

Abstract
Background
Topoisomerase II is essential for the maintenance of DNA integrity and the survival of proliferating cells. Topoisomerase II poisons, including etoposide and doxorubicin, inhibit enzyme-mediated DNA ligation causing the accumulation of double-stranded breaks and have been front-line drugs for the treatment of leukemia for many years. Voreloxin is a first-in-class anti-cancer quinolone derivative that intercalates DNA and inhibits topoisomerase II. The efficacy and mechanisms of action of voreloxin in acute myeloid leukaemia were addressed in this study.
Design and Methods
Primary acute myeloid leukemia blasts (n = 88) and myeloid cell lines were used in vitro to study voreloxin through viability assays to assess cell killing and synergy with other drugs. Apoptosis and cell cycling were assessed by flow cytometry. DNA relaxation assays were utilized to determine that voreloxin was active on topoisomerase II.
Results
The mean lethal dose 50% (LD50) (± standard deviation) of voreloxin for primary acute myeloid leukemia blasts was 2.30 μM (± 1.87). Synergy experiments between voreloxin and cytarabine identified synergism in 22 of 25 primary acute myeloid leukemia samples tested, with a mean combination index of 0.79. Apoptosis was shown to increase in a dose-dependent manner. Furthermore, voreloxin was active in the p53-null K562 cell line suggesting that the action of voreloxin is not affected by p53 status. The action of voreloxin on topoisomerase II was confirmed using a DNA relaxation assay.
Conclusions
Voreloxin may provide an interesting addition to the cache of drugs available for the treatment of acute myeloid leukemia, a disease with a poor long-term survival. In addition to its potent action as a single agent in dividing cells, the synergy we demonstrated between voreloxin and cytarabine recommends further investigation of this topoisomerase II inhibitor.
Keywords: voreloxin, topoisomerase II inhibitor, myeloid leukemia, synergism, cytarabine
Introduction
Acute myeloid leukemia (AML) remains a difficult disease to treat with particular problems in the elderly (over 60 years old) in whom complete remissions are seen in only 50–60% of patients treated with intensive chemotherapy1,2 and are usually transient with less than 10% of patients remaining in remission at 3 years.2 Many other elderly patients are unsuitable for intensive chemotherapy, due to frailty or comorbidity,1 illustrating the need for continuing development of novel treatments for AML.
Topoisomerases are involved in regulating the under-and over-winding of DNA. This process is essential for removing knots and tangles in the DNA that can be lethal to cells if not resolved.3,4 Type I and II topoisomerases (topo I and topo II) are distinguished by the number of DNA strands they cleave and the mechanism by which they alter the topological properties of DNA.3,4 Topo II functions as a homodimer, generating double-stranded breaks and regulating the super-coiling of the DNA before the breaks are rejoined3. In cells with insufficient topo II activity, DNA remains entangled resulting in reduced gene transcription.3 Conversely if a cell has excessive topo II activity the cleavage intermediates it forms with DNA can be converted into permanent strand breaks resulting in the loss of DNA integrity.3 High expression of topoisomerases during cell growth3 combined with the necessity of these enzymes for normal cell functioning make them viable targets for drug development in cancer.
Etoposide is a topo II poison that inhibits the ability of the enzyme to re-ligate cleaved DNA molecules3,5 while other topo II poisons act by stabilizing the transient complex composed of the DNA backbone and topo II resulting in the triggering of DNA damage checkpoints that halt mitosis.4,6–8 Topo II inhibition or poisoning has been successful in a wide range of contexts9 and drugs targeting these agents can be effectively combined with many other agents10 reinforcing the attraction of targeting these enzymes for drug development.
Voreloxin (SNS-595, AG-7352) is a naphthyridine analog that intercalates into DNA and results in poisoning the action of topo II.11 The intercalation of voreloxin into DNA is both replication-dependent and site-specific and results in double-stranded DNA damage, arrest of the cell cycle in G2 and subsequent apoptosis. Previous work has shown that voreloxin is not a P-gp substrate; it has antitumor activity in tumor cell lines, and in both solid tumor and hematologic xenograft models.11
Voreloxin is one of a number of emerging novel therapies of AML with which it is hoped to augment the current cytarabine and anthracycline-based approaches to treatment.12 Phase 2 clinical trials of voreloxin are currently underway in relapsed and refractory acute leukemias in combination with cytarabine (Ara-C) and also in ovarian cancer.13
The aims of this study were to determine the efficacy of voreloxin in AML cells in vitro as a single agent and in combination with cytarabine and to confirm the mechanism of action of this potentially important drug in leukemia cells.
Design and Methods
Cell culture
Myeloid cell lines NB4, HL-60 and K562 were maintained at concentrations between 2×105 and 1×106 cells/mL in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum (FBS), 50 U penicillin/mL and 50 μg streptomycin/mL. Bone marrow (n=26) or peripheral blood (n=62) samples were collected from patients with newly diagnosed AML with the patients’ informed consent using documentation approved by the Wales Multicentre Research Ethics Committee. The patients’ characteristics are shown in Table 1. Primary cells extracted from bone marrow were enriched by density gradient centrifugation with Histopaque (Sigma, Poole, UK) and were maintained in RPMI, with 10% FBS and 1% penicillin and streptomycin. All cultures were kept at 37°C, with 5% CO2. Cell viability was measured on a Vi-Cell XR cell counter (Beckman Coulter, High Wycombe, UK) and expansion of cultures was monitored by cell counts on the same machine.
Table 1.
Characteristics of primary samples used in vitro in this study.

Cytotoxicity assays
In vitro toxicity assays were performed on primary AML mononuclear cells over a 48 h period using a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H- tetrazolium, inner salt (MTS) cell proliferation assay (Promega UK Ltd.). Lethal doses (LD50: at which 50% of the cells were no longer viable) were calculated. Cells were treated with voreloxin (31.25 nM to 4 μM) and Ara-C (62.5 nM to 8 μM) by serial dilution and incubated for 48 h in a final volume of 90 μL. Following the incubation, 20 μL of MTS reagent were added and the reaction was incubated for a further 4 h. The absorbance of the reaction after this time was read by spectrophotometry at 490 nm and the percentage of viable cells calculated relative to untreated control cells in the same assay. IC50 values were calculated using Calcusyn software (Biosoft, Cambridge, UK).
Detection of apoptosis
Cells were treated with voreloxin at doubling concentrations between 0.0313 μM and 0.5 μM and etoposide at concentrations between 0.625 μM and 10 μM and incubated for 48 h at 37°C, 5% CO2 in a final volume of 1 mL. Annexin V positivity was determined in treated cells using a fluorescein isothiocyante-labeled Annexin V Apoptosis Detection Kit (Axxora Ltd, Nottingham, UK) according to the manufacturer’s instructions. Briefly, cells were washed in phosphate-buffered saline (PBS) and resuspended in the supplied binding buffer containing calcium chloride and incubated with annexin V-fluorescein thio-cyanate in the dark for 10 min. Untreated samples were also prepared in this manner. Cells were then washed with PBS and resuspended in the supplied binding buffer and 1 μg/mL propidium iodide added. All data was acquired on an Accuri C6 flow cytometer (St. Ives, Cambs, UK) and analyzed using CFlow Plus software. LD50 values were calculated using CalcuSyn software (Biosoft, Cambridge, UK). All experiments were performed in triplicate.
Additionally cell lines were incubated at 37°C in a humidified 5% CO2 atmosphere in the presence of voreloxin at doubling concentrations between 0.25 μM and 2 μM or etoposide at concentrations between 0.625 μM and 10 μM for 48 h. Cells were then harvested by centrifugation. Subsequently the cells were incubated for 1 h at 37°C in the presence of the PhiPhiLux™ G1D2 substrate (Calbiochem, Nottingham, UK). This substrate contains two fluorophores separated by a quenching linker sequence that is cleaved by active caspase-3. Once cleaved, the resulting products fluoresce green and can be quantified using flow cytometry. Data on the caspase-3 activation of the cells were collected in the same manner as data on annexin V positivity.
Synergy with cytarabine
Synergy between voreloxin and Ara-C was assessed in primary AML samples in vitro. The ratio of voreloxin to Ara-C was 1:2. This ratio was determined experimentally and kept constant throughout the subsequent experiments. Cells were incubated with each agent both separately and in combination for 48 h before assessment of cytotoxicity by an MTS assay as described above. Calcusyn software was used to determine whether any synergy existed between the agents, using the median effect method of Chou and Talalay.14
Cell cycle
Following 12 h of treatment with voreloxin (0.250 μM, 0.5 μM, 1 μM), 1×106 cells from the leukemia cell lines NB4 and HL-60 were harvested in 200 μL cold PBS, resuspended in 1 mL ice cold 70% ethanol and stored at −20°C. Prior to analysis by flow cytometry the stored cells were pelleted and resuspended in 40 μg/mL propidium iodide and 100 μg/mL RNase (DNase-free) in PBS and incubated at 37°C for 30 min. Each cell line was tested in triplicate.
DNA relaxation assay
Relaxation of supercoiled DNA is mediated by topoisomerases I and II.15,16 Isolated topoisomerase IIα (0.2 U) was incubated in reactions containing 50 mM TrisHCl (pH 8.0), 60 mM KCl, 10 mM MgCl2, 10 mM ATP, 0.5 mM DTT, 0.5 mM EDTA, 30 μg/mL bovine serum albumin, 2 μg pBR322 (Axxora (UK) Ltd., Bingham, Nottingham, UK) and voreloxin at concentrations of 0.0625, 0.125, 0.25, 0.5 and 1 μM for 30 min. Following incubation samples were electrophoresed in 1% w/v agarose in TBE buffer before being stained with ethidium bromide and photographed under ultraviolet light.
Results
Voreloxin can kill primary acute myeloid leukemia cells and myeloid cell lines through induction of apoptosis
The in vitro efficacy of voreloxin was tested in 88 mononuclear cell samples isolated from patients with newly diagnosed AML using an MTS assay method following incubation with the drug for 48 h. The cytotoxicity of voreloxin was compared to that of Ara-C over the same duration and in the same cohort of patients. In the primary samples voreloxin had a mean LD50 (the dose required to kill 50% of the cells treated) of 2.3 μM (± 1.9 μM) (Figure 1A). This was less than half the LD50 of Ara-C which was 4.9 μM (± 5 μM) in the same group of patients. These LD50 values for Ara-C and voreloxin are both within ranges reported to be achieved clinically: plasma concentrations of voreloxin of 1.0 μM or more are achievable, although concentrations greater than 10 μM are associated with side effects,17 while plasma concentrations between 54.5 μM18 and 115 μM19 have been reported for Ara-C in patients. The response to voreloxin in the primary samples did not correlate with response to Ara-C, age, sex, French-American-British classification, cytogenetic risk group or whether the patient had de novo or secondary AML (Table 1). In the myeloid cell lines NB4 and HL-60, voreloxin was compared to Ara-C and etoposide (Figure 1B). The mean LD50 for voreloxin in the cell lines was 0.59 μM ± 0.25 μM, that for Ara-C was 0.67 μM ± 0.44 μM and that for etopo-side was 0.27 μM ± 0.10 μM.
Figure 1.

Primary AML blasts from 88 patients were isolated at diagnosis and treated in vitro with (A) voreloxin and Ara-C for 48 h and LD50 values were calculated (the mean LD50 for voreloxin was 2.3 μM (±1.9) and for Ara-C was 4.90 μM (±5.0).) (B) Mean LD50 values for voreloxin, Ara-C and etoposide in myeloid cell lines NB4 and HL-60 following 48 h of treatment with each drug. The effects of each drug were tested using an MTS assay. Annexin V and propidium iodide (PI) positivity were measured by flow cytometry to determine whether (C) voreloxin or (D) etoposide resulted in induction of apoptosis following 48 h treatment of myeloid cell lines NB4 and HL-60 with a range of concentrations. Both voreloxin and etoposide resulted in significant increases in the proportion of cells undergoing apoptosis identified by positivity for annexin V and PI staining across all the concentrations tested. The LD50 for voreloxin in the cell lines was 0.203 μM for NB4 cells and 0.061 μM for HL-60 cells. The LD50 of etoposide was 0.78 μM in NB4 cells and 4.23 μM for HL-60 cells. Activation of caspase-3 was measured by flow cytometry to confirm the pathway of apoptosis initiation in NB4 and HL-60 cells treated for 48 h with (E) voreloxin and (F) etoposide. Both voreloxin and etoposide generated significant activation of caspase-3 in the cell lines at all concentrations tested. Significant differences (P<0.05) with respect to the untreated control samples are indicated in each case by *.
Positivity for both annexin V and propidium iodide is frequently identified by flow cytometry and used to determine whether cells are in the process of undergoing apoptosis.20,21 The human promyelocytic leukemia cell line NB4 and human AML cell line HL-60 were treated with voreloxin for 48 h and the proportion of annexin V and propidium iodide-positive (apoptotic) cells resulting from treatment with voreloxin was identified by flow cytometry (Figure 1C). HL-60 cells were more susceptible to the action of voreloxin with 50% apoptotic cells detectable after incubation with 0.06 μM voreloxin while 0.20 μM voreloxin were required to result in the same level of apoptotic NB4 cells being detected by this method. Etoposide, by comparison (Figure 1D), had LD50 values of 0.78 μM for NB4 cells and 4.23 μM for HL-60 cells over a 48 h period. These concentrations of etoposide are achievable clinically as plasma concentrations of 1.7 to 5.6 μM have been demonstrated.22 Comparing the action of voreloxin and etoposide in the cell lines showed that voreloxin was more effective (mean LD50 0.13 μM) than etopoisde (mean LD50 2.51 μM) over 48 h. The K562 human erythroid leukemic cell line was derived from a patient with chronic myelogenous leukemia in acute myeloid blast crisis.23 This cell line has no functional P53 gene as a result of an insertional mutation in exon 5 of the gene which causes a frameshift at transcription and loss of detectable protein expression.24 Voreloxin resulted in a dose-dependent increase in the percentage of cells on which annexin V and propidium iodide were detectable. The increase in apoptotic cells suggests that voreloxin is still capable of killing cells, even in the absence of functional p53 protein in the cells.
Caspase-3 activation was also measured in the same cell lines by flow cytometry following 48 h of treatment of the cell lines with voreloxin and etoposide at a range of concentrations (Figure 1E,F). Caspase-3 is activated by cell signaling pathways resulting either from mitochondrial cytochrome c release, or through pathways independent of cytochrome c release, ultimately resulting in the cell undergoing apoptosis.25,26 In both the cell lines tested we saw significant (P<0.01) elevation of the proportion of cells with activated caspase-3 with all concentrations of voreloxin used between 0.250 μM and 2 μM and with etoposide at concentrations between 0.625 μM and 10 μM.
Voreloxin results in accumulation of cells in the S and G2 phases of the cell cycle
NB4 and HL-60 cells were treated for 12 h with voreloxin and the percentages of cells in each phase of the cell cycle were determined by flow cytometry. Both NB4 cells (Figure 2A) and HL-60 cells (Figure 2B) showed a reduced proportion of cells in G1, increased cells in S phase and some increases in the G2 phase of the cell cycle. One way analysis of variance (ANOVA) of the cell cycle changes confirmed that the accumulations in the S and G2 phases were statistically significant (NB4, P=0.0001; HL-60, P=0.0009).
Figure 2.

The effect of voreloxin on cell cycle distribution was investigated by flow cytometry in (A) NB4 and (B) HL-60 cells following 12 h of treatment. Decreases in the proportions of cells in the G1 phase of the cell cycle and increases in the S and G2 phases were observed in both cell lines. The distribution of the cells through the cell cycle was tested by ANOVA and the changes found to be statistically significant (NB4 P<0.0001, HL-60 P=0.0009).
Voreloxin acts on topoisomerase II
Relaxed topological isomers of DNA migrate more slowly through agarose gels than the supercoiled species. Relaxation of a supercoiled DNA substrate is mediated by topo II.15,16 Using the supercoiled plasmid pBR322 as a substrate we demonstrated that in the presence of topoisomerase IIα in vitro the majority of the pBR322 was in a relaxed state (Figure 3) by visualization of its slower migration rate during electrophoresis through an agarose gel compared to the small amount of plasmid remaining supercoiled. Addition of voreloxin resulted in a concentration-dependent increase in the amount of supercoiled pBR322 visible in the agaorse gel and distinct from the relaxed plasmid due to its faster migration rate. These results confirm that voreloxin inhibits the activity of topo II.
Figure 3.
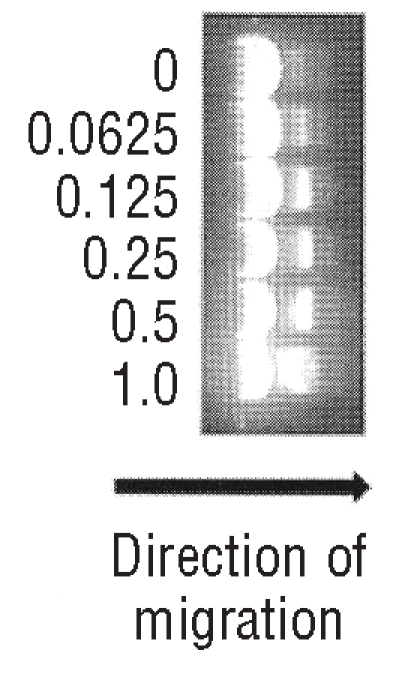
DNA relaxation is mediated by topoisomerases. We incubated a supercoiled plasmid, pBR322 with isolated topoisomerare II in the presence of increasing concentrations of voreloxin. DNA migrates through agarose gels more slowly in its relaxed state so the presence of increasing levels of super-coiled DNA with increasing voreloxin concentration (μM) illustrates that voreloxin is acting on the action of topoisomerase II.
Voreloxin acts in synergy with cytarabine
Primary AML samples from patients (n=25) were used to determine whether voreloxin and Ara-C act in synergy over 48 h. The molar ratio at which the agents were combined was voreloxin 1:2 Ara-C which was determined experimentally and kept constant throughout the experiments. The ratio of 2:1 Ara-C to voreloxin was based on the LD50 values of each drug singly: the drugs were combined at their LD50 values as the LD50 for Ara-C was approximately double that of voreloxin giving a ratio of 2:1. A combination index was derived for each patient’s sample, indicating whether the agents act in a synergistic (<0.9), additive (0.9-1.1) or antagonistic (>1.1) manner.14 The combination index for each patient, calculated at the LC50, is shown in Figure 4. All but three of the primary samples had a combination index of less than 1. The mean combination index was 0.7983 ± 1.152 indicating that synergy exists between the two agents. The three samples that did not demonstrate synergy were found to be from patients who responded very well to either Ara-C, voreloxin or to both agents when used separately. In these cases it may be possible to demonstrate synergy between the agents if the ratio between them were to be altered to allow for the apparently greater susceptibility to one of the agents. Additionally, synergy between voreloxin and Ara-C and also etoposide and Ara-C was investigated in the cell lines. The ratio of Ara-C and voreloxin was kept the same as that used for the primary samples. The different sensitivities of the cell lines to voreloxin may account for the different levels of synergy seen when combined with Ara-C at this ratio. The 1:1 ratio used to combine Ara-C and etoposide was based on the LD50 of Ara-C in the primary samples and previously published data suggesting that therapeutic effects of etoposide were seen at similar levels. Voreloxin and Ara-C showed synergy (combination index = 0.68±0.22) in NB4 cells, as did etoposide combined with Ara-C at a 1:1 ratio (combination index = 0.54±0.04). In contrast, in HL-60 cells the combination of Ara-C and voreloxin had an additive effect (combination index = 0.99±0.46) while the combination of Ara-C and etoposide had an antagonistic effect (combination index = 17.9±15.4).
Figure 4.

Synergy between voreloxin and Ara-C was investigated in 25 primary AML samples over 48 h in vitro. The ratio between voreloxin and Ara-C was kept constant at 2:1 at all times. This ratio was determined experimentally based on the relative sensitivity of primary samples to voreloxin and Ara-C. (A) Dose response curves to voreloxin, Ara-C and both drugs combined in a representative primary AML sample illustrating the different responses to voreloxin, Ara-C and both drugs in combination (B) Fractional effect plot where synergy (identified by a combination index <1) is demonstrated over a range of drug concentrations. (C) Combination index plot. The combination index at the LD50 of all 25 primary samples tested is shown. Synergy between the two agents was found in 22 of the 25 samples. The samples in which synergy was not shown were particularly sensitive to one of the agents when tested separately. The mean combination index was 0.798. (D) The combination index (CI) values for the combinations of voreloxin plus Ara-C and of Ara-C plus etoposide are shown for NB4 and HL-60 cells. The combination of Ara-C and either voreloxin or etoposide showed synergy in NB4 cells while in HL-60 cells there was an additive response when voreloxin and Ara-C were combined and antagonism when Ara-C and etoposide were used in combination.
Discussion
AML remains a difficult disease to treat and the majority of patients have a short survival.27,28 Topoisomerase inhibitors have been used in cancer treatment for many years,9 showing significant activity in a variety of human malignancies, and studies continue to identify new agents that target topo II with greater potency and reduced toxicity than previously used drugs.29
The cell killing activity of voreloxin was demonstrated in primary AML samples taken from patients at diagnosis. In these cells the dose-dependent activity was twice that of Ara-C in the same samples with a mean LD50 of 2.3 μM for voreloxin (n = 88). In myeloid cell lines the activity of voreloxin was compared to that of etoposide and was also found to be more effective in this comparison (LD50 of 132 nM for voreloxin and of 2.51 μM for etoposide). Etoposide was not used on the primary samples because of the paucity of the material available. The different sensitivity of primary AML blasts and AML cell lines to voreloxin likely results from the difference in growth dynamics between these cell types. Different sensitivities to treatment between primary cells and cell lines have previously been documented in osteosarcoma30 and cultured cell lines identified as putting more of their resources into proliferative functions than do primary cells.31 Myeloid cell lines actively grow in vitro and both the cell lines that were used in these experiments divide approximately once every 24 h while cells isolated from AML patients grow much more slowly in culture which may explain the difference we found in concentrations resulting in death induction between the cell types.
Voreloxin mediates apoptosis and retains some activity in cells with a deficiency of functional p53 protein. Previously published reports indicated that cells lacking p53 require higher doses of Ara-C to undergo apoptosis32 or are relatively unaffected by treatment with Ara-C33 while the efficacy of etoposide is dramatically reduced in p53-null cells in comparison with p53-expressing cells.34 The retention of voreloxin’s activity in cells not expressing p53 is relevant to the development of this agent as p53 is a member of a key pathway mediating cell death following DNA damage.32 Mutations in the tumor suppressor protein P53 gene are common in AML, particularly in therapy-related AML35,36 and the presence of a P53 mutation correlates with resistance to chemotherapy and a shorter survival.35 Novel agents whose mechanism of action do not require functional p53 will provide additional treatment options in p53-mutant patients who have a shorter survival.
The accumulation of cells treated with voreloxin in the S and G2 phases of the cell cycle is in agreement with previous reports of inhibition of topo II arresting mammalian cells in G27,37,38 or resulting in apoptosis of cells in S phase.7,8 The expression of topoisomerase II had been correlated to voreloxin-induced G2 cell cycle arrest.38 It has also been observed that sensitivity to topo II inhibitors increases in proportion to the proportion of cells in the S phase (etoposide)39 or in the S and G2 phases (teniposide)40 although another study in myelodysplastic syndromes found G0/G1 accumulation following treatment with etoposide.41 Ara-C, by comparison, results in G1 arrest in treated AML samples.42 Voreloxin has previously been shown to result in double-stranded breaks in DNA in acute lymphoblastic leukemia cells. The double-stranded breaks are mediated by topoisomerase II and are not repaired once topoisomerase II has been poisoned.38
The demonstration of synergy between Ara-C and voreloxin is consistent with the findings of previous studies using other topoisomerase inhibitors in combination with Ara-C. Specifically, etoposide and Ara-C showed synergy in non-Hodgkin’s lymphoma,43 topotecan combined with Ara-C was an effective combination in patients with myelodysplastic syndromes44 and fludarabine, Ara-C and topotecan were effectively combined in elderly AML patients.1 Synergy between voreloxin and Ara-C has been demonstrated both in leukemic cell lines and in an in vivo study.45
In summary, it has been shown that voreloxin inhibits the action of topo II and that, in vitro, it is more potent than both Ara-C and etoposide. It retains activity in the absence of a functional p53 protein, induces apoptosis and acts in synergy with Ara-C. Coupled with previous work demonstrating that voreloxin has similar degrees of activity in cell lines with high and low topoisomerase II levels, has potent activity in aggressive and drug-resistant tumor models,11 and does not result in significant induction of reactive oxygen species which have been linked to drug cardiotoxicity,38 the data we present here make a strong argument for continued investigation into the efficacy of voreloxin as a treatment for patients with AML. Clinical trials are currently underway in ovarian cancer and AML.
Footnotes
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Leoni F, Ciolli S, Nozzoli C, Santini V, Fanci R, Rossi FP. Fludarabine, cytarabine and topotecan (FLAT) as induction therapy for acute myeloid leukemia in the elderly: a preliminary report. Haematologica. 2001;86 (1):104. [PubMed] [Google Scholar]
- 2.Estey EH. How I treat older patients with AML. Blood. 2000;96(5):1670–3. [PubMed] [Google Scholar]
- 3.Deweese JE, Osheroff N. The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing. Nucleic Acids Res. 2009;37(3):738–48. doi: 10.1093/nar/gkn937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baxter J, Diffley JF. Topoisomerase II inactivation prevents the completion of DNA replication in budding yeast. Mol Cell. 2008;30(6):790–802. doi: 10.1016/j.molcel.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 5.Lage H, Helmbach H, Dietel M, Schadendorf D. Modulation of DNA topoisomerase II activity and expression in melanoma cells with acquired drug resistance. Br J Cancer. 2000;82(2):488–91. doi: 10.1054/bjoc.1999.0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jenkins JR. A proteomic approach to identifying new drug targets (potentiating topoisomerase II poisons) Br J Radiol. 2008;(Spec No 1):81. S69–S77. doi: 10.1259/bjr/76952340. [DOI] [PubMed] [Google Scholar]
- 7.Barry MA, Reynolds JE, Eastman A. Etoposide-induced apoptosis in human HL-60 cells is associated with intracellular acidification. Cancer Res. 1993;53(10 Suppl):2349–57. [PubMed] [Google Scholar]
- 8.Clark PI, Slevin ML. The clinical pharmacology of etoposide and teniposide. Clin Pharmacokinet. 1987;12(4):223–52. doi: 10.2165/00003088-198712040-00001. [DOI] [PubMed] [Google Scholar]
- 9.Hande KR. Etoposide: four decades of development of a topoisomerase II inhibitor. Eur J Cancer. 1998;34(10):1514–21. doi: 10.1016/s0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- 10.Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 2009;9(5):338–50. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoch U, Lynch J, Sato Y, Kashimoto S, Kajikawa F, Furutani Y, et al. Voreloxin, formerly SNS-595, has potent activity against a broad panel of cancer cell lines and in vivo tumor models. Cancer Chemother Pharmacol. 2009;64(1):53–65. doi: 10.1007/s00280-008-0850-3. [DOI] [PubMed] [Google Scholar]
- 12.Fathi AT, Karp JE. New agents in acute myeloid leukemia: beyond cytarabine and anthracyclines. Curr Oncol Rep. 2009;11 (5):346–52. doi: 10.1007/s11912-009-0047-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evanchik MJ, Allen D, Yoburn JC, Silverman JA, Hoch U. Metabolism of (+)−1,4-dihydro-7-(trans-3-methoxy-4-methylamino-1-pyrrolidinyl)-4-oxo-1-( 2-thiazolyl) −1,8-naphthyridine-3-carboxylic acid (voreloxin; formerly SNS-595), a novel replication-dependent DNA-damaging agent. Drug Metab Dispos. 2009;37(3):594–601. doi: 10.1124/dmd.108.023432. [DOI] [PubMed] [Google Scholar]
- 14.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 15.Barrett JF, Sutcliffe JA, Gootz TD. In vitro assays used to measure the activity of topoisomerases. Antimicrob Agents Chemother. 1990;34(1):1–7. doi: 10.1128/aac.34.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kellner U, Hutchinson L, Seidel A, Lage H, Danks MK, Dietel M, et al. Decreased drug accumulation in a mitoxantrone-resistant gastric carcinoma cell line in the absence of P-glycoprotein. Int J Cancer. 1997;71(5):817–24. doi: 10.1002/(sici)1097-0215(19970529)71:5<817::aid-ijc20>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 17.Advani RH, Hurwitz HI, Gordon MS, Ebbinghaus SW, Mendelson DS, Wakelee HA, et al. Voreloxin, a first-in-class anti-cancer quinolone derivative, in relapsed/refractory solid tumors: a report on two dosing schedules. Clin Cancer Res. 2010;16(7):2167–75. doi: 10.1158/1078-0432.CCR-09-2236. [DOI] [PubMed] [Google Scholar]
- 18.Burk M, Heyll A, Arning M, Volmer M, Fartash K, Schneider W. Pharmacokinetics of high-dose cytarabine and its deamination product – a reappraisal. Leuk Lymphoma. 1997;27(3–4):321–7. doi: 10.3109/10428199709059686. [DOI] [PubMed] [Google Scholar]
- 19.Capizzi RL, Yang JL, Cheng E, Bjornsson T, Sahasrabudhe D, Tan RS, et al. Alteration of the pharmacokinetics of high-dose ara-C by its metabolite, high ara-U in patients with acute leukemia. J Clin Oncol. 1983;1 (12):763–71. doi: 10.1200/JCO.1983.1.12.763. [DOI] [PubMed] [Google Scholar]
- 20.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phos-phatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods. 1995;184 (1):39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 21.van EM, Nieland LJ, Ramaekers FC, Schutte B, Reutelingsperger CP. Annexin V-affinity assay: a review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry. 1998;31(1):1–9. doi: 10.1002/(sici)1097-0320(19980101)31:1<1::aid-cyto1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 22.guilar Ponce JL, Flores-Picazo Y, Perez-Urizar J, Castaneda-Hernandez G, Zinser-Sierra JW, Duenas-Gonzalez A, et al. Bioavailability of etoposide after oral administration of the solution marketed for intravenous use: therapeutic and pharmacoeconomic perspectives. Arch Med Res. 1999;30(3):212–5. doi: 10.1016/s0188-0128(99)00014-7. [DOI] [PubMed] [Google Scholar]
- 23.Lozzio BB, Lozzio CB. Properties of the K562 cell line derived from a patient with chronic myeloid leukemia. Int J Cancer. 1977;19(1):136. doi: 10.1002/ijc.2910190119. [DOI] [PubMed] [Google Scholar]
- 24.Law JC, Ritke MK, Yalowich JC, Leder GH, Ferrell RE. Mutational inactivation of the p53 gene in the human erythroid leukemic K562 cell line. Leuk Res. 1993;17(12):1045–50. doi: 10.1016/0145-2126(93)90161-d. [DOI] [PubMed] [Google Scholar]
- 25.Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6(2):99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Lenardo MJ. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J Cell Sci. 2000;113(5):753–7. doi: 10.1242/jcs.113.5.753. [DOI] [PubMed] [Google Scholar]
- 27.Robak T, Wrzesien-Kus A. The search for optimal treatment in relapsed and refractory acute myeloid leukemia. Leuk Lymphoma. 2002;43(2):281–91. doi: 10.1080/10428190290006053. [DOI] [PubMed] [Google Scholar]
- 28.Estey E. New drugs in acute myeloid leukemia. Semin Oncol. 2008;35(4):439–48. doi: 10.1053/j.seminoncol.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 29.Hande KR. Topoisomerase II inhibitors. Update Cancer Ther. 2008;3:13–26. [Google Scholar]
- 30.Graat HC, Witlox MA, Schagen FH, Kaspers GJ, Helder MN, Bras J, et al. Different susceptibility of osteosarcoma cell lines and primary cells to treatment with oncolytic adenovirus and doxorubicin or cisplatin. Br J Cancer. 2006;94(12):1837–44. doi: 10.1038/sj.bjc.6603189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pan C, Kumar C, Bohl S, Klingmueller U, Mann M. Comparative proteomic phenotyping of cell lines and primary cells to assess preservation of cell type-specific functions. Mol Cell Proteomics. 2009;8(3):443–50. doi: 10.1074/mcp.M800258-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Truong T, Sun G, Doorly M, Wang JY, Schwartz MA. Modulation of DNA damage-induced apoptosis by cell adhesion is independently mediated by p53 and c-Abl. Proc Natl Acad Sci USA. 2003;100(18):10281–6. doi: 10.1073/pnas.1635435100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanno S, Higurashi A, Watanabe Y, Shouji A, Asou K, Ishikawa M. Susceptibility to cytosine arabinoside (Ara-C)-induced cyto-toxicity in human leukemia cell lines. Toxicol Lett. 2004;152(2):149–58. doi: 10.1016/j.toxlet.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 34.Attardi LD, de VA, Jacks T. Activation of the p53-dependent G1 checkpoint response in mouse embryo fibroblasts depends on the specific DNA damage inducer. Oncogene. 2004;23(4):973–80. doi: 10.1038/sj.onc.1207026. [DOI] [PubMed] [Google Scholar]
- 35.Wattel E, Preudhomme C, Hecquet B, Vanrumbeke M, Quesnel B, Dervite I, et al. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood. 1994;84 (9):3148–57. [PubMed] [Google Scholar]
- 36.Ben-Yehuda D, Krichevsky S, Caspi O, Rund D, Polliack A, Abeliovich D, et al. Microsatellite instability and p53 mutations in therapy-related leukemia suggest mutator phenotype. Blood. 1996;88(11):4296–303. [PubMed] [Google Scholar]
- 37.Downes CS, Clarke DJ, Mullinger AM, Gimenez-Abian JF, Creighton AM, Johnson RT. A topoisomerase II-dependent G2 cycle checkpoint in mammalian cells. Nature. 1994;372(6505):467–70. doi: 10.1038/372467a0. [DOI] [PubMed] [Google Scholar]
- 38.Hawtin RE, Stockett DE, Byl JA, McDowell RS, Nguyen T, Arkin MR, et al. Voreloxin is an anticancer quinolone derivative that intercalates DNA and poisons topoisomerase II. PLoS One. 2010;5(4):e10186. doi: 10.1371/journal.pone.0010186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chow KC, Ross WE. Topoisomerase-specific drug sensitivity in relation to cell cycle progression. Mol Cell Biol. 1987;7(9):3119–23. doi: 10.1128/mcb.7.9.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Del BG, Skierski JS, Darzynkiewicz Z. The concentration-dependent diversity of effects of DNA topoisomerase I and II inhibitors on the cell cycle of HL-60 cells. Exp Cell Res. 1991;195(2):485–91. doi: 10.1016/0014-4827(91)90400-o. [DOI] [PubMed] [Google Scholar]
- 41.Doll DC, Kasper LM, Taetle R, List AF. Treatment with low-dose oral etoposide in patients with myelodysplastic syndromes. Leuk Res. 1998;22(1):7–12. doi: 10.1016/s0145-2126(97)00149-5. [DOI] [PubMed] [Google Scholar]
- 42.Banker DE, Groudine M, Willman CL, Norwood T, Appelbaum FR. Cell cycle perturbations in acute myeloid leukemia samples following in vitro exposures to therapeutic agents. Leuk Res. 1998;22(3):221–39. doi: 10.1016/s0145-2126(97)00174-4. [DOI] [PubMed] [Google Scholar]
- 43.Gentet JC, Patte C, Quintana E, Bergeron C, Rubie H, Pein F, et al. Phase II study of cytarabine and etoposide in children with refractory or relapsed non-Hodgkin’s lymphoma: a study of the French Society of Pediatric Oncology. J Clin Oncol. 1990;8(4):661–5. doi: 10.1200/JCO.1990.8.4.661. [DOI] [PubMed] [Google Scholar]
- 44.Pagano L, Mele L, Voso MT, Chiusolo P, Putzulu R, Mazzotta S, et al. The association of topotecan and cytarabine in the treatment of secondary or relapsed acute myeloid leukemia. Haematologica. 2001;86 (4):440–1. [PubMed] [Google Scholar]
- 45.Scatena CD, Kumer JL, Arbitrario JP, Howlett AR, Hawtin RE, Fox JA, et al. Voreloxin, a first-in-class anticancer quinolone derivative, acts synergistically with cytarabine in vitro and induces bone marrow aplasia in vivo. Cancer Chemother Pharmacol. 2010;66(5):881–8. doi: 10.1007/s00280-009-1234-z. [DOI] [PMC free article] [PubMed] [Google Scholar]