

Abstract
P7 is a small membrane protein that is essential for the infectivity of hepatitis C virus. Solution-state NMR experiments on p7 in DHPC micelles, including hydrogen/deuterium exchange, paramagnetic relaxation enhancement and bicelle ‘q-titration’; demonstrate that the protein has a range of dynamic properties and distinct structural segments. These data along with residual dipolar couplings yield a secondary structure model of p7. We were able to confirm previous proposals that the protein has two transmembrane segments with a short interhelical loop containing the two basic residues K33 and R35. The 63-amino acid protein has a remarkably complex structure made up of 7 identifiable sections, four of which are helical segments with different tilt angles and dynamics. A solid-state NMR two-dimensional separated local field spectrum of p7 aligned in phospholipid bilayers provided the tilt angles of two of these segments. A preliminary structural model of p7 derived from these NMR data is presented.
Keywords: Hepatitis C Virus, p7, membrane protein, secondary structure, nuclear magnetic resonance
1. Introduction
Hepatitis C Virus (HCV), a member of the Hepacivirus Flaviviridae family of viruses, infects more than 170 million people worldwide. Infection by HCV is persistent, often leading to chronic hepatitis resulting in cirrhosis and hepatocellular carcinoma [1]. Since there is currently no vaccine for the virus and the limited available treatments are ineffective and costly [2; 3; 4; 5], studies of its proteins are essential in order to advance their potential as targets of anti-viral drugs. In HCV, a single stranded RNA of 9.6 kb is translated into a 3,000 amino acid polypeptide that is proteolytically cleaved into 10 mature proteins; 3 structural proteins (E1, E2 and core), 6 non-structural proteins (NS2-NS5) and a small 63 amino acid membrane protein (p7) that is the subject of the studies described in this article [6; 7]. p7 appears to have characteristics of a viroporin, a class of small virus-encoded hydrophobic proteins that oligomerize and form ion channels, modifying membrane permeability [8]. Although p7 does not appear to be directly involved in the replication of the virus, its role in the late steps of virus particle assembly and release make it essential to the HCV lifecycle [9; 10]. It has been proposed that p7 has a hairpin structure with a short loop containing two highly conserved basic residues between two transmembrane segments [11; 12]. It has been shown to exhibit channel activity in planar phospholipid bilayers and that this activity can be blocked by a variety of compounds including amantadine [13], amiloride [14] and a number of imminosugar derivatives [15], demonstrating its potential as a target for drugs. Recently, structural analysis of p7 by single-particle electron microscopy yielded a density map with resolution of ~16Å [16].
NMR spectroscopy is capable of describing the dynamics of p7 and determining its structure at atomic resolution. The challenge is in performing these studies in lipid environments that resemble those of native biological membranes. Previously, we described the expression, purification, and sample preparation of p7 in micelle and bilayer environments suitable for nuclear magnetic resonance (NMR) spectroscopy. The two-dimensional 1H-15N heteronuclear single quantum coherence (HSQC) spectrum of uniformly 15N labeled full-length p7 in DHPC (1,2-dihexyl-1-sn-glycero-3-phosphocholine) micelles is well resolved, and is a starting point for informative solution-state NMR experiments. Heteronuclear 1H-15N nuclear Overhauser effect (NOE) measurements are sensitive indicators of local dynamics of the polypeptide backbone [17]. The hydrogen/deuterium exchange of amide sites can be monitored for individual residues and provide information about solvent accessibility and structural stability [18]. The measurement of paramagnetic relaxation enhancement (PRE) with the use of paramagnetic probes measures the surface accessibility of individual residues [19; 20]. We find the ‘q-titration’ experiment, where the ratio of long chain to short chain lipids that assemble into bicelles is systematically varied, useful in characterizing the global and local backbone dynamics of membrane proteins [21]. The chemical shifts of backbone resonances that differ from random-coil values are useful indicators of secondary structure [22]. In addition, the measurement of residual dipolar couplings (RDCs) from weakly aligned samples in stressed polyacrylamide gels can provide angular information that provides definitive structural information about the length, relative orientation, presence of kinks, and other features of α-helices [23; 24; 25]. Although each of these methods has limitations, taken together they provide a consistent picture of the secondary structure of p7 in DHPC micelles. Combined with initial solid-state NMR results on p7 in magnetically aligned phospholipid bilayers, we present a preliminary model of the architecture of p7 in membranes environments.
2. Materials and Methods
2.1 Protein preparation
The expression and purification of isotopically labeled p7 protein was performed as previously described [26]. The DNA sequence corresponding to p7 of HCV genotype J4 was ligated into the expression vector pHLV containing the fusion partner TrpΔLE [27; 28]. The fusion protein was overexpressed in E. coli. The standard M9 minimal media included 15N-labeled ammonium sulfate and/or 13C-labeled glucose to produce uniformly isotopically labeled samples (Cambridge Isotope Laboratories, www.isotope.com). The cells were harvested by centrifugation and lysed using sonication; the inclusion bodies were then isolated and solubilized in 6 M guanidine hydrochloride and Tris buffer. The fusion protein was purified by nickel affinity chromatography. The eluted polypeptide was dialyzed against water, lyophilized, and cleaved at the single Met residue using cyanogen bromide in 70% formic acid. The cleaved material was dialyzed and then lyophilized. The polypeptide with the sequence corresponding to p7 was separated from the fusion partner by size exclusion FPLC in sodium dodecyl sulfate (SDS) and sodium phosphate buffer. The sample of p7 was then exhaustively dialyzed to remove SDS and then lyophilized. The dried powder was stored at 4°C prior to sample preparation for the NMR experiments.
2.2 NMR Sample Preparation
The samples for solution-state NMR were prepared by dissolving the protein powder in a 0.4 M solution of DHPC (Avanti Polar Lipids, Inc., www.avantilipids.com). The sample was vortexed until clear, and then diluted with water and deuterium oxide (D2O) to make the final concentration 125 mM DHPC and 10% D2O. The pH of the sample was adjusted to 4.0 with the addition of a small amount of 0.1 N NaOH, and then transferred to a standard 5 mm outer diameter NMR tube.
The samples for the deuterium fractionation experiments were prepared by first lyophilizing the NMR sample, redissolving it in various ratios of D2O to H2O, and sonicating it for 1 hour before transferring it back to the NMR tube. The 1H/15N HSQC spectra were recorded immediately. Samples were made with D2O concentrations of 10, 25, 50, 75, 90, 95 and 99%.
The samples for paramagnetic relaxation enhancement experiments were prepared by adding chelated manganese to the protein/micelle solution. Mn (II) was chelated by dissolving manganese sulfate in a solution of 0.5 M ethylenediaminetetraacetic acid (EDTA) at pH 8.0. After a white precipitate formed, the mixture was centrifuged, and the pellet containing the chelated metal Mn2+-EDTA was washed twice with methanol and once with ethanol. The dried complex was weighed, dissolved in 1 M HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer (pH 8.3), and added to the NMR sample at a final concentration of 10 mM.
The ‘q-titration’ experiments were performed by adding long-chain lipid, 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) (Avanti), to a previously prepared DHPC micelle sample. The sample was removed from the NMR tube and placed in a microcentrifuge tube. The long-chain lipid was added in powder form and the sample was heating to 42°C and vortexed until the lipid dissolved; subsequently, the sample was subjected to several temperature cycles (42°C/4°C) with vortexing to ensure homogenous bicelle formation. The pH of the sample was adjusted to 4.0, and the sample returned to the NMR tube.
The weakly aligned samples used for the RDC measurements were prepared by soaking the protein/micelle solution into dried acrylamide as described previously [29; 30]. A gel containing 5% total acrylamide and 0.15% N,N’-methylenebisacrylamide was cast in a medium-walled 3.5 mm NMR tube. The polymerization reaction was initiated by the addition of 0.05% ammonium persulfate and 0.5% N,N,N’,N’-tetramethylenediamine. The gel was removed from the tube and cut to a length of 3.0 cm. The gels were dialyzed in water for two days and then dried in an oven set to 37°C for several days. The protein-containing solution was added to the dried gel in a symmetric NMR microtube (Shigemi Inc., www.shigeminmr.com). Compression of the gel was achieved by limiting the length of the gel during the swelling step using a plunger set at the desired height of 2.1 cm. The tube was sealed with teflon tape and left to soak for 24 hours at 37°C.
The sample for solid-state NMR was prepared using the previously described protocol [31]. Purified p7 protein was dissolved in 9.5 milligrams of 1,2-di-O-hexyl-sn-glycero-3-phosphocholine (6-O-PC; in 100 ul of water with vortexing. Once dissolved, the protein/lipid micelle mixture was added to 45.6 mg of 14-O-PC in 100 ul of water. The sample containing a lipid/volume concentration of 28% and q of 3.2 was then subjected to temperature cycles (42°C/4°C) with vortexing until the mixture became clear and displayed the proper phase transition associated with bicelle formation. The pH was adjusted to 4.0 with the addition of a small amount of 0.1 NaOH.
2.3 NMR Spectroscopy
The solution-state NMR experiments were performed on a Bruker Avance II 800 MHz spectrometer (www.bruker-biospin.com) using a triple-resonance probe (1H/13C/15N) with three-axis pulsed field gradients at 50°C. The spectra were referenced to the 1H resonance of water at 4.70 ppm. For deuterium fractionation and bicelle titration experiments the fast 1H-15N HSQC pulse sequence [32] was performed with 1024 t2 points and 256 t1 points. Backbone resonance assignments were made using a standard three-dimensional HNCA experiment on uniformly 15N/13C protein samples [33]. For 1H-15N heteronuclear NOE measurements interleaved data was recorded with and without 3 seconds of saturation of the 1H resonances between scans using the pulse sequence described by Farrow et al [17].
Backbone amide J coupling values were measured for an isotropic sample and a weakly aligned sample using a modified 1H-15N in-phase/anti-phase (IPAP) HSQC pulse program (Ishii et al., 2001). Residual dipolar coupling values were measured by calculating the difference of in-phase anti-phase JD-splitting for the two samples. The RDCs were fit with a sinusoid in Matlab (Mathworks, www.mathworks.com) using a sliding window method to find the best fits.
The solid-state experiments were performed on a 700 MHz Bruker Avance spectrometer with a homebuilt double-resonance probe with a solenoid coil and strip shield [34]. The two-dimensional magic sandwich based separated local field spectroscopy (SAMMY) spectrum [35] was obtained at 42°C with 1024 t2 points and 80 t1 points.
The NMR data was processed using NMRPipe [36] and the figures prepared using Sparky (T.D. Goddard and D.G. Kneller, SPARKY 3, University of California, San Francisco).
3. Results
3.1 Assignment of p7
The backbone amide resonances in the two-dimensional 1H-15N HSQC spectrum of uniformly 15N-labeled p7 in DHPC micelles shown in Figure 1 have been assigned, as indicated by the annotations of the amino acid residues. The spectrum is remarkably well resolved for a helical membrane protein in micelles as a result of the successful optimization of protein purification, sample preparation, and experimental conditions [26]. The resonances were assigned using a combination of two- and three-dimensional experiments on uniformly and selectively isotopically labeled protein samples. The through-bond connectivities identified from a three-dimensional HNCA experiment are shown in Figure 2. In this experiment, the amide nitrogen hydrogen chemical shifts are correlated with those of the intra-residue alpha carbon and preceding (n-1) inter-residue alpha carbon to yield a spectral representation where the peaks from neighboring residues can be correlated to provide sequential resonance assignments. These results were supplemented with through-space connectivities identified in a two-dimensional HSQC-NOESY spectrum obtained on a uniformly 15N labeled sample and two-dimensional HSQC spectra obtained on selectively 15N-labeled samples to yield the complete set of assignments shown in Figure 1. Fortunately, we were able to assign all of the protein resonances without invoking other triple-resonance experiments because the spectra of p7 are extraordinarily well resolved for a helical protein.
Fig. 1.
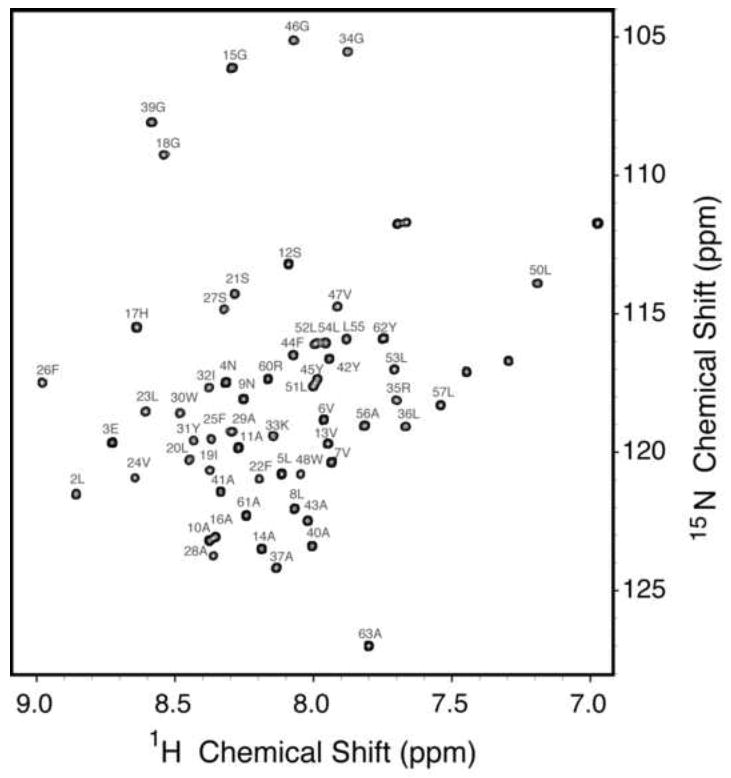
Two-dimensional 1H-15N HSQC solution-state NMR spectrum of uniformly 15N-labeled HCV p7 in DHPC micelles at pH 4.0 and 50 °C. The spectrum is fully assigned with each of the backbone amides labeled.
Fig. 2.
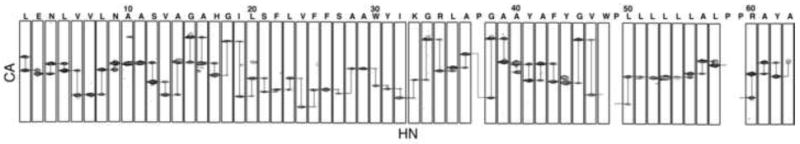
Assigned strip plots from the three-dimensional HNCA spectrum of p7. The connectivities of neighboring amino acids are shown, demonstrating how the protein resonances were assigned.
3.2 Deuterium/hydrogen fractionation
Deuterium/hydrogen solvent exchange properties of the backbone amide sites of p7 in DHPC micelles were characterized by comparing resonance intensities in two-dimensional 1H-15N HSQC spectra obtained on samples containing various ratios of H2O and D2O. The spectrum of a sample with 75% D2O (Figure 3B) contains many resonances, although those from residues in the inter-helical loop region are clearly diminished in intensity. Notably, resonances from residues near the N- and C- termini of the protein, although reduced in intensity, can be observed. In a spectrum obtained on a sample with 90% D2O (data not shown) only seven signals are present, and they are assigned to the vicinal residues 25F and 26F and the unusual stretch of leucines 50, 51, 52, 53, and 54, suggesting that these residues are part of stable structural elements.
Fig. 3.
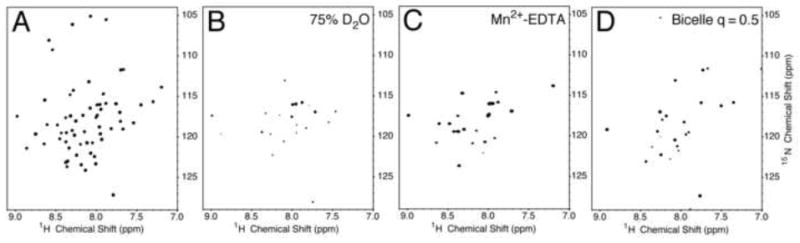
1H-15N HSQC spectra of uniformly 15N labeled HCV p7. (A) p7 in micelles in H2O. (B) p7 in micelles in D2O. (C) p7 in micelles with Mn2+-EDTA. (D) p7 in isotropic bicelles. The micelle samples contained 125 mM DHPC. For deuterium exchange experiments the sample was prepared in 75% D2O. Mn2+-EDTA at a concentration of 10 mM was added to the sample for PRE experiments. The isotropic bicelle sample contained DMPC and DHPC (q = 0.5).
3.3 Paramagnetic relaxation enhancement measurements using Mn2+-EDTA
Paramagnetic relaxation enhancement monitors solvent accessibility of residues, since chelated manganese added to the solution does not partition into the hydrophobic interior of the micelles; therefore, it only affects the relaxation properties of residues exposed to the aqueous solvent reducing their intensity through line broadening. The addition of Mn2+ complexed to EDTA demonstrates that residues in both of the termini and the loop region are exposed to the aqueous solution, on the exterior of the micelle. As a result, only resonances from residues in the trans-membrane helical regions are observed in the spectrum shown in Figure 3C.
3.4 Bicelle titration experiments
A series of two-dimensional 1H-15N HSQC spectra were used to monitor the effects of increasing q values of the bicelles on p7. The ‘q-titration’ experiment was initiated with a p7-containing bicelle sample with a q value of 0.1. As the long-chain lipid, DMPC, was added to the solution, the differential broadening of some resonances was observed in the spectrum. In the spectrum (Figure 3D) obtained from a sample with q = 0.5 there is a sharp contrast between resonances from residues in the trans-membrane helices and those of the termini and loop regions. The resonances from signals in the trans-membrane helices are broadened out completely as a consequence of their slow correlation time. Interestingly, the signals from residues 2-16 are not affected to the same extent, suggesting that there are some internal motions affecting this region of the protein.
3.5 Analysis of the data
The experimental data obtained on p7 in DHPC micelles are plotted as a function of residue number in Figure 4. Organized in this way, they demonstrate that the prevailing view of p7 as a simple ‘hairpin’ of two trans-membrane helices is too simplistic. Seven distinct structural segments of the protein identified based on the experimental data are shown schematically at the top of the Figure. Information about the structure and dynamics of the segments are contained in the NMR data and can be separated through interpretation and comparisons.
Fig. 4.
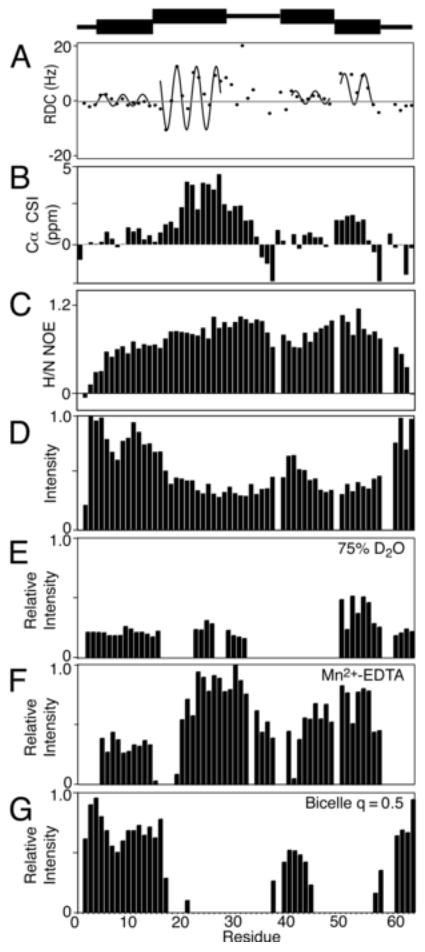
Secondary structure plots of p7 in DHPC micelles. (A) 1H-15N Residual Dipolar Couplings are plotted with the dipolar waves superimposed. (B) Cα chemical shift index values, (C) 1H-15N Heteronuclear NOEs. (D) 1H-15N HSQC intensities. (E) Relative intensity plots for a sample containing 75% D2O (F) PRE data from a sample containing 10 mM Mn2+-EDTA (G) Intensities from an isotropic bicelle sample with q = 0.5 (G). A schematic representation of the seven structural elements of p7 is at the top of the Figure.
The most informative results are in Figure 4A, which is a plot of 1H-15N RDCs as a function of residue number. The dots are the experimental data points, and the sinusoids that have been fit to the regular oscillations of the data represent Dipolar Waves that define the helical secondary structure.
The RDCs were measured from a weakly aligned sample of uniformly 15N labeled p7 in DHPC micelles in a compressed 5% acrylic acid charged gel. Based on the structural periodicity of the α-helix, 3.6 residues per turn, a set of sinusoidal waves was fit to the data and identified four helical segments, two in each of the trans-membrane helices predicted on the basis of hydropathy plots. The first dipolar wave starts at residue 6 and continues to residue 16 where there is a marked discontinuity in phase and amplitude separating it from the second helical segment between residues 17 and 27. There is an irregular pattern of RDCs between residues 28 and 36, which could not be fit with a sinusoidal wave with low RMSD. This is characteristic of an inter-helical loop, as observed in other bitopic membrane proteins. The C-terminal portion of p7, 41 to 57, is also made up of two helical segments that differ in their relative orientations. The second of these is clearly delineated by the two prolines that flank it, P49 and P58. The five residues at the N- and C- termini appear to be unstructured and mobile based on their near-zero RDC values. The RDC values of helices A and C are uniformly small, most likely due to internal motions of these helical segments.
Several other sets of data are plotted and demonstrate the consistency of the measurements. The chemical shifts of the alpha carbon resonances measured from the HNCA experiment are plotted as a function of residue number in Figure 4B. Deviation from the chemical shift index (CSI) random coil values are consistent with the presence of the alpha helices identified by the dipolar waves in Figure 4A. These data, along with the values from the 1H-15N heteronuclear-NOE measurements (Figure 4C) and the resonance intensities as a function of residues number plot (Figure 4D), suggest that the protein consists of helical segments that constitute two trans-membrane helices separated by a short, structured inter-helical loop.
The deuterium fractionation studies also identified the secondary structure elements of the protein, since hydrogens that participate in stable hydrogen bonds, like those in trans-membrane helices, preferentially bind 1H over 2H. The relative intensity plot of the sample in 75% deuterium oxide suggests that some secondary structure is present at the N-terminus (Figure 4E). This is confirmed by the paramagnetic perturbation data, which has a similar profile (Figure 4F). One notable difference in the two plots is the reduced intensity of the third helical segment in the presence of deuterium oxide, which suggests that the third helical segment is mobile and/or exposed to the aqueous solution to facilitate exchange of amide hydrogens.
The bicelle ‘q-titration’ experiment also provided information about the secondary structure of p7. By increasing the q value of the bicelle to 0.5 it is clear that the majority of residues in trans-membrane helices are immobilized in the bicelle environment and that other residues have sufficient local motions to give observable resonances in solution-state NMR experiments.
Taken together, these data indicate that p7 has a remarkably complex structure. It contains four helical segments within the two trans-membrane helices predicted by hydropathy plots. A representation of the correlated secondary structure data of p7 is shown above the plots in Figure 4 illustrating the seven distinct sections of the protein. It shows that the N- and C- termini as well as the loop containing the charged residues K33 and R35 are short segments and highlights the four helical elements of the protein sequence.
4. Discussion
The acquisition of high-resolution NMR spectra of highly pure isotopically labeled p7 in DHPC micelles provided a great deal of information about the structure and dynamics of the protein. We measured the residual dipolar couplings of weakly aligned protein in a polyacrylamide gel providing reliable data on the helical boundaries of the protein in a phospholipid environment.
These results will also contribute angular constraints for determining the full three-dimensional structure of p7. To develop a more detailed characterization of the helical segments a combination of NMR experiments were performed to look at the structural elements of the protein over a large range of timescales. By using deuterium exchange, paramagnetic relaxation enhancement and bicelle titration experiments we were able to show that the four helical segments have differences in their segmental motions even though they all reside within the hydrophobic environment of the micelle. For instance, a small portion of the N-terminal trans-membrane helix, residues 5 through 15, was found to be helical and buried in the lipid micelle but its mobility is not restricted in the formation of larger bicelles. The same is true for the third helical segment. But unlike the first helical segment, where the mobility probably results from an absence of membrane anchoring residues, the dynamics of this helix may be related to its proximity to the short interhelical loop, and amplified by the presence of a helix-breaking proline at residue 37. The second helical segment, also attached to the loop, does not show evidence of internal dynamics most likely because it is anchored at the polar/apolar interface of the lipid micelle by a tryptophan (W30) and a tyrosine (Y31). The fourth helical segment is also rigid, indicated by the remaining signals in the deuterium exchange experiments corresponding to the string of leucines in the sequence. These findings demonstrate that a hydropathy plot is not sufficient for predicting secondary structure of a protein, and that solution-state NMR can provide unique information about protein structure and dynamics.
A solid-state NMR separated local field spectrum was obtained for p7 in magnetically-aligned 14-O-PC/6-O-PC bilayers (Figure 5). The signal intensity in the two-dimensional spectrum between 80 and 110 ppm shows that the protein is aligned in the phospholipid bilayer. As described previously [37], an overlay of simulated polar index slant angle (PISA) “wheels” indicate the protein contains helices that are approximately 10° and 25° tilted from the bilayer normal. The secondary structure data demonstrates that the helix tilted 10° away from the bilayer normal consists of residues L50 through L57. Our results show that the two predicted transmembrane helices are of different lengths, and since the second helix is shorter it requires a smaller tilt angle to span the membrane. On the other hand, the longer helix can accommodate a larger tilt angle, possibly in the second helical segment. A representation of p7 in the bilayer that is consistent with the secondary structure derived from the solution-state NMR experiments obtained on micelle and isotropic bicelle samples and the solid-state NMR data obtained on a bilayer sample is shown in Figure 6. The protein has four helical segments that vary in length and dynamic properties. The interhelical loop is shown pointing toward the endoplasmic reticulum (ER) lumen and the two termini protruding out into the cytosol, as suggested in previous reports [11; 12]. This illustration serves as a model for the secondary structure of p7 in a membrane environment and sets the stage for determining the three-dimensional structure of the protein.
Fig. 5.
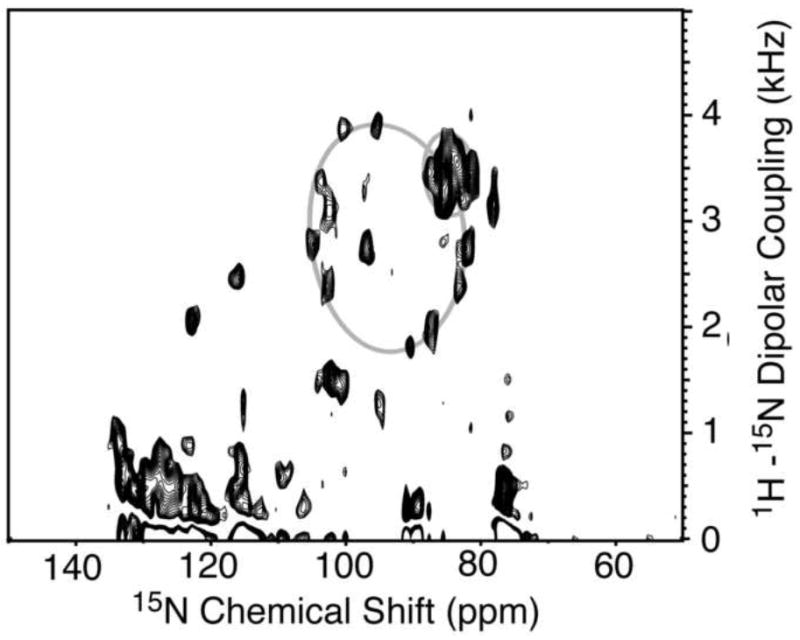
Two-dimensional solid-state SAMMY spectrum of uniform 15N-labeled p7 in magnetically-aligned 14-O-PC/6-O-PC bilayers (q = 3.2, w/v = 28%). The spectrum was obtained at 42°C at 700 MHz. Simulated PISA wheels of tilt angles 10° and 25° are superimposed on the spectrum.
Fig. 6.
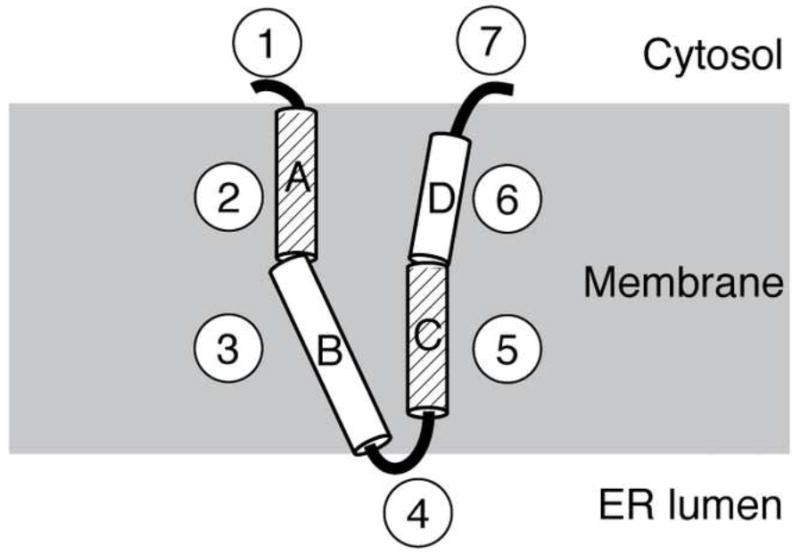
Schematic structural representation of p7 in a phospholipid bilayer. Each of the seven structural segments is shown, with the four helical segments labeled A through D. The dynamic helices, A and C, are differentiated by their diagonal line pattern. The loop containing the two basic residues is shown facing the ER lumen while the N- and C-termini are shown exposed to the cytosol. The helical segments labeled B and D are shown tilted at 25 and 10°, respectively.
p7 is not the first protein from the viroporin family to be studied in detail. A considerable amount of information has been learned about the structure and dynamics of other viroporins including vpu from HIV-1 and M2 from the influenza virus. Although the structures of these two proteins are quite different from p7, they share a number of structural and functional characteristics. We know that viroporins play a crucial role in the lifecycle of the viruses that encode them and that they make attractive targets for therapeutics. Like Vpu and M2 proteins, the channel activity of p7 is blocked by the addition of amantadine. By determining the mechanism by which this inhibition occurs through a combination of structural and functional studies it may be possible to identify additional agents that block multiple viroporins and affect the infectivity of these viruses.
Acknowledgments
We thank Yanwen Mai for assistance with the preparation of the protein samples. This research was supported by grants from the National Institutes of Health and a gift from Gilead Sciences. It utilized the Biomedical Technology Resource for NMR Molecular Imaging of Proteins at the University of California, San Diego, which is supported by grant P41EB002031.
Abbreviations
- HCV
Hepatitis C Virus
- NMR
nuclear magnetic resonance
- DHPC
(1,2-dihexyl-1-sn-glycero-3-phosphocholine)NOE, nuclear Overhauser effect
- NOE
nuclear Overhauser effect
- PRE
paramagnetic relaxation enhancement
- RDC
residual dipolar couplings
- SDS
sodium dodecyl sulfate
- D2O
deuterium oxide
- EDTA
ethylenediaminetetraacetic acid
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- 6-O-PC
1,2-di-O-hexyl-sn-glycero-3-phosphocholine
- IPAP
in-phase/anti-phase
- CSI
chemical shift index
- PISA
polar index slant angle
- ER
endoplasmic reticulum
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Choo QL, Richman KH, Han JH, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby R, Barr PJ, et al. Genetic organization and diversity of the hepatitis C virus. Proc Natl Acad Sci U S A. 1991;88:2451–5. doi: 10.1073/pnas.88.6.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McHutchison JG, Gordon SC, Schiff ER, Shiffman ML, Lee WM, Rustgi VK, Goodman ZD, Ling MH, Cort S, Albrecht JK. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N Engl J Med. 1998;339:1485–92. doi: 10.1056/NEJM199811193392101. [DOI] [PubMed] [Google Scholar]
- 3.Hadziyannis SJ, Sette H, Jr, Morgan TR, Balan V, Diago M, Marcellin P, Ramadori G, Bodenheimer H, Jr, Bernstein D, Rizzetto M, Zeuzem S, Pockros PJ, Lin A, Ackrill AM. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med. 2004;140:346–55. doi: 10.7326/0003-4819-140-5-200403020-00010. [DOI] [PubMed] [Google Scholar]
- 4.Di Bisceglie AM, Conjeevaram HS, Fried MW, Sallie R, Park Y, Yurdaydin C, Swain M, Kleiner DE, Mahaney K, Hoofnagle JH. Ribavirin as therapy for chronic hepatitis C. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:897–903. doi: 10.7326/0003-4819-123-12-199512150-00001. [DOI] [PubMed] [Google Scholar]
- 5.Davis GL, Esteban-Mur R, Rustgi V, Hoefs J, Gordon SC, Trepo C, Shiffman ML, Zeuzem S, Craxi A, Ling MH, Albrecht J. Interferon alfa-2b alone or in combination with ribavirin for the treatment of relapse of chronic hepatitis C. International Hepatitis Interventional Therapy Group. N Engl J Med. 1998;339:1493–9. doi: 10.1056/NEJM199811193392102. [DOI] [PubMed] [Google Scholar]
- 6.Selby MJ, Glazer E, Masiarz F, Houghton M. Complex processing and protein:protein interactions in the E2:NS2 region of HCV. Virology. 1994;204:114–22. doi: 10.1006/viro.1994.1515. [DOI] [PubMed] [Google Scholar]
- 7.Lin C, Lindenbach BD, Pragai BM, McCourt DW, Rice CM. Processing in the hepatitis C virus E2-NS2 region: identification of p7 and two distinct E2-specific products with different C termini. J Virol. 1994;68:5063–73. doi: 10.1128/jvi.68.8.5063-5073.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez ME, Carrasco L. Viroporins. FEBS Lett. 2003;552:28–34. doi: 10.1016/s0014-5793(03)00780-4. [DOI] [PubMed] [Google Scholar]
- 9.Sakai A, Claire MS, Faulk K, Govindarajan S, Emerson SU, Purcell RH, Bukh J. The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc Natl Acad Sci U S A. 2003;100:11646–51. doi: 10.1073/pnas.1834545100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harada T, Tautz N, Thiel HJ. E2-p7 region of the bovine viral diarrhea virus polyprotein: processing and functional studies. J Virol. 2000;74:9498–506. doi: 10.1128/jvi.74.20.9498-9506.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carrere-Kremer S, Montpellier-Pala C, Cocquerel L, Wychowski C, Penin F, Dubuisson J. Subcellular localization and topology of the p7 polypeptide of hepatitis C virus. J Virol. 2002;76:3720–30. doi: 10.1128/JVI.76.8.3720-3730.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patargias G, Zitzmann N, Dwek R, Fischer WB. Protein-protein interactions: modeling the hepatitis C virus ion channel p7. J Med Chem. 2006;49:648–55. doi: 10.1021/jm050721e. [DOI] [PubMed] [Google Scholar]
- 13.Griffin SD, Beales LP, Clarke DS, Worsfold O, Evans SD, Jaeger J, Harris MP, Rowlands DJ. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 2003;535:34–8. doi: 10.1016/s0014-5793(02)03851-6. [DOI] [PubMed] [Google Scholar]
- 14.Pavlovic D, Neville DC, Argaud O, Blumberg B, Dwek RA, Fischer WB, Zitzmann N. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc Natl Acad Sci U S A. 2003;100:6104–8. doi: 10.1073/pnas.1031527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Premkumar A, Wilson L, Ewart GD, Gage PW. Cation-selective ion channels formed by p7 of hepatitis C virus are blocked by hexamethylene amiloride. FEBS Lett. 2004;557:99–103. doi: 10.1016/s0014-5793(03)01453-4. [DOI] [PubMed] [Google Scholar]
- 16.Luik P, Chew C, Aittoniemi J, Chang J, Wentworth P, Jr, Dwek RA, Biggin PC, Venien-Bryan C, Zitzmann N. The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc Natl Acad Sci U S A. 2009;106:12712–6. doi: 10.1073/pnas.0905966106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 18.Veglia G, Zeri AC, Ma C, Opella SJ. Deuterium/hydrogen exchange factors measured by solution nuclear magnetic resonance spectroscopy as indicators of the structure and topology of membrane proteins. Biophys J. 2002;82:2176–83. doi: 10.1016/s0006-3495(02)75564-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lau TL, Partridge AW, Ginsberg MH, Ulmer TS. Structure of the integrin beta3 transmembrane segment in phospholipid bicelles and detergent micelles. Biochemistry. 2008;47:4008–16. doi: 10.1021/bi800107a. [DOI] [PubMed] [Google Scholar]
- 20.Iwahara J, Schwieters CD, Clore GM. Ensemble approach for NMR structure refinement against (1)H paramagnetic relaxation enhancement data arising from a flexible paramagnetic group attached to a macromolecule. J Am Chem Soc. 2004;126:5879–96. doi: 10.1021/ja031580d. [DOI] [PubMed] [Google Scholar]
- 21.Vold RR, Prosser RS, Deese AJ. Isotropic solutions of phospholipid bicelles: a new membrane mimetic for high-resolution NMR studies of polypeptides. J Biomol NMR. 1997;9:329–35. doi: 10.1023/a:1018643312309. [DOI] [PubMed] [Google Scholar]
- 22.Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–80. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 23.Mesleh MF, Lee S, Veglia G, Thiriot DS, Marassi FM, Opella SJ. Dipolar waves map the structure and topology of helices in membrane proteins. J Am Chem Soc. 2003;125:8928–35. doi: 10.1021/ja034211q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mesleh MF, Opella SJ. Dipolar Waves as NMR maps of helices in proteins. J Magn Reson. 2003;163:288–99. doi: 10.1016/s1090-7807(03)00119-8. [DOI] [PubMed] [Google Scholar]
- 25.Mesleh MF, Veglia G, DeSilva TM, Marassi FM, Opella SJ. Dipolar waves as NMR maps of protein structure. J Am Chem Soc. 2002;124:4206–7. doi: 10.1021/ja0178665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cook GA, Steffer S, Opella SJ. Expression and Purification of the Membrane Protein p7 from HCV. Peptide Science. 2010 doi: 10.1002/bip.21453. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Estephan R, Englander J, Arshava B, Samples KL, Becker JM, Naider F. Biosynthesis and NMR analysis of a 73-residue domain of a Saccharomyces cerevisiae G protein-coupled receptor. Biochemistry. 2005;44:11795–810. doi: 10.1021/bi0507231. [DOI] [PubMed] [Google Scholar]
- 28.Miozzari GF, Yanofsky C. Translation of the leader region of the Escherichia coli tryptophan operon. J Bacteriol. 1978;133:1457–66. doi: 10.1128/jb.133.3.1457-1466.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones DH, Opella SJ. Weak alignment of membrane proteins in stressed polyacrylamide gels. J Magn Reson. 2004;171:258–69. doi: 10.1016/j.jmr.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 30.Ishii Y, Markus MA, Tycko R. Controlling residual dipolar couplings in high-resolution NMR of proteins by strain induced alignment in a gel. J Biomol NMR. 2001;21:141–51. doi: 10.1023/a:1012417721455. [DOI] [PubMed] [Google Scholar]
- 31.De Angelis AA, Opella SJ. Bicelle samples for solid-state NMR of membrane proteins. Nat Protoc. 2007;2:2332–8. doi: 10.1038/nprot.2007.329. [DOI] [PubMed] [Google Scholar]
- 32.Mori S, Abeygunawardana C, Johnson MO, van Zijl PC. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J Magn Reson B. 1995;108:94–8. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
- 33.Grzesiek S, Bax A. Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J Biomol NMR. 1993;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- 34.Wu CH, Grant CV, Cook GA, Park SH, Opella SJ. A strip shield improves the efficiency of a solenoid coil in probes for high field solid-state NMR of lossy biological samples. J Magn Reson. 2009;200:74–80. doi: 10.1016/j.jmr.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nevzorov AA, Opella SJ. A “magic sandwich” pulse sequence with reduced offset dependence for high-resolution separated local field spectroscopy. J Magn Reson. 2003;164:182–6. doi: 10.1016/s1090-7807(03)00240-4. [DOI] [PubMed] [Google Scholar]
- 36.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 37.Cook GA, Opella SJ. NMR studies of p7 protein from hepatitis C virus. Eur Biophys J. 2009 doi: 10.1007/s00249-009-0533-y. [DOI] [PMC free article] [PubMed] [Google Scholar]