

Abstract
Previous studies have implicated a role of heterotrimeric Gαi proteins in lipopolysaccharide (LPS) -induced inflammatory responses. We hypothesized that Toll-like receptor (TLR) signaling regulates Gαi proteins, which are anti-inflammatory in endotoxemia and polymicrobial sepsis. Raw 264.7 cells were stimulated with LPS and the Gαi-GTP protein complex was immunoprecipitated with a Gαi protein activation assay. In subsequent in vivo studies, the Gαi protein inhibitor pertussis toxin (PTx) or Gi protein agonist mastoparan (MP-7) were administrated prior to endotoxemia. LPS-induced pro-inflammatory cytokines and mortality were determined. To examine the role of Gαi2 in sepsis, Gαi2 (−/−) and wildtype (WT) mice were subjected to cecal ligation and puncture (CLP) and monitored every 24 hours for 120 hours. Other mice were sacrificed 24 hours after CLP. Peritoneal fluid, blood, and tissue samples were collected. Plasma pro-inflammatory cytokine production, bacterial load in peritoneal fluid, blood and lung tissue, myeloperoxidase (MPO) activity in lung and liver and different immune cell populations in spleen were studied. We found that Gαi proteins are rapidly activated by LPS followed by rapid inactivation. These studies provide the first direct evidence that Gαi proteins are modulated by TLR signaling. In following studies, PTx augmented LPS-induced plasma TNFα, IL-6, whereas MP-7 suppressed LPS-induced TNFα and decreased LPS-induced mortality. In sepsis studies, the survival rate post-CLP was significantly decreased in the Gαi2 (−/−) mice compared to WT mice. CLP-induced plasma TNFα, IL-6, bacterial load in peritoneal fluid, blood and lung tissue and lung and liver MPO activity were significantly increased in Gαi2 (−/−) compared to WT mice. Gαi2 (−/−) mice also exhibited increased Th1 and Th2 responses compared to WT mice. Taken together, Gαi proteins are activated by LPS and negatively regulate endotoxemia and sepsis. Understanding the role of Gαi2 protein in regulation of the inflammatory response in sepsis may provide novel targets for treatment of sepsis.
Keywords: Gαi protein, cecal ligation and puncture, LPS, pertussis toxin, mastoparan
1. Introduction
Sepsis leading to septic shock and multiple organ failure is the leading cause of death in intensive care units and remains a major health problem in the United States and worldwide [1,2]. Sepsis is a systemic maladaptive host response to invasive infection. The interaction of bacterial components such as LPS, peptidoglycan, lipoteichoic acid, and exotoxins with macrophages, monocytes, endothelial cells or other host cells induces the release of inflammatory mediators that play a major role in the pathophysiology of septic shock [3,4]
Toll-like receptors (TLR) expressed on host cells play critical roles in the recognition of pathogen-associated molecular patterns and damage-associated molecular patterns. TLRs are an evolutionarily conserved family of pattern recognition receptors that mediate anti-microbial responses [5]. TLR4 recognizes the Gram-negative bacteria cell wall component LPS by forming a multi-molecular protein complex with CD14 and the secreted protein MD-2 [6]. The G protein coupled receptor chemokine receptor CXCR4 co-localizes with TLR4 in lipid rafts and has been implicated in the initial steps of LPS recognition and signaling [7]. Binding to TLR4 leads to activation of a series of signaling proteins leading to activation of transcription factors and pro-inflammatory gene expression [8].
Previous studies have implicated heterotrimeric Gi proteins in signaling leading to inflammatory mediator production induced by LPS. It has been shown that Gαi proteins are co-immunoprecipitated with the CD14 receptor [9]. Mastoparan, a Gi protein agonist suppressed LPS- induced cytokine production in human monocytes, human dermal microvessel endothelial cells, primary murine macrophages, and RAW 264.7 cells [9–11]. In contrast, pertussis toxin (PTx), a Gαi protein inhibitor, or dominant negative Gαi3 or Gαi2 minigene plasmid blocked TLR4 induced ERK1/2 activation [12]. On the other hand, constitutively active Gαi2 and Gαi3 plasmids potentiated TLR4-induced ERK1/2 phosphorylation [12]. An anti-inflammatory role for ERK 1/2 MAP kinase and PI3 kinase has been reported [13–16]. Splenocytes from Gαi2(−/−) mice but not the Gαi1/3(−/−) mice exhibited significantly higher cytokine and chemokine production in response to LPS stimulation [17]. The in vivo inflammatory response of Gαi2(−/−) mice to LPS challenge was significantly augmented, as measured by increased plasma TNFα as well as lung and liver leukosequestration relative to WT mice [18]. These findings together suggest an anti-inflammatory role of Gi protein.
To date, how Gαi proteins are involved in TLR signaling and the role of Gαi2 protein in polymicrobial sepsis induced inflammation has not been investigated. We employed a Gαi protein activation assay to demonstrate that activation of Gαi proteins are modulated by LPS stimulation. In subsequent in vivo studies, the Gαi protein inhibitor PTx and agonist MP-7 modulated acute endotoxemia. By employing Gαi2(−/−) mice, we demonstrated that Gαi2 negatively regulates polymicrobial sepsis induced by cecal ligation and puncture (CLP). Understanding the role of Gαi2 in polymicrobial sepsis may provide a novel therapeutic target to sepsis.
2. Materials and methods
2.1 Mice
Gαi2(−/−) mice and littermate WT mice with 129Sv background were generated by breeding heterozygous animals as described previously [18]. Studies employed 5 to 8 week old Gαi2(−/−) and age matched WT mice and 7 to 8 week old CD-1 mice. The original knockout mice were obtained from Dr. Lutz Birnbaumer. (NIH, Research Triangle Park, NC). Gαi2(−/−) mice at 5 to 8 weeks old appear healthy with normal colon and there is no diarrhea. There are no significant difference in fecal bacterial colony counts. Fecal peritonitis induced inflammatory response with fecal preparations derived from WT and Gαi2(−/−) mice were not significantly different (data not shown). The latter observations suggest no change in gut microflora virulence between WT and Gαi2(−/−) mice. The investigations conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and operated under the approval of the institutional animal care and use committee.
2.2 Cell culture and immunoprecipitation
RAW 264.7 cells were grown in Dulbecco’s modified Eagle’s medium (Gibco Invitrogen Corporation, Carlsbad, CA) supplemented with heat inactivated 10% fetal bovine serum (Cellgro Mediatech Inc., Herndon, VA), 2% Penicillin/streptomycin (BioWhittaker Inc., Walkersville, MD) in 150 cm2 tissue culture flasks and maintained at 37°C in 5% CO2, 95% air. The confluent cells were detached using 0.05% trypsin-EDTA (Gibco Invitrogen Corporation, Carlsbad, CA) and passaged every 2–3 days. RAW 264.7 cells within 20 passages were used for experiments.
RAW 264.7 cells were stimulated with LPS (500ng/ml, ultra pure LPS from Escherichia coli O111:B4, List Laboratories, Campbell, CA) for various periods of time. Activated Gαi protein was immunoprecipitated with a Gαi protein activation assay kit (New East Biosciences, Malvern PA). Briefly, cells were lysed with lysis buffer provided in the kit and Gαi-GTP complex was immunoprecipitated with an antibody that specifically bound to the Gαi-GTP complex. The immunoprecipitated proteins were subjected to Western blot with anti-Gαi protein antibody.
2.3 Endotoxemia
Endotoxemia was induced by intraperitoneal injection of LPS (125mg/kg for survival study and 25mg/kg for plasma TNFα and IL-6 levels). PTx (4mg/kg) or B-oligomer (4mg/kg) was given i.p. to CD-1 mice. Thirty minutes later, LPS (25mg/kg) was administered i.p. One and six hours later plasma was taken for TNFα and IL-6 measurements. A second group of mice received vehicle (PBS) or Mastoparan 7 (6 mg/kg) i.p and was challenged 1 hour later with LPS (25mg/kg) i.p.. One hour after LPS challenge, mice were sacrificed and plasma TNFα was determined by ELISA. A third group of mice received vehicle (PBS) or Mastoparan 7 (6 mg/kg) i.p and challenged 1 hour later with LPS (125mg/kg) i.p.. Survival was monitored every 6 hours for 48 hours. After 48 hours, the mice that survived were sacrificed.
2.4 Cecal ligation and puncture
Cecal ligation and puncture (CLP) was performed in Gαi2(−/−) mice and littermate WT mice as previously described [19]. Specifically, mice were anaesthetized with ether and a midline incision was made below the diaphragm to expose the cecum. The cecum was ligated at the cecal junction with a 5-0 silk suture without interrupting intestinal continuity and punctured twice with a 22-gauge needle. The cecum was returned to the abdomen, and the incision was closed in layers with a 5-0 silk suture and wound clips. After the procedure, the animals were fluid resuscitated with sterile saline injected subcutaneously. Sham operation was performed the same as CLP except for the ligation and puncture of the cecum.
Fifteen mice per group of WT and Gαi2(−/−) were subjected to CLP and fifteen mice per group of WT and nine mice per group of Gαi2(−/−) were subjected to a sham operation. Mouse survival was monitored every 24 hours for a total of 120 hours. For the mediator studies 3–6 mice per group were subjected to sham and CLP and sacrificed after twenty four hours. The plasma was collected and analyzed for TNFα and IL-6 production. The lung and liver were collected to determine myeloperoxidase activity. Peritoneal fluid, blood and lung tissue were collected to determine bacterial load. The spleen was isolated and different cell populations in spleen were determined by flow cytometry analysis (see below).
2.5 Assay for TNFα and IL-6 production
TNFα and IL-6 production were measured using an enzyme-linked immunosorbant assay (ELISA) with mouse TNFα or IL-6 ELISA kits (eBioscience, San Diego, CA).
2.6 Bacterial Load
To determine the bacterial load in the peritoneum, the peritoneal cavity was lavaged with 5 ml of sterile PBS and diluted 1000 fold with sterile PBS. To determine the bacterial load in the blood, 100 μl of blood was collected and diluted 100 fold with sterile PBS. To determine the pulmonary bacterial load, the lungs were harvested and equal amounts of wet tissue were homogenized and diluted with 1 ml PBS. One hundred microliters of each dilution were plated on chocolate agar plates (Fisher Scientific, Pittsburgh, PA) and incubated at 37°C for 24 hours under aerobic conditions. Colony-forming units (CFU) were counted. Results were expressed as CFU per milliliter or gram of wet tissue.
2.7 Measurement of myeloperoxidase activity
Myeloperoxidase activity was determined in the lung and liver as an index of neutrophil accumulation as previously described [19]. Tissues were homogenized in a solution containing 0.5% hexa-decyl-trimethylammonium bromide dissolved in 10mM potassium phosphate buffer (pH 7.0) and were centrifuged for 30 min at 20,000×g at 4°C. An aliquot of the supernatant was allowed to react with a solution of tera-methyl-benzidine (1.6mM) and 0.1 mM H2O2. The rate of change in absorbance was measured by spectrophotometry at 650 nm. Myeloperoxidase activity was defined as the quantity of enzyme degrading 1 μmol hydrogen peroxide/min at 37°C and was expressed in units per 100 mg of tissue.
2.8 Flow cytometry analysis of splenocytes
Splenocytes were harvested from Gαi2(−/−) and littermate WT mice as described previously [17,18]. Briefly, spleen tissue was dispersed and passed through a 70μm nylon mesh to obtain single cell suspensions. Red blood cells were removed by adding red blood cells lysis buffer (eBioscience, San Diego, CA) for 2 min at room temperature. Cells were stained with the following antibodies (eBioscience, San Diego, CA): FITC-anti-mouse CD4 (L3T4) antibody for CD4+ cells, APC- anti-mouse CD8a (Ly-2) antibody for CD8+ cells, FITC-anti-mouse MHCII and PE-anti-mouse CD11c antibodies for dendritic cells. Dead cells were excluded by 7-AAD staining. For analysis of different subsets of CD4+ T cells, Foxp3 mouse regulatory T cell staining kit was purchased from eBiosciences. Splenocytes were stained with FITC-anti-mouse CD4 (L3T4) antibody or FITC-anti-mouse CD4 (L3T4) together with APC-anti-mouse CD25 antibodies. Cells were then fixed and permeabilized and stained with the following antibodies: PE-anti-mouse T-Bet antibody for Th1 cells, PE-anti-mouse Gata-3 antibody for Th2 cells, PE-anti-mouse RORγ(t) for Th17 cells and PE-anti-mouse Foxp3 for T-reg cells. Cells were analyzed on a FACSCalibur flow cytometer (BD Pharmingen). Data were analyzed with CellQuest Pro. software.
2.9 Statistical analysis
Data are expressed as the mean ± standard error of the mean (SEM). Statistical significance was determined by analysis of variance (ANOVA) with Fisher’s probable least-squares difference test or Log-rank (Mantel-Cox) Test using GraphPad Prism software. A p< 0.05 value was considered statistically significant.
3. RESULTS
3.1 Heterotrimeric Gαi proteins are modulated by LPS
To determine if there was an effect of LPS on Gαi proteins activation, RAW 264.7 cells were stimulated with LPS (500ng/ml) for selected periods of time and activated Gαi proteins were immunoprecipitated. Before immunoprecipitation, the input proteins were immune bloted with Gαi protein to show equal amounts of protein were loaded for immunoprecipitation. Gαi proteins were rapidly activated by LPS within 2 minutes of stimulation followed thereafter by rapid deactivation (Fig. 1A). The statistical analysis was performed by densitometry scan (p<0.05, N=7, Fig. 1B). Further immune blotting with specific Gαi2 and Gαi3 antibodies suggest that both Gαi2 and Gαi3 protein were activated by LPS (data not shown).
Figure 1. LPS modulated Gαi proteins activation.
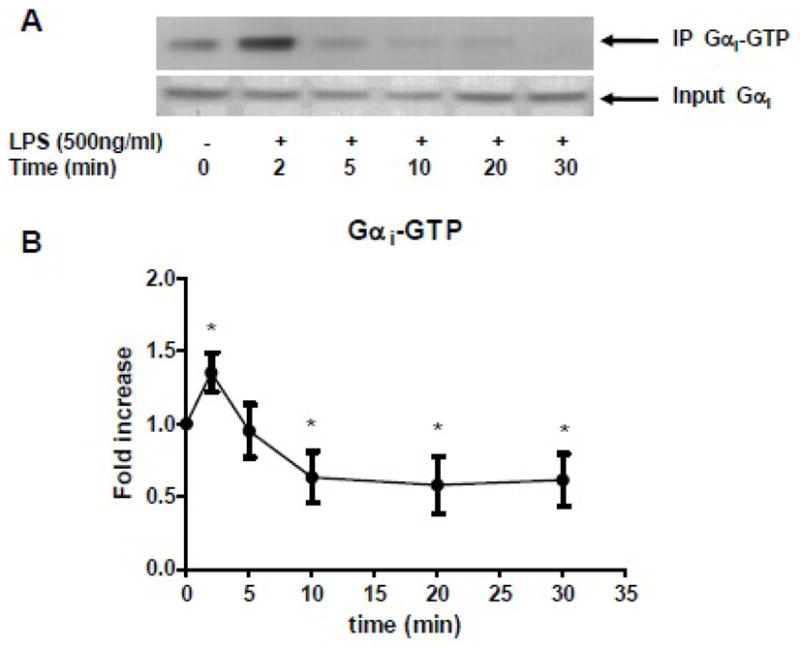
RAW 264.7 cells were stimulated with LPS (500ng/ml). (A) Gαi-GTP complex was immunoprecipitated and subjected to Western blot (top gel). The total Gαi proteins were shown in bottom gel. (B) The statistical analysis was performed on densitometry scan. Data are representative of seven independent experiments. *, p<0.05 compared to basal group.
3.2 Gαi proteins inhibitor PTx and agonist MP-7 modulates acute endotoxemia
To determine the role of Gαi in vivo, PTx (4mg/kg) or MP-7 (6 mg/kg) was injected intraperitoneally in to CD-1 mice prior to the challenge with LPS. LPS induced an increase in plasma inflammatory mediators and survival rates were determined. PTx significantly increased LPS-induced increases in plasma TNFα (2.1±0.4 fold at 1 hour and 2.3±0.1 fold at 6 hour, p<0.05) and IL-6 (3.3±0.3 fold at 1 hour and 1.9±0.1 fold at 6 hour, p<0.05). The negative control B-oligomer group did not show an increased mediator release compared to the PBS treated group (Fig. 2A, 2B). In contrast, the Gαi protein agonist MP-7 decreased LPS-induced plasma TNFα production (82±12% decrease, p<0.05, Fig. 2C). In MP-7 treated mice, challenged with LPS, 67% of mice survived while none of vehicle (PBS) treated mice survived (p<0.05, Fig. 2D).
Figure 2. Effect of PTx and MP-7 on acute endotoxemia induced inflammatory cytokine production and mortality.

CD-1 mice were injected with PTx (4mg/kg) or the inactive B-oligomer (4 mg/kg) 30 min prior to challenge with LPS (25mg/kg). LPS induced plasma TNFα (A) and IL-6 (B) production were determined by ELISA (4 mice/group). MP-7 (6 mg/kg) was also given to CD-1 mice 1 hour prior to challenge with LPS. LPS induced plasma TNFα (C, 4 mice/group ) and survival of mice (D, 10–12 mice/group) was determined. *, p<0.05 compared to control group; #, p<0.05 compared to LPS stimulation group. N/A: not available. Statistical analysis of survival studies was determined with “Log-rank (Mantel-Cox) Test” using GraphPad Prism software.
3.3 Gαi2(−/−) mice exhibit increased mortality in response to CLP
Gαi2 (−/−) and littermate WT mice were subjected to CLP and monitored every 24 hours for 120 hours. Both sham operated Gαi2 (−/−) and WT mice survived 120 hours. Thirty-three percent of WT mice survived out to 120 hours. However, by 96 hours none of the Gαi2 (−/−) mice survived (Fig. 3).
Figure 3. Effect of Gαi2 genetic deletion on survival in mice following CLP.

Gαi2 (−/−) and WT mice were subjected to CLP (n=15/group) and sham operation (n=9–15/group). Survival was monitored every 24 hours for a total of 120 hours. Statistics were determined with “Log-rank (Mantel-Cox) Test” using GraphPad Prism software. *, p<0.05 compared to WT CLP group.
3.4 Gαi2(−/−) mice exhibit increased plasma inflammatory cytokines in response to CLP
To determine the effect of the genetic deletion of Gαi2 on plasma inflammatory cytokines, Gαi2 (−/−) and littermate WT mice were subjected to CLP and sacrificed twenty-four hours later. Plasma TNFα and IL-6 production were determined by ELISA. In both sham operated Gαi2 (−/−) mice and WT mice, plasma TNFα and IL-6 levels were very low. CLP significantly increased plasma TNFα and IL-6 production. CLP-induced plasma TNFα and IL-6 were significantly increased (14±7 and 33±15 fold respectively, p<0.05) in Gαi2 (−/−) compared to WT mice (Fig. 4A, 4B).
Figure 4. Effect of Gαi2 genetic deletion on CLP-induced plasma TNFα and IL-6 production.

Gαi2 (−/−) and WT mice were subjected to CLP and sham operation. Twenty-four hours after CLP, mice were sacrificed. Plasma TNFα (A) and IL-6 (B) levels were measured by ELISA. Data represent means ± SE from 3–5 animals. *, p<0.05 compared to sham group; #, p<0.05 compared to WT CLP group.
3.5 Gαi2(−/−) mice exhibit impaired bacterial clearance following CLP
To determine the effect of the genetic deletion of Gαi2 on bacterial clearance following CLP, Gαi2 (−/−) and littermate WT mice were subjected to sham and CLP operation. Twenty-four hours after CLP, mice were sacrificed. Bacterial load in peritoneal fluid, blood and lung tissue were determined by colony count. Both sham operated Gαi2 (−/−) and WT mice did not have bacteria in peritoneal fluid, blood and lung tissue. Gαi2 (−/−) mice exhibited impaired bacterial clearance as evidenced by increased bacterial load in peritoneal fluid, blood and lung tissue (41±18 fold, 10±5 fold, and 13±5 fold, respectively, p<0.05) compared to WT mice following CLP (Fig.5A, 5B, 5C).
Figure 5. Effect of Gαi2 genetic deletion on bacterial clearance following CLP.

Gαi2 (−/−) and WT mice were subjected to CLP and sham operation. Twenty-four hours after CLP, mice were sacrificed. Bacterial loads in peritoneal fluid (A), blood (B) and lung tissue (C) were determined by colony count. Data represent means ± SE from 5–6 animals. *, p<0.05 compared to sham group; #, p<0.05 compared to WT CLP group.
3.6 Gαi2(−/−) mice exhibit increased MPO activity in response to CLP
Gαi2 (−/−) and littermate WT mice were subjected to sham and CLP operation. Twenty-four hours after CLP, mice were sacrificed. MPO activity was determined in lung and liver tissue. CLP-induced lung and liver MPO activity were significantly increased (2±0.3 and 7±2 fold respectively, p<0.05) in Gαi2 (−/−) compared to WT mice (Fig. 6A, 6B).
Figure 6. Effect of Gαi2 genetic deletion on CLP-induced lung and liver myeloperoxidase activity.

Gαi2 (−/−) and WT mice were subjected to CLP and sham operation. Twenty-four hours after CLP, mice were sacrificed. Lung (A) and liver (B) myeloperoxidase activity was determined. Data represent means ± SE from 3 mice. #, p<0.05 compared to WT CLP group.
3.7 Gαi2(−/−) mice exhibit increased Th1 and Th2 response following CLP
Previous studies demonstrated that the splenocytes from Gαi2 (−/−) mice exhibit augmented IFNγ and IL-12 production compared to WT mice in response to LPS and heat killed Staphylococcus aureus stimulation [17]. These data suggest that Gαi2 protein may play a role in Th1 differentiation. To determine the effect of the genetic deletion of Gαi2 on Th1 and Th2 responses, Gαi2 (−/−) and littermate WT mice were subjected to sham and CLP and sacrificed twenty-four hours later. The spleen was isolated and the different cell populations in spleen were determined by Flow Cytometry analysis. There were no significant differences between Gαi2 (−/−) and WT mice regarding CD4+ cells (Fig. 7A). CD8+ cells were decreased in both sham and CLP Gαi2 (−/−) mice (27±5% and 28±5% respectively, p<0.05, Fig. 7B). Dendritic cells were increased in sham operated Gαi2 (−/−) mice compared to WT mice (1.8±0.1 fold, p<0.05), while there was no significant differences between Gαi2 (−/−) and WT mice in response to CLP (Fig.7C). In sham operated Gαi2 (−/−) and WT mice, Th1 and Th2 cells were not significantly different. However, both Th1 and Th2 cells were increased (2.2±0.2 fold and 1.7±0.1 fold respectively, p<0.05) in Gαi2 (−/−) mice compared to WT mice in response to CLP (Fig. 7D, 7E). Sham operated Gαi2 (−/−) mice exhibited increased Th17 cells compared to WT mice (1.2±0.1 fold, p<0.05), while there was no significant difference in Th17 cells between Gαi2 (−/−) mice and WT mice in response to CLP (Fig. 7F). CLP increased T-reg cells in both Gαi2 (−/−) and WT mice (1.5±0.1 fold and 1.6±0.1 fold respectively, p<0.05), and there was no significant differences in T-reg cells between Gαi2 (−/−) and WT mice (Fig. 7G).
Figure 7. Effect of Gαi2 genetic deletion on immune cell population following CLP.

Gαi2 (−/−) and WT mice were subjected to CLP and sham operation. Twenty-four hours after CLP, mice were sacrificed. The spleens were isolated and CD4+ cells (A), CD8+ cells (B), dendritic cells (C) were determined by Flow Cytometry analysis. The subsets of CD4+ T cells including Th1 cells (D), Th2 cells (E), Th17 cells (F) and T-reg cells (G) were analyzed. Data are representative of 4–5 independent experiments. *, p<0.05 compared to sham group; #, p<0.05 compared to WT CLP group; **, p<0.05 compared to Gαi2 (−/−) sham.
4. DISCUSSION
These studies provide the first evidence that activation of Gαi proteins are modulated by LPS stimulation. We found that Gαi proteins are rapidly activated by LPS followed by rapid inactivation. In vivo studies with acute endotoxemia demonstrated that the Gαi protein inhibitor PTx and agonist MP-7 modulate LPS-induced inflammatory responses and mortality. In the sepsis studies, we found that Gαi2(−/−) mice exhibited increased mortality, plasma TNFα and IL-6 production, bacterial load, pulmonary and hepatic MPO activity, and Th1 and Th2 cells in response to CLP. These data support the notion that Gαi2 proteins are an important negative regulator of inflammation in polymicrobial sepsis.
Our previous studies demonstrated that PTx, dominant negative Gαi3 or Gαi2 minigene plasmid blocked TLR4 induced ERK1/2 activation. Constitutively active Gαi2 and Gαi3 plasmids potentiated TLR4-induced ERK1/2 activation [12]. Since Gαi protein activation mediates ERK 1/2 activation [20], these studies suggested that Gαi proteins were involved in regulating TLR4-mediated signaling. Our studies provide the first direct evidence that LPS activates Gαi proteins in vitro. The Gαi proteins were activated rapidly suggesting that potential autocrine pathways of GPCR activation may not be involved in the activation of Gαi protein. Triantafilou et al. have reported that LPS directly binds CXCR4 and activates CXCR4 induced signaling [21]. It is possible that LPS-induced Gαi protein activation through direct activation of CXCR4. It has also been reported that CXCR4 co-localize with TLR4 in lipid rafts and are involved in LPS recognition and signaling [7]. Our previous studies demonstrated that blockade of Gαi protein activation inhibits TLR4 induced ERK 1/2 activation [12]. Therefore, it is possible that TLR4 transactivates CXCR4 and its cognate Gαi protein. It is also possible that Gαi proteins directly couples to TLR4 or adaptor proteins in the TLR4 signaling cascade. However, these possibilities require further investigation.
Previous studies demonstrated that PTx and MP-7 modulate LPS-induced inflammatory responses in vitro. PTx increased LPS-induced TNFα production in murine macrophages [22]. Our current studies demonstrate that PTx pretreatment of mice increases LPS-induced plasma TNFα and IL-6 production in vivo. Recent studies suggested that PTx not only inhibits Gαi protein activation, it also activates TLR4 through the B-oligomer component [23]. We therefore employed the B-oligomer as a control for the PTx holotoxin. We observed that PTx significantly increased LPS induced inflammatory cytokines while the same concentration of B-oligomer did not have similar augmenting effects. PTx without LPS did not induce TNFα production. Therefore it is reasonable to conclude the effect of PTx on LPS-induced TNFα and IL-6 production is due to its inhibition of Gαi protein activation properties. Previous studies have demonstrated that the Gαi protein agonist MP-7 protected rats from endotoxmia [9]. Our data agree with previous findings that MP-7 decreased LPS-induced plasma TNFα production and mortality in mice. Taken together PTx and MP-7 data support our hypothesis and extend previous findings that Gαi protein negatively regulates LPS-induced inflammatory responses.
The availability of Gαi2(−/−) mice has allowed us to further study the role of Gαi2 protein in vivo and in vitro. In previous studies splenocytes from Gαi2(−/−) mice exhibited significantly higher cytokine and chemokine production in response to LPS stimulation, while Gαi1/3(−/−) mice did not have such an effect [17]. Also LPS-induced plasma TNFα, leukosequestration in the lung and liver were significantly increased in Gαi2(−/−) mice compared to WT mice [18]. Therefore we focused further studies on Gαi2(−/−) mice instead of Gαi1/3 (−/−) mice. The present studies extend previous studies by employing a polymicrobial sepsis model. Our findings demonstrate that Gαi2 proteins also negatively regulate the CLP-induced inflammatory response. We demonstrated that CLP-induced inflammatory cytokines were increased in Gαi2(−/−) mice compared to WT mice. Plasma inflammatory cytokine levels, especially IL-6 levels have been implicated as an indicator of sepsis severity and correlate with sepsis outcome [24]. Gαi2(−/−) mice exhibited increased bacterial load in peritoneal fluid, blood, and lung tissue which may also be the result of impaired chemotaxis. Chemokine receptors are GPCRs and many signal through Gαi [25]. However other G proteins as well as adaptor protein β-arrestins also modulate chemotaxis [26]. It is possible that a deficiency of Gαi may have resulted in impaired recruitment of neutrophils into the peritoneal cavity which would be consistent with the enhanced colony counts in the Gαi2(−/−) mice during sepsis. However increased leukosequestration and bacterial load were observed in the tissue of septic Gαi2(−/−) mice suggesting that chemotaxis was not impaired. Therefore it is possible that altered kinetics of recruitment of cells and/or impaired phagocytosis and bacterial killing activity may account for the increased bacterial load in the Gαi2(−/−) mice.
A limitation of interpreting data from Gαi2(−/−) mice bred on the 129sv background is that they are prone to develop inflammatory bowel disease (IBD) [27]. However the incidence of IBD manifests itself in older mice at 13 weeks of age whereas the animal employed in our study were 5–8 weeks of age. There was no evidence of diarrhea and histology of the colon is normal. Additionally, fecal preparations from either Gαi2(−/−) or WT mice induced equivalent inflammatory responses to fecal peritonitis in recipient normal mice suggesting no difference in virulence of gut microflora (data not shown). Nevertheless the possibility that subclinical colitis at an earlier age in the Gαi2(−/−) mice cannot be excluded.
It has also been demonstrated that CXCR4 is in a “LPS activation cluster” involved in the initial steps of LPS recognition and signaling [7]. CXCR4 blocking antibodies increased TLR4 activation of NFκB [28]. Thus data also suggested that activation of the CXCR4-Gαi signaling pathway maybe anti-inflammatory. The Gi protein coupled ERK 1/2 signaling pathway regulates LPS-induced signaling ]. Activation of Gi protein with transforming growth factor-β (TGF-β) activates ERK 1/2 which can suppress NFκB and p38 signaling and consequently negatively regulate the LPS-induced inflammatory responses ]. It has also been reported that ERK 1/2 negatively regulates p38 signaling through up-regulation of MAP kinase phosphatase (MKP)-1, a serine/threonine phosphatase ]. MKP-1 decreases pro- and anti-inflammatory cytokines in innate immune responses and endotoxic shock ]. These studies suggest that ligands that activate Gi proteins and ERK 1/2 are candidates for negative regulation of LPS-induced signaling pathways.
Recent studies suggest that T lymphocytes and dendritic cells are important in the pathogenesis of sepsis [29,30]. There is a significant loss of T lymphocytes, dendritic cells and increase of T-reg cells during sepsis [29–32]. The T cell subset Th 17 cells also are involved in bacterial clearance [33]. Our current studies also investigated the effect of Gαi2 protein expression on different immune cell populations during sepsis. We observed increased T-reg cells in the spleens of WT mice following CLP, which agrees with previous findings [29,30]. CD8+ cells were decreased in both sham and CLP Gαi2(−/−) mice whereas dendritic cells were increased only in sham Gαi2(−/−) mice. These data suggested that Gαi2 differentially regulates adaptive immune cell populations. We further observed that Th1 and Th2 cells were increased in the spleens of Gαi2(−/−) mice compared to WT mice following CLP. These data agree with our previous finding that splenocytes of Gαi2(−/−) mice produce more IFNγ and IL-12 production in response to LPS in vitro [17]. There were no differences between WT and Gαi2(−/−) mice on Th17 cells and T-reg cells following CLP although Th17 cells were increased in sham Gαi2(−/−) mice. These data suggest that Gαi2 protein negatively regulates both Th1 and Th2 cells following CLP.
Taken together our composite data demonstrate that Gαi proteins are regulated by LPS signaling and Gαi2 is anti-inflammatory in polymicrobial sepsis.
Acknowledgments
This work was supported in part by GM27673 (JAC), GM027673-S1 (JAC), GM67202 (BZ) and AI079248 (HF).
Abbreviations
- ANOVA
analysis of variance
- CLP
cecal ligation and puncture
- FCS
fetal calf serum
- GPCR
G protein coupled receptors
- IL
interleukin
- LPS
lipopolysaccharide
- MP-7
Mastoparan
- MPO
myeloperoxidase
- PTx
Pertussis toxin
- TLR
Toll-like receptor
- TNF
tumor necrosis factor
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Weycker D, Akhras KS, Edelsberg J, Angus DC, Oster G. Long-term mortality and medical care charges in patients with severe sepsis. Crit Care Med. 2003;31:2316–23. doi: 10.1097/01.CCM.0000085178.80226.0B. [DOI] [PubMed] [Google Scholar]
- 3.Karima R, Matsumoto S, Higashi H, Matsushima K. The molecular pathogenesis of endotoxic shock and organ failure. Mol Med Today. 1999;5:123–32. doi: 10.1016/s1357-4310(98)01430-0. [DOI] [PubMed] [Google Scholar]
- 4.Bone RC. Why sepsis trials fail. JAMA. 1996;276:565–6. [PubMed] [Google Scholar]
- 5.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 6.Akashi S, Shimazu R, Ogata H, Nagai Y, Takeda K, Kimoto M, Miyake K. Cutting edge: cell surface expression and lipopolysaccharide signaling via the toll-like receptor 4-MD-2 complex on mouse peritoneal macrophages. J Immunol. 2000;164:3471–5. doi: 10.4049/jimmunol.164.7.3471. [DOI] [PubMed] [Google Scholar]
- 7.Triantafilou K, Triantafilou M, Dedrick RL. A CD14-independent LPS receptor cluster. Nat Immunol. 2001;2:338–45. doi: 10.1038/86342. [DOI] [PubMed] [Google Scholar]
- 8.Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 9.Solomon KR, Kurt-Jones EA, Saladino RA, Stack AM, Dunn IF, Ferretti M, Golenbock D, Fleisher GR, Finberg RW. Heterotrimeric G proteins physically associated with the lipopolysaccharide receptor CD14 modulate both in vivo and in vitro responses to lipopolysaccharide. J Clin Invest. 1998;102:2019–27. doi: 10.1172/JCI4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lentschat A, Karahashi H, Michelsen KS, Thomas LS, Zhang W, Vogel SN, Arditi M. Mastoparan. a G protein agonist peptide, differentially modulates TLR4- and TLR2-mediated signaling in human endothelial cells and murine macrophages. J Immunol. 2005;174:4252–61. doi: 10.4049/jimmunol.174.7.4252. [DOI] [PubMed] [Google Scholar]
- 11.Böcker U, Manigold T, Watson JM, Singer MV, Rossol S. Regulation of Staphylococcus aureus-mediated activation of interleukin-18 in peripheral blood mononuclear cells. Eur Cytokine Netw. 2001;12:631–8. [PubMed] [Google Scholar]
- 12.Fan H, Peck OM, Tempel GE, Halushka PV, Cook JA. Toll-like receptor 4 coupled GI protein signaling pathways regulate extracellular signal-regulated kinase phosphorylation and AP-1 activation independent of NFkappaB activation. Shock. 2004;22:57–62. doi: 10.1097/01.shk.0000129759.58490.d6. [DOI] [PubMed] [Google Scholar]
- 13.Xiao YQ, Freire-de-Lima CG, Janssen WJ, Morimoto K, Lyu D, Bratton DL, Henson PM. Oxidants selectively reverse TGF-beta suppression of proinflammatory mediator production. J Immunol. 2006;176:1209–17. doi: 10.4049/jimmunol.176.2.1209. [DOI] [PubMed] [Google Scholar]
- 14.Xiao YQ, Malcolm K, Worthen GS, Gardai S, Schiemann WP, Fadok VA, Bratton DL, Henson PM. Cross-talk between ERK and p38 MAPK mediates selective suppression of pro-inflammatory cytokines by transforming growth factor-beta. J Biol Chem. 2002;277:14884–93. doi: 10.1074/jbc.M111718200. [DOI] [PubMed] [Google Scholar]
- 15.Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA. 15Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci U S A. 2006;103:2274–9. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Q, Wang X, Nelin LD, Yao Y, Matta R, Manson ME, Baliga RS, Meng X, Smith CV, Bauer JA, Chang CH, Liu Y. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med. 2006;203:131–40. doi: 10.1084/jem.20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan H, Williams DL, Zingarelli B, Breuel KF, Teti G, Tempel GE, Spicher K, Boulay G, Birnbaumer L, Halushka PV, Cook JA. Differential regulation of lipopolysaccharide and Gram-positive bacteria induced cytokine and chemokine production in splenocytes by Galphai proteins. Biochim Biophys Acta. 2006;1763:1051–8. doi: 10.1016/j.bbamcr.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Fan H, Zingarelli B, Peck OM, Teti G, Tempel GE, Halushka PV, Spicher K, Boulay G, Birnbaumer L, Cook JA. Lipopolysaccharide- and gram-positive bacteria-induced cellular inflammatory responses: role of heterotrimeric Galpha(i) proteins. Am J Physiol Cell Physiol. 2005;289:C293–301. doi: 10.1152/ajpcell.00394.2004. [DOI] [PubMed] [Google Scholar]
- 19.Fan H, Bitto A, Zingarelli B, Luttrell LM, Borg K, Halushka PV, Cook JA. Beta-arrestin 2 negatively regulates sepsis-induced inflammation. Immunology. 2010;130:344–51. doi: 10.1111/j.1365-2567.2009.03185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Della Rocca GJ, van Biesen T, Daaka Y, Luttrell DK, Luttrell LM, Lefkowitz RJ. Ras-dependent mitogen-activated protein kinase activation by G protein-coupled receptors, Convergence of Gi- and Gq-mediated pathways on calcium/calmodulin, Pyk2, and Src kinase. J Biol Chem. 1997;272:19125–32. doi: 10.1074/jbc.272.31.19125. [DOI] [PubMed] [Google Scholar]
- 21.Triantafilou M, Lepper PM, Briault CD, Ahmed MA, Dmochowski JM, Schumann C, Triantafilou K. Chemokine receptor 4 (CXCR4) is part of the lipopolysaccharide “sensing apparatus”. Eur J Immunol. 2008;38:192–203. doi: 10.1002/eji.200636821. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Morrison DC. Pertussis toxin-sensitive factor differentially regulates lipopolysaccharide-induced tumor necrosis factor-alpha and nitric oxide production in mouse peritoneal macrophages. J Immunol. 1993;150:1011–8. [PubMed] [Google Scholar]
- 23.Nishida M, Suda R, Nagamatsu Y, Tanabe S, Onohara N, Nakaya M, Kanaho Y, Shibata T, Uchida K, Sumimoto H, Sato Y, Kurose H. Pertussis toxin up-regulates angiotensin type 1 receptors through Toll-like receptor 4-mediated Rac activation. J Biol Chem. 2010;285:15268–77. doi: 10.1074/jbc.M109.076232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock. 2002;17:463–7. doi: 10.1097/00024382-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 25.Allen SJ, Crown SE, Handel TM. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol. 2007;25:787–820. doi: 10.1146/annurev.immunol.24.021605.090529. [DOI] [PubMed] [Google Scholar]
- 26.DeFea KA. Stop that cell! Beta-arrestin-dependent chemotaxis: a tale of localized actin assembly and receptor desensitization. Annu Rev Physiol. 2007;69:535–60. doi: 10.1146/annurev.physiol.69.022405.154804. [DOI] [PubMed] [Google Scholar]
- 27.Rudolph U, Finegold MJ, Rich SS, Harriman GR, Srinivasan Y, Brabet P, Boulay G, Bradley A, Birnbaumer L. Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat Genet. 1995;2:143–50. doi: 10.1038/ng0695-143. [DOI] [PubMed] [Google Scholar]
- 28.Kishore SP, Bungum MK, Platt JL, Brunn GJ. Selective suppression of Toll-like receptor 4 activation by chemokine receptor 4. FEBS Lett. 2005;579:699–704. doi: 10.1016/j.febslet.2004.12.047. [DOI] [PubMed] [Google Scholar]
- 29.Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6:813–22. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]
- 30.Scumpia PO, McAuliffe PF, O'Malley KA, Ungaro R, Uchida T, Matsumoto T, Remick DG, Clare-Salzler MJ, Moldawer LL, Efron PA. CD11c+ dendritic cells are required for survival in murine polymicrobial sepsis. J Immunol. 2005;175:3282–6. doi: 10.4049/jimmunol.175.5.3282. [DOI] [PubMed] [Google Scholar]
- 31.Efron PA, Martins A, Minnich D, Tinsley K, Ungaro R, Bahjat FR, Hotchkiss R, Clare-Salzler M, Moldawer LL. Characterization of the systemic loss of dendritic cells in murine lymph nodes during polymicrobial sepsis. J Immunol. 2004;173:3035–43. doi: 10.4049/jimmunol.173.5.3035. [DOI] [PubMed] [Google Scholar]
- 32.Scumpia PO, Delano MJ, Kelly KM, O'Malley KA, Efron PA, McAuliffe PF, Brusko T, Ungaro R, Barker T, Wynn JL, Atkinson MA, Reeves WH, Salzler MJ, Moldawer LL. Increased natural CD4+CD25+ regulatory T cells and their suppressor activity do not contribute to mortality in murine polymicrobial sepsis. J Immunol. 2006;177:7943–9. doi: 10.4049/jimmunol.177.11.7943. [DOI] [PubMed] [Google Scholar]
- 33.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–88. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]