4. J. Biol. Chem. 278, 38254–38259 [DOI] [PubMed] [Google Scholar]
- Smith S. B., Watada H., German M. S. (2004). Neurogenin3 activates the islet differentiation program while repressing its own expression. Mol. Endocrinol. 18, 142–149 [DOI] [PubMed] [Google Scholar]
- Villasenor A., Chong D. C., Cleaver O. (2008). Biphasic Ngn3 expression in the developing pancreas. Dev. Dyn. 237, 3270–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. N., Kloppel G., Bouwens L. (1995). Duct- to islet-cell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia 38, 1405–1411 [DOI] [PubMed] [Google Scholar]
- Wang S., Hecksher-Sorensen J., Xu Y., Zhao A., Dor Y., Rosenberg L., Serup P., Gu G. (2008). Myt1 and Ngn3 form a feed-forward expression loop to promote endocrine islet cell differentiation. Dev. Biol. 317, 531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Jensen J. N., Seymour P. A., Hsu W., Dor Y., Sander M., Magnuson M. A., Serup P., Gu G. (2009). Sustained Neurog3 expression in hormone-expressing islet cells is required for endocrine maturation and function. Proc. Natl. Acad. Sci. USA 106, 9715–9720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watada H., Scheel D. W., Leung J., German M. S. (2003). Distinct gene expression programs function in progenitor and mature islet cells. J. Biol. Chem. 278, 17130–17140 [DOI] [PubMed] [Google Scholar]
- Wilson M. E., Scheel D., German M. S. (2003). Gene expression cascades in pancreatic development. Mech. Dev. 120, 65–80 [DOI] [PubMed] [Google Scholar]
- Wilson M. E., Yang K. Y., Kalousova A., Lau J., Kosaka Y., Lynn F. C., Wang J., Mrejen C., Episkopou V., Clevers H. C., et al. (2005). The HMG Box transcription factor Sox4 contributes to the development of the endocrine pancreas. Diabetes 54, 3402–3409 [DOI] [PubMed] [Google Scholar]
- Xu X., D’Hoker J., Stange G., Bonne S., De Leu N., Xiao X., Van de Casteele M., Mellitzer G., Ling Z., Pipeleers D., et al. (2008). Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 132, 197–207 [DOI] [PubMed] [Google Scholar]
- Yamaoka T., Idehara C., Yano M., Matsushita T., Yamada T., Ii S., Moritani M., Hata J., Sugino H., Noji S., et al. (1998). Hypoplasia of pancreatic islets in transgenic mice expressing activin receptor mutants. J. Clin. Invest. 102, 294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. W., Model P., Heintz N. (1997). Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat. Biotechnol. 15, 859–865 [DOI] [PubMed] [Google Scholar]