

Abstract
DDX3, a DEAD box protein family member, appears to promote the progression of some cancers, which may partly result from its impedance of death receptor-mediated apoptosis. We found that another mechanism by which DDX3 may aid cancer progression is by promoting increased levels of the transcription factor Snail. Snail represses expression of cellular adhesion proteins, leading to increased cell migration and metastasis of many types of cancer. Knockdown of DDX3 levels by shRNA reduced basal levels of Snail in HeLa cells and MCF-7 cells, and this was associated with reduced cell proliferation and migration. Snail protein and mRNA levels were increased by treatment with the HDAC inhibitors sodium butyrate or trichostatin A, and these increases were attenuated in cells with DDX3 knocked down. Treatment of cells with camptothecin was discovered to increase Snail protein levels, and this increase was diminished in cells with DDX3 knocked down. Analysis of 31 patient glioblastoma multiforme (GBM) samples revealed a significant correlation between the levels of DDX3 and Snail. Thus, DDX3 is required for basal Snail expression and increases in Snail induced by HDAC inhibitors or camptothecin, indicating that this action of DDX3 may contribute to its promotion of the progression of some cancers.
Keywords: DDX3, Snail, HDAC, camptothecin, metastasis
1. Introduction
Snail is a rapidly turning-over transcription factor that has been shown to be a particularly important regulator of the development and progression of cancer [1]. These actions derive from the essential role of Snail in driving epithelial-mesenchymal transition (EMT), a crucial process in embryonic development [2] that also promotes cancer metastasis [3, 4]. A key underlying mechanism for this effect is that Snail suppresses the expression of several cellular adhesion proteins, such as E-cadherin [3, 4]. Suppression by Snail of E-cadherin expression is associated with EMT, which promotes the invasive phase of carcinoma [5, 6].
Despite these crucial actions of Snail, few mechanisms that regulate Snail have been identified. One regulatory mechanism is the phosphorylation of Snail by glycogen synthase kinase-3β (GSK3β) at two sites, one promoting nuclear export of Snail, which is mediated by a CRM1-dependent mechanism [7], and the other site promoting Snail ubiquitination that leads to its degradation by the proteasome [8]. Conversely, Snail levels are increased by treatment of cells with inhibitors of GSK3 or with histone deacetylase (HDAC) inhibitors [9], and γ-irradiation stabilizes Snail by inhibiting its nuclear export [10]. Considering the critical actions of Snail in development and cancer, it is crucial to gain a better understanding of cellular mechanisms that regulate Snail.
Recently, several findings linked cancer progression to DDX3, a member of the DEAD box family of proteins named for a conserved D-E-A-D sequence [11, 12]. There is limited knowledge about the functions of DDX3, which was originally linked to actions of the hepatitis C and human immunodeficiency viruses [13-15]. DDX3 is a nucleo-cytoplasmic shuttling protein [14, 16] that can associate with the Sp1 transcription factor in the nucleus and contribute to regulating cyclin D1 and p21 levels [17, 18]. Evidence that DDX3 may promote cancer includes reports that DDX3 levels are increased in hepatocellular carcinomas [19], carcinogen treatment of MCF cells increased DDX3 levels [20], and DDX3 impedes death receptor-induced apoptosis [21]. These findings raised the possibility that DDX3 may have regulatory influences on the development, progression, or treatment of certain cancers. Furthermore, recent findings that overexpression of DDX3 induced EMT [20] and that DDX3 associates with the Snail regulator GSK3β [21] prompted us to examine if DDX3 has a role in modulating Snail.
Herein we report that knocking down the cellular level of DDX3 reduced basal Snail levels and the increase in Snail expression induced by HDAC inhibitors. Furthermore, Snail levels also were found to be increased by the topoisomerase inhibitor camptothecin, and this increase in Snail was dependent on DDX3. Furthermore, the levels of DDX3 and Snail were significantly correlated in a panel of glioblastoma multiforme (GBM) tumor samples. These findings suggest that DDX3 may promote cancer progression in part by supporting Snail expression.
2. Materials and methods
2.1. Cell culture and materials
HeLa and MDA-MB-231 cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 100 U/ml penicillin, 100 μg/ml streptomycin, and 15 mM HEPES, in humidified, 37°C chambers with 5% CO2. MCF-7 cells were grown in the same conditions as HeLa cells with the addition of 10 μg/ml insulin. Human neuroblastoma SH-SY5Y cells were grown in 50% Minimum Essential Medium Eagle (MEM) (Cellgro, Herndon, VA) and 50% Kaighn's Modification of Ham's F-12 (ATCC) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Nuclei were extracted from cells using a nuclear extraction kit according to the manufacturer's instructions (Active Motif, Carlsbad, CA). Cell proliferation was measured using the CellTiter 96R AQueous one solution cell proliferation kit according to the manufacturer's protocol (Promega, Madison, WI). For the wound healing assay, monolayers of mock-shRNA and DDX3 knockdown MCF-7 cells in 6 well plates were scratched along a diameter of the well with a sterile p200 pipette tip, followed by a wash to remove debris. Images were taken at 10× magnification.
Sources of chemicals were: sodium butyrate, trichostatin A, nicotinamide, valproic acid, lithium chloride, camptothecin, cytosine β-D-arabino-furanoside (Ara-C) (Sigma, St. Louis, MO), bleomycin, leptomycin B, and etoposide (Alexis Biochemicals, San Diego, CA). The primers for PCR were from integrated DNA Technologies (Coralville, IA). The following sources provided antibodies: β-actin (Sigma), CREB, acetyl-histone H3, histone H3, ATM, phosphorylated-serine-1981-ATM, Snail (Cell Signaling Technology, Beverly, MA), and p53, E-cadherin (Transduction Laboratories, Lexington, KY). DDX3 antibodies were prepared in the laboratory of Dr. T. Zhou [22]. GSK3β-HRP conjugated antibody and all horseradish peroxidase (HRP)-conjugated secondary reagents were purchased from Southern Biotechnology Associates (Birmingham, AL).
2.2. siRNA and expression
Lentiviral mediated shRNA was performed using shRNA lentiviral (pLKO.1-puro) plasmids (Sigma). The oligonucleotides containing the DDX3 target sequence that were used are: sequence #1, 5′-CCGGCCCTGCCAAACAAGCTAATATCTCGAGATATTAGCTTGTTTGGCAGGGTTTT and sequence #4, 5′-CCGGCGCTTGGAACAGGAACTCTTTCTCGAGAAAGAGTTCCTGTTCCAAGCGTTTT One 100 mm dish of 293FT cells (Invitrogen) was co-transfected with 3 μg of the pLKO.1-puro plasmids plus 3 μg each of the packaging vectors pLP1, pLP2, and pLP/VSVG (Invitrogen) using lipofectamine 2000. The media was changed approximately 16 hr after transfection, and the cells were cultured an additional 48–72 hr. The media was then collected, centrifuged at 3000rpm for 5 min, and filtered through a 0.45 μm filter. Experimental cells were incubated with the virus-containing medium overnight in 6-well plates, the media was changed, and cells incubated for 24 hr. Cells were transferred to 100 mm dishes and infected cells were selected by incubation in puromycin (1 μg/ml).
For RT-PCR analysis, RNA was isolated using Trizol (Invitrogen) according to manufacturer's protocol. The RNA was quantified using a Nano-drop spectrophotometer. The RT-PCR reaction was performed using the ImProm-II™ Reverse Transcription System (Promega) according to the manufacturer's protocol. To analyze Snail RNA, 200 ng of RNA was used in the reaction with the following primers 5′-TCCCGGGCAATTTAACAATG-3′ and 5′-TGGGAGACACATCGGTCAGA-3′ for 32 cycles. To analyze 18S RNA, 200 ng of RNA was used with the following primers 5′-GAGCGAAAGCATTTGCCAAG-3′ and 5′-GGCATCGTTTATGGTCGGAA-3′ for 20 cycles. The resulting cDNA was then visualized on a 2% agarose gel containing ethidium bromide and imaged on a multi-imager (Bio-Rad). For mRNA quantitation, real-time PCR was performed with the LightCycler system (Roche Molecular Biochemicals) using the SYBR green fluorophore.
2.3. Immunoblotting
Cells were washed twice with PBS, and lysed in lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1 mM sodium orthovanadate, 100 μM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml pepstatin, 50 mM NaF, 1 nM okadaic acid, 1% Triton X-100, and 10% glycerol). The lysates were incubated for 30 min on ice, centrifuged at 20,800×g for 15 min, and supernatants were collected. To immunoblot acetylated histones, cells were harvested, washed twice with PBS, and resuspended in triton extraction buffer containing 0.5% TritonX100 (v/v), 2 mM phenylmethylsulfonyl fluoride (PMSF), and 0.02% (w/v) NaN3. Lysed cells were incubated on ice for 10 min at 4°C, then washed with extraction buffer and centrifuged. The pellet was resuspended in 0.2 N HCl, incubated overnight at 4°C, and the supernatant was used for immunoblotting. Protein concentrations were determined using the bicinchoninic method (Pierce, Rockford, IL). Cell lysates were mixed with Laemmli sample buffer and placed in a boiling water bath for 5 min. Proteins were resolved in SDS-polyacrylamide gels, and transferred to nitrocellulose. Blots were probed with the indicated antibodies, and were developed using horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit IgG, followed by detection with enhanced chemiluminescence.
2.4. Human GBM tissue samples
The Brain Tumor Tissue Core Facility of the UAB Brain Tumor SPORE provided patient tumor specimens that had been collected and archived under an IRB approved protocol (X050415007). Freshly resected glioblastoma multiforme (GBM) tissues were aseptically debrided of blood clots and any charred or obviously necrotic portions, cut into 75-100 mg samples and snap-frozen in sterile cryovials in liquid nitrogen. Tissues were stored in the vapor phase of liquid nitrogen until used. Distribution of patients by race and gender are shown in Table 1. The average age of these patients at surgery was 57.7 ± 13.1 years. All but five of the tumor specimens were from an initial resection. The five exceptions (Figure 7: #2, 12, 15, 29, 31) were derived from surgical resections of recurrent GBM.
TABLE 1. Characteristics of GBM donors.
| Gender | American Indian or Alaskan native | Asian or Pacific Islander | Black, not of Hispanic origin | Hispanic | White, not of Hispanic origin | Other or unknown | Total |
|---|---|---|---|---|---|---|---|
| Female | 0 | 0 | 2 | 0 | 10 | 0 | 12 |
| Male | 0 | 0 | 1 | 0 | 13 | 5 | 19 |
| Unknown | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| Total | 0 | 0 | 3 | 0 | 23 | 5 | 32 |
Fig. 7. Correlation between DDX3 and Snail levels in human glioblastoma samples.
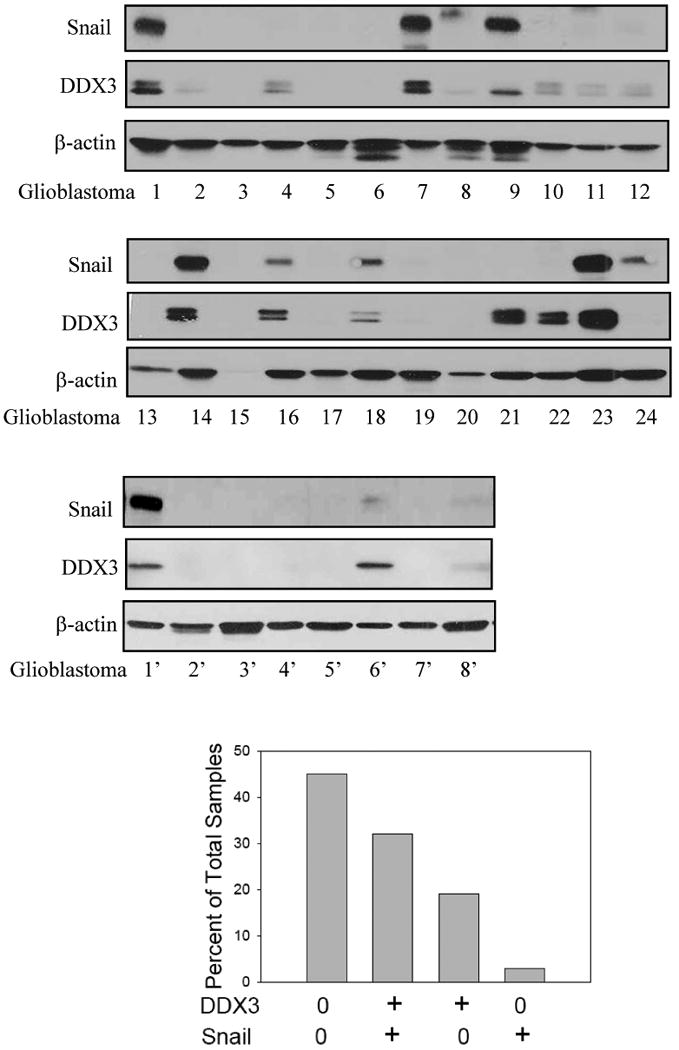
Snail and DDX3 levels were measured in 32 glioblastoma samples. Sample 15 was excluded from the analysis because of the absence of the β-actin loading control. The relative relationships between the expression of DDX3 and Snail were calculated as a percent of total human glioblastoma samples analyzed. Evaluation by the Chi-Square test demonstrated a significant correlation (p=0.001) between Snail and DDX3 levels.
3. Results
3.1. HDAC inhibitors increase the expression level of Snail
Snail levels were compared in nine cell lines and were found to be highest in HeLa cells and MDA-MB-231 cells (Figure 1A). We tested if HDAC inhibitors altered Snail levels in each of the two cell lines with the highest Snail levels. Nuclear levels of Snail were increased in HeLa cells (Figure 1B) and MDA-MB-231 cells (Figure 1C) following 3 hr treatments with the HDAC inhibitors valproic acid (5 mM), sodium butyrate (5 mM) (NaBu) or trichostatin A (1 μM) (TSA), but not nicotinamide (5 mM), and nicotinamide did not alter the response to TSA. This indicates Snail regulation by Class I or II HDACs inhibited by valproic acid, NaBu, and TSA, but not by the Class III sirtuins inhibited by nicotinamide [23]. Pretreatment with the protein synthesis inhibitor cycloheximide (40 μg/ml) for 1 hr completely blocked the increased Snail levels induced by treatment with HDAC inhibitors (Figure 1D). None of the HDAC inhibitors affected the level of DDX3. We examined the effects of HDAC inhibitors on Snail mRNA levels. Treatment with valproic acid, NaBu or TSA each increased Snail mRNA levels in HeLa, SH-SY5Y and MCF-7 cells, although the response to valproic acid was much less than the others (Figure 1E).
Fig. 1. Treatment with HDAC inhibitors increases Snail expression levels.
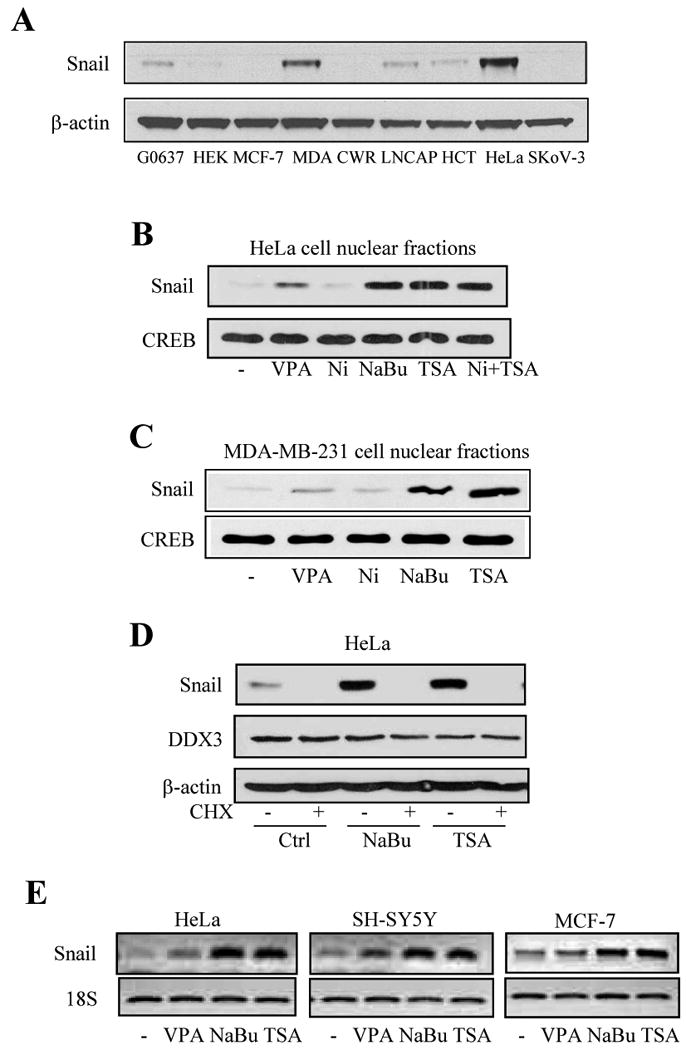
(A) Comparison of Snail levels in GM0637, HEK293, MCF-7, MDA-MB-231, CWR22RV1, LNCAP, HCT116, HeLa, SKOV-3 cells. (B) HeLa cells or (C) MDA-MB-231 cells were treated with 5 mM valproic acid (VPA), 5 mM nicotinamide (Ni), 5 mM sodium butyrate (NaBu), 1 μM trichostatin A (TSA), or nicotinamide with trichostatin A for 3 hr. Nuclear fractions were prepared and Snail and CREB were measured by immunoblot analysis. Light film exposures were used to allow detection of up-regulated levels of Snail following treatments with HDAC inhibitors. (D) HeLa cells were pretreated with cycloheximide (CHX; 40 μg/ml) for 1 hr, followed by NaBu or TSA treatment for 3 hr, and Snail, DDX3, and β-actin levels were immunoblotted. (E) Snail and 18S mRNA levels were measured in HeLa, SH-SY5Y, and MCF-7 cells after treatment with VPA, NaBu, or TSA for 3 hr.
3.2. Knockdown of DDX3 reduces Snail and alters cell morphology, proliferation and migration
In accordance with a previous report that DDX3 suppressed E-cadherin expression in MCF-10A cells [20], knocking down DDX3 increased E-cadherin levels in both HeLa cells and MCF-7 cells (Figure 2A). This suggested that DDX3 may reduce Snail levels since Snail is a transcriptional repressor of E-cadherin [1, 3, 4]. In support of this hypothesis, Snail levels were decreased in both HeLa and MCF-7 cells with DDX3 knocked down (Figure 2A). Using two different shRNA sequences to knockdown DDX3 levels in MCF-7 cells revealed that the Snail level was reduced by an amount that corresponded to the extent of knockdown of the level of DDX3 (Figure 2B). Thus, down-regulation of DDX3 caused a corresponding decrease in the basal level of Snail. We were also interested in testing if over-expression of DDX3 altered Snail levels. However, we were unable to generate HeLa, MCF-7, HEK293, NIH3T3, or SH-SY5Y cells that stably over-express DDX3, and transient over-expression of DDX3 in MCF-7 cells resulted in only a 2-fold increase in the level of DDX3. This modest DDX3 over-expression in MCF-7 cells did not increase basal or stimulated levels of Snail (Supplemental Figure 1). Thus, although DDX3 is required to support Snail levels, a modest increase in the level of DDX3 is not alone sufficient to increase the level of Snail.
Fig. 2. DDX3 knockdown alters Snail levels and cell morphology, proliferation, and migration.
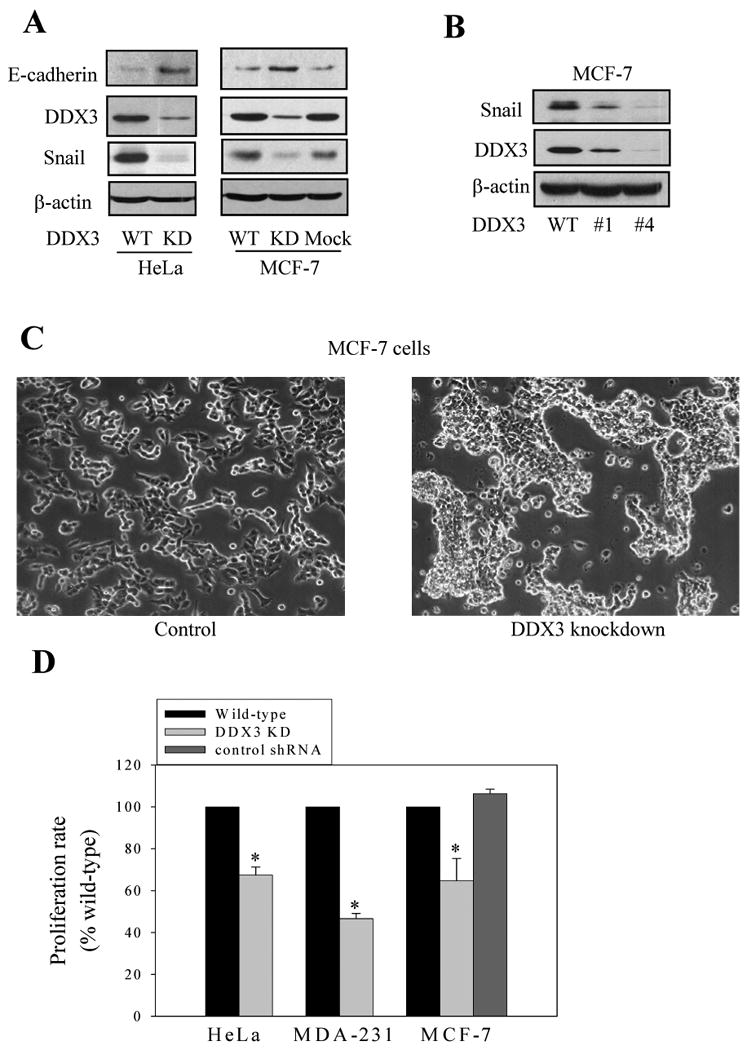
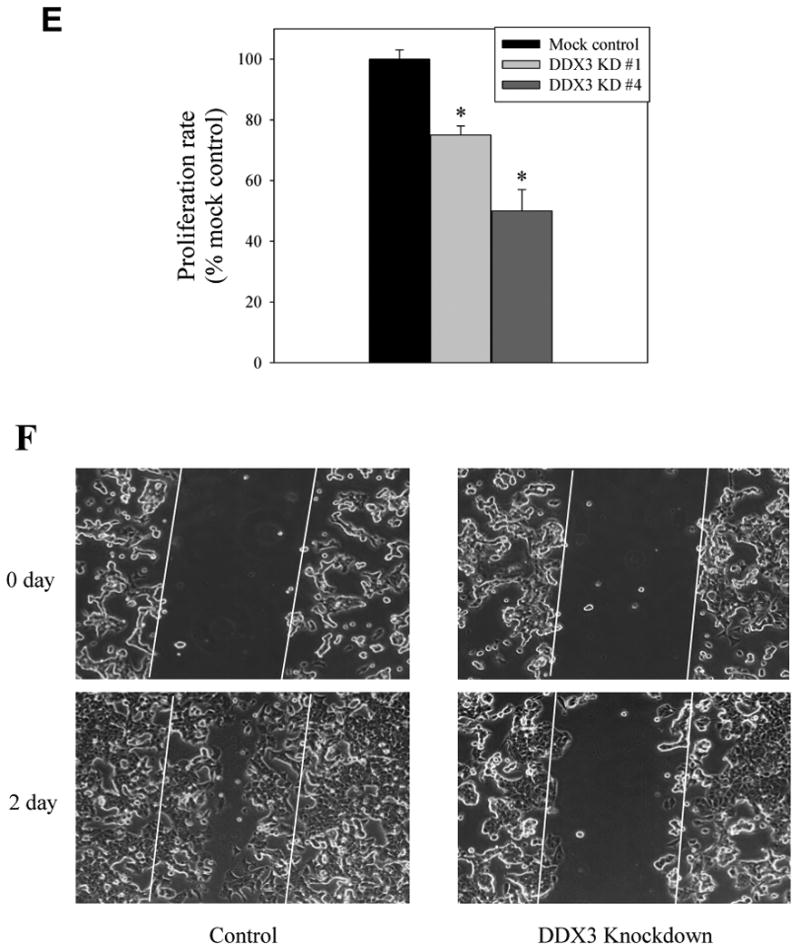
(A) E-cadherin, DDX3, Snail, and β-actin levels were immunoblotted in HeLa and MCF-7 wild-type (WT), DDX3 knockdown (KD), or mock knockdown (using a control shRNA) cells. Dark exposures of films were used to allow detection of low levels of Snail in cells with DDX3 knocked down. (B) DDX3 was knocked down by two different sequences of shRNA in MCF-7 cells, and DDX3 and Snail levels were immunoblotted. (C) Morphology of mock-shRNA (Control) and DDX3 knockdown (using clone #4 described in Figure 2B) MCF-7 cells. (D) Proliferation was measured in wild-type and DDX3 knockdown (KD; using clone #4 described in Figure 2B) HeLa cells (3000 cells), MDA-MB-231 cells (1000 cells), and MCF-7 cells (3000 cells; using clone #4 described in Figure 2B), along with a mock-shRNA control. Means ± SEM, n=3, *p < 0.05 compared with wild-type cells. E. Proliferation was measured in MCF-7 cells with control mock shRNA or clones #1 or #4 DDX3 knockdown described in Figure 2B. Means ± SEM, n=3, *p < 0.05. F. The wound healing assay was performed in mock-shRNA (Control) and DDX3 knockdown (using clone #4 described in Figure 2B) MCF-7 cells. The scratch was implemented and the cells were imaged immediately and after incubation for 2 days.
Snail regulates EMT, and examination of the morphology of MCF-7 cells in which DDX3 had been knocked down demonstrated morphological characteristics consistent with increased cellular adhesion, which correlated with the reduced Snail levels induced by knocking down DDX3 (Figure 2C). Snail also promotes cell proliferation [24], and the proliferation of HeLa cells, MDA-MB-231 cells, and MCF-7 cells was reduced by knocking down DDX3 levels (Figure 2D). The anti-proliferative effect correlated with the extent of DDX3 knockdown (Figure 2E). Thus, Snail and its regulation of E-cadherin and cellular characteristics are supported by DDX3.
Since Snail regulates cellular migration, the wound healing assay was performed to test if cell migration is regulated by DDX3. Mock-shRNA control MCF-7 cells migrated into the scratched area within 48 h (Figure 2F). In contrast, MCF-7 cells with DDX3 knocked down only minimally migrated into the scratched area. Thus, the reduced levels of Snail in cells with DDX3 knocked down correlates with reduced cell migration.
3.3. DDX3 is required for HDAC inhibitor-induced Snail expression
Since the basal level of Snail is supported by DDX3, we tested if DDX3 is required for HDAC inhibitors to increase Snail expression. The increased Snail levels induced by treating MCF-7 cells with NaBu or TSA were greatly diminished in cells with DDX3 knocked down (Figure 3A), indicating that DDX3 is required for HDAC inhibitor-induced increases in Snail. Measurements of Snail mRNA levels confirmed that increases induced by treatment with NaBu or TSA were diminished in cells with DDX3 knocked down (Figure 3B). Snail mRNA measured by qRT-PCR demonstrated increases induced by NaBu and TSA treatment were 28-fold and 26-fold, respectively, and these increases were attenuated by ∼60% in cells with DDX3 knocked down (Figure 3C). NaBu-induced acetylation of histone H3 was unaffected by knocking down DDX3, excluding the possibility that DDX3 knockdown inhibited histone acetylation (Figure 3D).
Fig. 3. DDX3 is required for HDAC inhibitor-induced Snail expression.
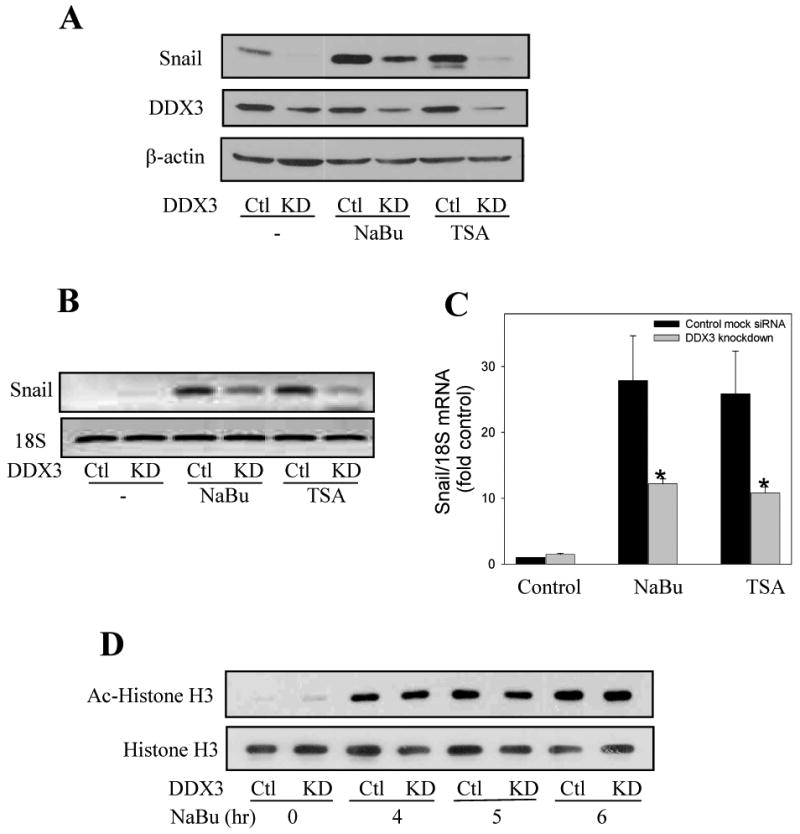

(A) Mock-shRNA (Ctl) or DDX3 knockdown (KD) MCF-7 cells were treated with HDAC inhibitors 5 mM NaBu, or 1 μM TSA for 4 hr, and then the levels of Snail, DDX3 and β-actin were measured. (B) Snail and 18S mRNA levels were measured in mock-shRNA (Ctl) or DDX3 knockdown MCF-7 cells after treatment with 5 mM NaBu or 1 μM TSA for 3 hr. (C) Real time PCR was used to measure Snail and 18S mRNA levels in MCF-7 mock-shRNA and DDX3 knockdown cells after treatment with 5 mM NaBu or 1 μM TSA for 3 hr. Quantitative values are from NaBu or TSA treatment of DDX3 knockdown cells compared with mock-shRNA MCF-7 cells Means ± SEM, n=3, *p < 0.05. (D) Acetyl-histone H3 and total histone H3 levels were immunoblotted after mock-shRNA or DDX3 knockdown MCF-7 cells were treated with 5 mM NaBu for 4, 5, or 6 hr. (E) Mock-shRNA and DDX3 knockdown MCF-7 cells were treated with lithium (20 mM) for 30 min, 5.5 hr, with lithium for 30 min followed by the addition of 10 μM MG132 during the last 5 hr, or MG132 for 5 hr, and DDX3, Snail, phospho-serine9-GSK3β, and β-actin levels were measured. (F) Mock-shRNA and DDX3 knockdown MCF-7 cells as described in Figure 3E were pretreated with lithium (20 mM) for 30 min, followed by treatment with 5 mM NaBu or 1 μM TSA for 4 hr, and Snail and β-actin levels were measured.
Snail phosphorylation by GSK3 promotes its proteosomal degradation [8], and GSK3 inhibitors were shown to increase Snail by both translational [25] and post-translational regulation [8]. Therefore, we tested if the inhibitory effect of knocking down DDX3 on Snail levels could be due to promoting the actions of GSK3. Inhibition of GSK3 by pretreatment of MCF-7 cells with the GSK3 inhibitor lithium for 30 min or 5.5 hr alone had little effect on Snail levels in wild-type or DDX3 knocked down MCF-7 cells, but augmented Snail accumulation in cells treated with the proteasome inhibitor MG132 (Figure 3E). Increased serine9-phosphorylated GSK3β in lithium-treated cells confirmed that lithium inhibited GSK3 [26]. Pretreatment with lithium to inhibit GSK3 did not alter either the increases in Snail induced by NaBu or TSA or the reduction evident in cells with DDX3 knocked down (Figure 3F), altogether indicating that the effect of DDX3 on HDAC inhibitor-induced Snail expression is independent of GSK3.
3.4. Topoisomerase inhibitors but not other DNA damaging agents increase Snail expression level
Because HDAC inhibitors can cause DNA damage [27, 28], we tested if Snail levels were influenced by other DNA damaging agents. These experiments showed that treatment with bleomycin, H2O2, or Ara-C did not increase, and in some cases decreased, Snail levels, although all of these agents caused DNA damage as indicated by increased levels of p53 (Supplemental Figure 2). However, Snail levels increased after treatment with the topoisomerase II inhibitor etoposide (Figure 4A), and treatment with the topoisomerase I inhibitor camptothecin (10 μM) increased Snail levels in MCF-7 cells (Figure 4B), SH-SY5Y cells (Figure 4C), and MDA-MD-231 cells, which express mutant p53, (Figure 4D) in a time-dependent manner that matched p53 accumulation. Nuclear Snail, as well as nuclear p53, levels increased after camptothecin treatment, whereas cytosolic Snail and p53 levels were very low (Figure 4E). The camptothecin-induced increase in Snail protein level was mirrored by increased Snail mRNA levels in both MCF-7 and SH-SY5Y cells (Figure 5A). qRT-PCR measurements demonstrated that camptothecin treatment increased Snail mRNA levels by 22-fold in MCF-7 cells after treatment for 4 hr (Figure 5B). Since camptothecin-induced DNA damage activates the transcription factor p53, we tested if p53 regulates camptothecin-induced increased Snail expression in H1299 p53-null cells that inducibly express p53 [29]. Camptothecin (10 μM) treatment increased Snail levels equivalently in H1299 cells with or without induced expression of p53 (Figure 5C), indicating that p53 does not mediate the upregulation of Snail caused by camptothecin treatment.
Fig. 4. Topoisomerase inhibitors increase Snail expression.
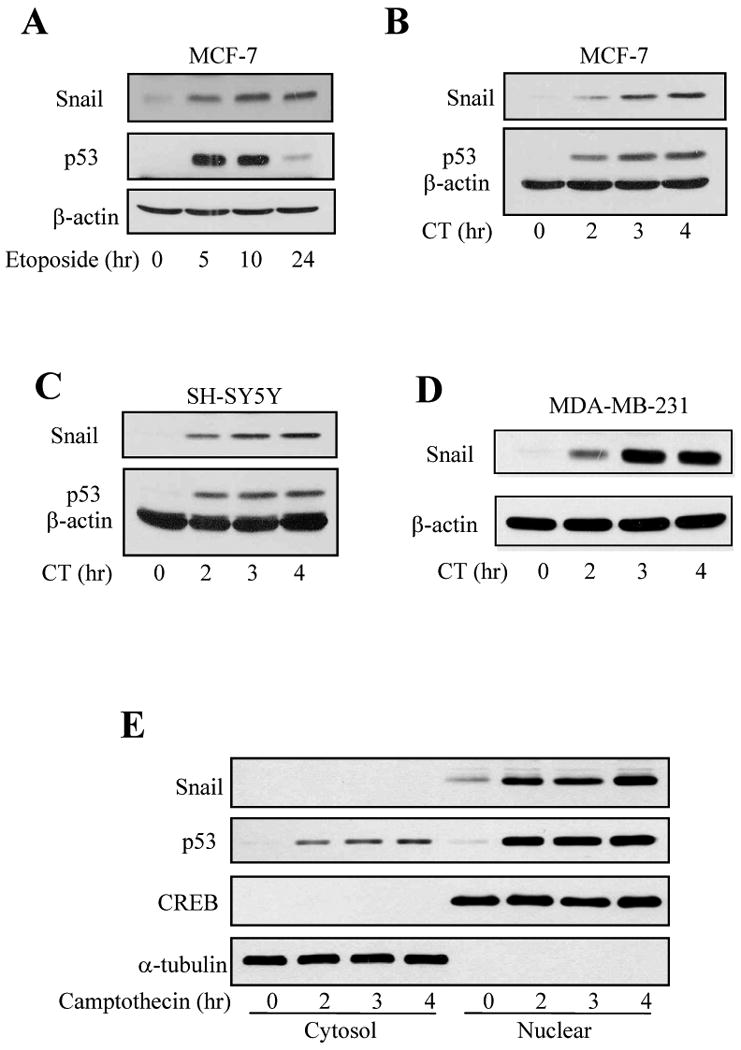
(A) MCF-7 cells were treated with 100 μM etoposide for 5-24 hr, and the levels of Snail, p53, and β-actin were immunoblotted. (B) MCF-7, (C) SH-SY5Y, or (D) MDA-MB-231 cells, which express mutant p53, were treated with 10 μM camptothecin for 2, 3, or 4 hr following incubation in serum-free media for 2 hr, and then Snail, p53, and β-actin levels were immunoblotted. (E) Cytosolic and nuclear p53 and Snail levels were measured after MCF-7 cells were treated with 10 μM camptothecin for 2, 3, or 4 hr. CREB was immunoblotted as a nuclear marker and α-tubulin as a cytosolic marker.
Fig. 5. Camptothecin increases Snail mRNA levels.
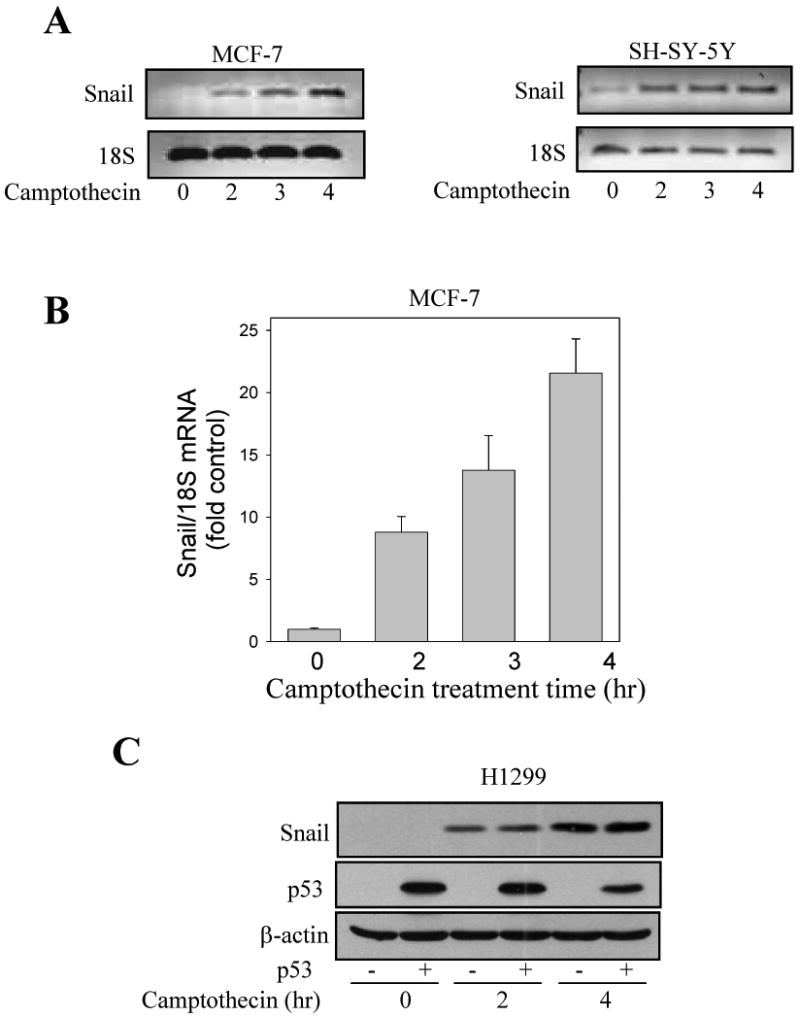
(A) Snail and 18S mRNA levels were measured in MCF-7 and SH-SY5Y cells after treatment with 10 μM camptothecin for 2, 3, or 4 hr. (B) Real time PCR was used to measure Snail and 18S mRNA levels in MCF-7 cells after treatment with 10 μM camptothecin. Means ± SEM, n=3. (C) H1299 p53-null cells were used, with or without inducibly expressing p53 for 24 hr, followed by treatment with 10 μM camptothecin for 2 or 4 hr, and Snail, p53, and β-actin levels were measured.
3.5. DDX3 knockdown reduces Snail levels induced by camptothecin
We tested if the camptothecin-induced increase in Snail levels was dependent on DDX3. Treatment of MCF-7 cells with 10 μM camptothecin increased the level of Snail, but this was attenuated in cells with DDX3 knocked down (Figure 6A). The level of GSK3β was used as a loading control and to demonstrate that neither camptothecin nor DDX3 regulated GSK3β levels. Measurements of Snail mRNA revealed increases induced by camptothecin treatment, but knocking down DDX3 did not alter the camptothecin-induced increase in the Snail mRNA level (Figure 6B), indicating that DDX3 supports the accumulation or stabilization of Snail protein instead of transcriptional expression after camptothecin treatment.
Fig. 6. DDX3 knockdown-induced inhibition of camptothecin-induced Snail is a post-translational effect.
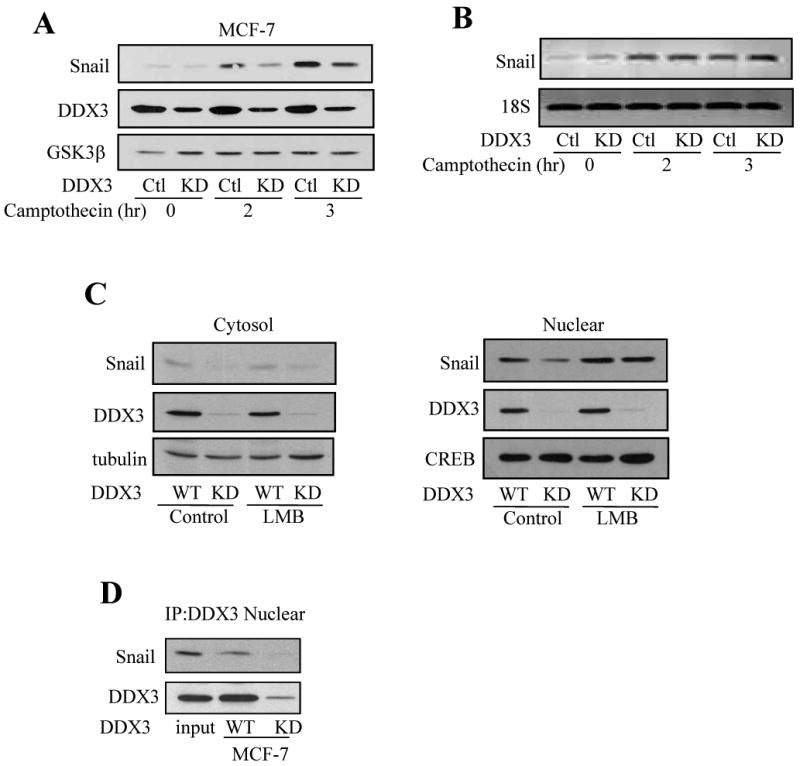
(A) Mock-shRNA (Ctl) and DDX3 knockdown (KD) MCF-7 cells were treated with 10 μM camptothecin for 2 or 3 hr, and Snail, DDX3, and GSK3β in cell lysates were immunoblotted. (B) Snail and 18S mRNA levels were measured in mock-shRNA or DDX3 knockdown MCF-7 cells after treatment with 10 μM camptothecin for 2 or 3 hr. (C) Cytosolic and nuclear fractions were prepared from wild-type (WT) or DDX3 knockdown MCF-7 cells after treatment with or without leptomycin B (20 ng/ml) for 5 hr, and the levels of Snail, and DDX3 in both fractions were measured. Fractions were also immunoblotted with the cytosolic marker tubulin and the nuclear marker CREB. (D) Nuclear fractions were prepared from wild-type and DDX3 knocked down MCF-7 cells, DDX3 was immunoprecipitated, and DDX3 and Snail were immunoblotted.
Since Snail can be stabilized by inhibiting its nuclear export, which is mediated by a CRM1-dependent mechanism [7], we tested if the depletion of nuclear Snail caused by DDX3 knockdown was affected by treatment with leptomycin B, an inhibitor of CRM1-mediated nuclear export. After leptomycin B treatment (20 ng/ml; 5 hr), the nuclear Snail level slightly increased in wild-type MCF-7 cells, but in cells with DDX3 knocked down the depleted nuclear level of Snail was replenished to nearly the level attained in wild-type cells (Figure 6C). This suggested that knocking down DDX3 facilitated the nuclear export of Snail which was inhibited by leptomycin B, indicating that DDX3 promotes retention of Snail in the nucleus to support its accumulation. The possibility that this may be due to an interaction between DDX3 and Snail in the nucleus was examined by measuring their co-immunoprecipitation from nuclear fractions. In wild-type MCF-7 cells, Snail was detected in immunoprecipitants of DDX3, whereas as a control no Snail was detected after immunoprecipitating DDX3 from MCF-7 cells with DDX3 knocked down Figure (6D).
3.6. DDX3 and Snail levels in human GBM
To test if DDX3 may influence Snail in gliomas, we measured DDX3 and Snail levels in glioblastoma multiforme (GBM) tumor biopsy samples. Analysis of 31 samples revealed a statistically significant (p=0.001; Chi-Square analysis) correlation between DDX3 and Snail (Figure 7). Ten samples expressed high levels of both DDX3 and Snail, and neither DDX3 nor Snail was detected in 14 samples. DDX3 but not Snail was detected in 6 samples, indicating that DDX3 is not sufficient alone to increase Snail expression. Only 1 sample (#24; primary GBM, 51 yo WM) expressed Snail but not DDX3, indicating that only rarely does Snail accumulate in the absence of DDX3.
4. Discussion
Snail is a transcription factor with an important role in regulating cancer progression, especially invasion and metastasis [1], but few mechanisms have been identified that regulate Snail. This study found that DDX3 is essential for maintenance or upregulation of Snail levels, as both basal levels of Snail and increased Snail levels induced by treatment with HDAC inhibitors or by camptothecin were reduced in cells with the level of DDX3 knocked down. Furthermore, the levels of Snail and DDX3 were significantly correlated in a panel of glioblastoma samples. These findings demonstrate that DDX3 contributes to supporting the levels of Snail, which may have a regulatory role during cancer progression.
This study first found that DDX3 contributes to supporting basal levels of Snail, as knockdown of DDX3 in both breast cancer cells and cervical cancer cells reduced Snail levels in the absence of other treatments. Cells with DDX3 levels knocked down displayed significantly lower proliferation rates than control cells, cell adhesion appeared to be increased, as was the level of E-cadherin, and cell motility was greatly decreased after DDX3 knockdown. These results complement a previous report that overexpression of DDX3 induced EMT and increased motility and invasion [20]. Conversely, it was difficult to obtain cells with high levels of over-expressed DDX3, suggesting that over-expressed DDX3 is unstable or toxic. A modest increase of DDX3 after over-expression in MCF-7 cells did not increase Snail levels, indicating that DDX3 alone is not sufficient to increase Snail levels, consistent with findings in GBM that DDX3 is expressed in some cells in the absence of detectable Snail, whereas Snail expression requires DDX3. Together with previous evidence linking DDX3 to signaling events important in carcinogenesis and chemotherapy [18, 20, 21], these results further strengthen this connection and suggest that supporting Snail expression contributes to the capacity of DDX3 to regulate cancer progression.
Treatment with HDAC inhibitors caused rapid and large increases in Snail levels in several types of cells, and these increases correlated with increased Snail mRNA levels. This extends a previous report that the HDAC inhibitor TSA increased Snail levels in MCF-7 cells [9]. Depletion of DDX3 greatly reduced HDAC inhibitor-induced increases in Snail protein and mRNA levels, indicating that DDX3 supports the transcriptional upregulation of Snail in the presence of HDAC inhibitors. Moreover, Snail expression and protein levels supported by DDX3 was independent of GSK3, an enzyme reported to regulate both Snail stability and expression [7-9]. Along with previous reports that DDX3 is partially nuclear and associates with the Sp1 transcription factor [14, 16-18], these findings raise the possibility that nuclear DDX3 modulates the transcriptional machinery regulating Snail expression, the subject of future investigations.
Since HDAC inhibitors can induce DNA damage and cell death in many types of cancer cells [31, 32], and γ-irradiation increased Snail levels [10], we tested if the increased expression of Snail induced by HDAC inhibitors may be related to DNA damage. This was tested by measuring Snail levels after treatments with several agents that induce DNA damage by different mechanisms. We found that the topoisomerase inhibitors camptothecin and etoposide increased Snail levels, but increased Snail expression was not a general response to DNA damage because other DNA damaging treatments, including bleomycin, H2O2, and Ara-C, did not increase Snail levels. In agreement with this selective up-regulation of Snail, the camptothecin-induced upregulation of Snail was independent of p53. Increased Snail expression induced by the chemotherapeutic agent camptothecin [30] was robust and evident in several cell types, a disturbing finding since it raises the possibility that cancer cells surviving camptothecin-induced death during chemotherapy may up-regulate Snail levels and thereby increase the potential for metastasis. Thus, not only does Snail counteract apoptotic programs activated by DNA damaging agents [31, 32], but Snail is upregulated following camptothecin treatment. Knocking down DDX3 reduced the camptothecin-induced upregulation of Snail, but surprisingly considering our results with HDAC inhibitors, this was not a transcriptional effect of DDX3 but only was evident upon measuring Snail protein levels. This depletion of Snail with DDX3 knocked down was reversed by blocking nuclear export of Snail, raising the possibility that DDX3 may promote Snail stabilization by reducing its nuclear export to the proteasome. Thus, DDX3 supports Snail expression, as evident in basal conditions and in the responses to HDAC inhibitors. However, camptothecin-induced up-regulation of Snail expression eliminated the effect of DDX3 on Snail expression, but in this condition DDX3 affected the protein levels of Snail, possibly due to the direct interaction of DDX3 with Snail in the nucleus evident in co-immunoprecipitation experiments.
Analysis of human GBM tissue samples further supported the hypothesis that DDX3 contributes to increased Snail levels. Samples with high Snail levels always expressed high DDX3 levels. In this group of GBM tumors, Snail was only rarely (1 of 31 samples) detected in the absence of DDX3, although DDX3 could be detected in the absence of Snail, suggesting DDX3 is not sufficient, but may be required, to support Snail protein accumulation.
In summary, the results reported here reveal that DDX3 supports the expression of Snail, a transcription factor important in cancer progression. By supporting increased cellular Snail levels, DDX3 influences cell proliferation and motility. Thus, these findings add to the evidence that DDX3 plays important roles in processes that are associated with carcinogenesis and chemotherapy.
Supplementary Material
Figure 1. DDX3 over-expression in MCF-7 cells.
DDX3 was transiently over-expressed in MCF-7 cells. (A) Adenovirus-mediated DDX3 over-expression did not increase the basal level of Snail. (B) DDX3 over-expression did not alter increases in Snail induced by a 3 hr treatment with 1 μM TSA, 5 mM sodium butyrate, or 10 μM camptothecin. Adenovirus-mediated over-expression of green fluorescence protein (GFP) did not alter DDX3 or Snail levels.
Figure 2. Snail levels after treatment with DNA damaging agents.
SH-SY5Y cells were treated with (A) 50-200 μM bleomycin for 6 hr, (B) 100 μM H2O2 for 2, 3, or 4 hr, or (C) 5-20 μM Ara-C for 24 hr, and the levels of Snail, p53, and β-actin were immunoblotted.
Acknowledgments
We thank Dr. Xinbin Chen for the H1299 cells. This research was supported by grants P50CA089019, MH038752, and CA097247 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155–166. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 2.Carver EA, Jiang R, Lan Y, Oram KF, Gridley T. The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol Cell Biol. 2001;21:8184–8188. doi: 10.1128/MCB.21.23.8184-8188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, García De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 4.Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 5.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 6.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Domínguez D, Montserrat-Sentís B, Virgós-Soler A, Guaita S, Grueso J, Porta M, Puig I, Baulida J, Francí C, García de Herreros A. Phosphorylation regulates the subcellular location and activity of the snail transcriptional repressor. Mol Cell Biol. 2003;23:5078–5089. doi: 10.1128/MCB.23.14.5078-5089.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 9.Palmer MB, Majumder P, Green MR, Wade PA, Boss JM. A 3′ enhancer controls snail expression in melanoma cells. Cancer Res. 2007;67:6113–6120. doi: 10.1158/0008-5472.CAN-06-4256. [DOI] [PubMed] [Google Scholar]
- 10.Escrivà M, Peiró S, Herranz N, Villagrasa P, Dave N, Montserrat-Sentís B, Murray SA, Francí C, Gridley T, Virtanen I, García de Herreros A. Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation-induced apoptosis. Mol Cell Biol. 2008;28:1528–1540. doi: 10.1128/MCB.02061-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdelhaleem M, Maltais L, Wain H. The human DDX and DHX gene families of putative RNA helicases. Genomics. 2003;81:618–622. doi: 10.1016/s0888-7543(03)00049-1. [DOI] [PubMed] [Google Scholar]
- 12.Rocak S, Linder P. DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol. 2004;5:232–241. doi: 10.1038/nrm1335. [DOI] [PubMed] [Google Scholar]
- 13.Owsianka AM, Patel AH. Hepatitis C virus core protein interacts with a human DEAD box protein DDX3. Virology. 1999;257:330–340. doi: 10.1006/viro.1999.9659. [DOI] [PubMed] [Google Scholar]
- 14.Yedavalli VS, Neuveut C, Chi YH, Kleiman L, Jeang KT. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell. 2004;119:381–392. doi: 10.1016/j.cell.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 15.Ariumi Y, Kuroki M, Abe K, Dansako H, Ikeda M, Wakita T, Kato N. DDX3 DEAD-Box RNA helicase is required for hepatitis C virus RNA replication. J Virol. 2007;81:13922–13926. doi: 10.1128/JVI.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sekiguchi T, Iida H, Fukumura J, Nishimoto T. Human DDX3Y, the Y-encoded isoform of RNA helicase DDX3, rescues a hamster temperature-sensitive ET24 mutant cell line with a DDX3X mutation. Exp Cell Res. 2004;300:213–222. doi: 10.1016/j.yexcr.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Chang PC, Chi CW, Chau GY, Li FY, Tsai YH, Wu JC, Lee YH. DDX3, a DEAD box RNA helicase, is deregulated in hepatitis virus-associated hepatocellular carcinoma and is involved in cell growth control. Oncogene. 2006;25:1991–2003. doi: 10.1038/sj.onc.1209239. [DOI] [PubMed] [Google Scholar]
- 18.Chao CH, Chen CM, Cheng PL, Shih JW, Tsou AP, Lee YH. DDX3, a DEAD box RNA helicase with tumor growth-suppressive property and transcriptional regulation activity of the p21waf1/cip1 promoter, is a candidate tumor suppressor. Cancer Res. 2006;66:6579–6588. doi: 10.1158/0008-5472.CAN-05-2415. [DOI] [PubMed] [Google Scholar]
- 19.Huang JS, Chao CC, Su TL, Yeh SH, Chen DS, Chen CT, Chen PJ, Jou YS. Diverse cellular transformation capability of overexpressed genes in human hepatocellular carcinoma. Biochem Biophys Res Commun. 2004;315:950–958. doi: 10.1016/j.bbrc.2004.01.151. [DOI] [PubMed] [Google Scholar]
- 20.Botlagunta M, Vesuna F, Mironchik Y, Raman A, Lisok A, Winnard P, Jr, Mukadam S, Van Diest P, Chen JH, Farabaugh P, Patel AH, Raman V. Oncogenic role of DDX3 in breast cancer biogenesis. Oncogene. 2008;27:3912–322. doi: 10.1038/onc.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun M, Song L, Li Y, Zhou T, Jope RS. Identification of an anti-apoptotic protein complex at death receptors. Cell Death Differ. 2008;15:1887–1900. doi: 10.1038/cdd.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Wang H, Wang Z, Makhija S, Buchsbaum D, Lobuglio A, Kimberly R, Zhou T. Inducible resistance of tumor cells to tumor necrosis factor-related apoptosis-inducing ligand receptor 2-mediated apoptosis by generation of a blockade at the death domain function. Cancer Res. 2006;66:8520–8528. doi: 10.1158/0008-5472.CAN-05-4364. [DOI] [PubMed] [Google Scholar]
- 23.Carew JS, Giles FJ, Nawrocki ST. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008;269:7–17. doi: 10.1016/j.canlet.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 24.Roy HK, Iversen P, Hart J, Liu Y, Koetsier JL, Kim Y, Kunte DP, Madugula M, Backman V, Wali RK. Down-regulation of SNAIL suppresses MIN mouse tumorigenesis: modulation of apoptosis, proliferation, and fractal dimension. Mol Cancer Ther. 2004;3:1159–1165. [PubMed] [Google Scholar]
- 25.Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol. 2005;168:29–33. doi: 10.1083/jcb.200409067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3β phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- 27.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci USA. 2000;97:10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li GC, Zhang X, Pan TJ, Chen Z, Ye ZQ. Histone deacetylase inhibitor trichostatin A inhibits the growth of bladder cancer cells through induction of p21WAF1 and G1 cell cycle arrest. Int J Urol. 2006;13:581–586. doi: 10.1111/j.1442-2042.2006.01344.x. [DOI] [PubMed] [Google Scholar]
- 29.Zhu J, Zhang S, Jiang J, Chen X. Definition of the p53 functional domains necessary for inducing apoptosis. J Biol Chem. 2000;275:39927–39934. doi: 10.1074/jbc.M005676200. [DOI] [PubMed] [Google Scholar]
- 30.Liu LF. DNA topoisomerase poisons as antitumor drugs. Annu Rev Biochem. 1989;58:351–375. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- 31.Kajita M, McClinic KN, Wade PA. Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol Cell Biol. 2004;24:7559–7566. doi: 10.1128/MCB.24.17.7559-7566.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vega S, Morales AV, Ocaña OH, Valdés F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18:1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1. DDX3 over-expression in MCF-7 cells.
DDX3 was transiently over-expressed in MCF-7 cells. (A) Adenovirus-mediated DDX3 over-expression did not increase the basal level of Snail. (B) DDX3 over-expression did not alter increases in Snail induced by a 3 hr treatment with 1 μM TSA, 5 mM sodium butyrate, or 10 μM camptothecin. Adenovirus-mediated over-expression of green fluorescence protein (GFP) did not alter DDX3 or Snail levels.
Figure 2. Snail levels after treatment with DNA damaging agents.
SH-SY5Y cells were treated with (A) 50-200 μM bleomycin for 6 hr, (B) 100 μM H2O2 for 2, 3, or 4 hr, or (C) 5-20 μM Ara-C for 24 hr, and the levels of Snail, p53, and β-actin were immunoblotted.