

Abstract
Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelinating disease (TMEV-IDD) serves as virus-induced model of chronic progressive multiple sclerosis. Infection of susceptible SJL/J mice leads to life-long CNS virus persistence and a progressive autoimmune demyelinating disease mediated by myelin-specific T cells activated via epitope spreading. In contrast, virus is rapidly cleared by a robust CTL response in TMEV-IDD-resistant C57BL/6 mice. We investigated whether differential induction of regulatory T cells (Tregs) controls susceptibility to TMEV-IDD. Infection of disease-susceptible SJL/J, but not B6 mice, leads to rapid activation and expansion of Tregs resulting in an unfavorable CNS ratio of Treg:Teffector cells. In addition, anti-CD25-induced inactivation of Tregs in susceptible SJL/J, but not resistant B6, mice results in significantly decreased clinical disease concomitant with enhanced anti-viral CD4+, CD8+ and antibody responses resulting in decreased CNS viral titers. This is the first demonstration that virus-induced Treg activation regulates susceptibility to autoimmune disease differentially in susceptible and resistant strains of mice and provides a new mechanistic explanation for the etiology of infection-induced autoimmunity.
Keywords: T regulatory cells, CD8+ T cells, Theiler’s virus, demyelination, Multiple sclerosis, chronic viral infection
1. Introduction
The induction of autoimmune diseases, including MS, is considered to be a product of genetics, environment and/or aberrant immune regulation [1,2]. Numerous genes have been linked to MS disease susceptibility with MHC genes being the most strongly associated with disease [3,4]. However, the observation that monozygotic twins have only a 30–35% disease concordance rate strongly suggests that environmental factors (infection) are involved in disease induction [5]. Multiple infectious agents have been linked to susceptibility to MS, with the most recent candidate virus being EBV, although no specific infectious agent has been exclusively linked to MS [2].
Theiler’s murine encephalomyelitis virus (TMEV) is a natural endemic pathogen of mice that causes an induced demyelinating disease (TMEV-IDD) in susceptible strains of mice (e.g., SJL/J, H2KS) that resembles the primary progressive form of MS [2]. TMEV infection results in a life-long persistent virus infection of the CNS leading to development of a chronic T cell-mediated autoimmune demyelinating disease triggered via de novo activation of CD4+ T cell responses to endogenous myelin epitopes in the inflamed CNS (i.e. epitope spreading) [6–8]. Genetic susceptibility to TMEV-IDD is controlled by multiple genes with MHC class I genes playing a predominant role [9–12]. Resistant strains of mice, (e.g., C57BL/6 mice, H2Db) rapidly clear the virus and are protected from TMEV-IDD [13,14]. Thus, TMEV-IDD serves as an instructive model for investigating the interplay of genes, infection and immunoregulation of innate and adaptive immune responses in induction and progression of virus-induced CNS autoimmunity.
It was initially hypothesized that C57BL/6 mice were disease resistant due to their ability to mount a strong and effective CD8+ T cell response that rapidly cleared the viral infection [15,16]. Subsequent results demonstrated the importance of a potent and early CD8+ T cell response in TMEV clearance [17,18]. CD8+ T cells from TMEV-IDD resistant C57BL/6 mice respond to three CD8 epitopes (VP2121–131, VP2165–173, VP2110–120) and T cells specific for all three epitopes produce IFN-γ and efficiently lyse virus-infected target cells [19]. CD8+ T cells from disease-susceptible SJL/J mice also respond to three capsid protein epitopes - the immunodominant epitope - VP3159–166, and two subdominant epitopes - VP3173–181 and VP111–20. CD8+ T cells specific for the two VP3 epitopes produce IFN-γ and lyse virus-infected cells, while CD8+ T cells specific for the VP111–20 epitope produce IFN-γ, but are non-lytic [10,19–21]. However, SJL/J CD8+ T cells have been shown to be decreased in frequency and are less efficient in virus clearance in comparison to CTL responses in disease-resistant C57BL/6 mice [10]. For this reason we wanted to determine if the CD8+ T cell response was differentially regulated in disease-susceptible SJL/J mice vs. disease-resistant C57BL/6 mice.
Natural regulatory T cells (nTregs - CD4+CD25+Foxp3+), which develop in the thymus, have been shown to limit the activation, trafficking, and effector functions of both CD4+ and CD8+ T cells [22–24]. The transcription factor Foxp3 is sufficient to allow for suppressive function in Tregs and therefore is often used as the most reliable marker for Tregs [25]. While Tregs have been demonstrated to be critically important for protection from autoimmune disease, their role in infectious disease remains elusive. Tregs have been shown to be detrimental to viral clearance mechanisms in several chronic viral infections including hepatitis C virus, herpes simplex virus, Friend leukemia virus and, most recently, human immunodeficiency virus [23,26–30]. In these studies it was shown that patients with chronic infection had a higher numbers of Tregs and in some cases that these Tregs were potent suppressors of anti-viral responses [23,26,31]. Given the differential roles of Tregs in autoimmune disease vs. immunity to infectious diseases, we sought to determine if the differential genetic susceptibility of SJL/J as compared to C57BL/6 mice to a TMEV-induced autoimmune disease was related to heightened activation of Tregs in susceptible mice capable of dampening the virus-specific CTL response allowing CNS virus persistence facilitating subsequent development of CD4+ T cell responses to myelin antigens via epitope spreading.
We employed multiple approaches to assess the potential differential activation and function of Tregs in controlling susceptibility to TMEV-IDD. These included determination of: (a) the direct effects of TMEV infection on the numbers, anatomic location and function of Tregs in SJL/J vs. C57BL/6 mice, and the effects of Treg depletion/inactivation using anti-CD25 (b) and anti-glucocorticoid-induced tumor necrosis factor receptor (GITR) (c) monoclonal antibodies [32–37] prior to infection on clinical disease progression and virus-specific immune responses. The results show that Tregs significantly expand in TMEV-infected disease-susceptible SJL/J as compared to C57BL/6 mice leading to an elevated Treg:Teffector cell ratio in the CNS. In addition, Treg inactivation in disease-susceptible SJL/J mice results in significantly delayed disease progression, increased anti-TMEV CD4, CD8 and antibody responses and decreased CNS virus loads. In contrast, TMEV-specific immune responses were not enhanced by anti-CD25 mAb treatment of disease-resistant C57BL/6 mice. Collectively, our findings indicate that susceptibility to a virus-induced autoimmune disease can be regulated by differential responses of Tregs to the infectious agent which may be an important factor in regulating genetic susceptibility to various human autoimmune diseases.
2. Materials and Methods
2.1. Mice
SJL/J mice were purchased from Harlan Laboratories, Bethesda, MD. C57BL/6 mice were purchased from Jackson Laboratories, Bar Harbor, ME. Foxp3-GFP transgenic mice on a mixed genetic background were obtained from the Rudensky laboratory at the University of Washington (Seattle, WA) [25] and then backcrossed for 10 generations onto both the SJL/J and C57BL/6 strains. All mice were housed in the Center for Comparative Medicine, Northwestern University, Chicago, IL, under the guidelines of the Animal Care and Use Committee (ACUC).
2.2. Peptides
All synthetic peptides were obtained from Genemed Synthesis, San Francisco, CA. These included: TMEV peptides VP2121–130 (FHAGSLLVFM), VP2165–173 (TGYRYDSRT), VP3110–120 (NFLFVFTGAAM), VP3159–166 (FNFTAPFI), VP3173–181 (QTSYTSPTI), VP111–20 (SNDDASVDFV), and VP270–86 (WTSQEAFSHIRIPLP), as well as SV40 Epitope I (SAINNYAQKL). All peptides were purified by HPLC and were certified to be ≥ 95% pure.
2.3 TMEV Inoculation and Clinical Evaluation of TMEV-induced Demyelinating Disease (TMEV-IDD)
Mice were infected by i.c. inoculation with 5.0–10×106 PFU of strain BeAn 8386 in a volume of 30 μl serum-free Dulbecco’s Modified Eagle’s Medium. For long-term experiments, mice were monitored every 3–5 days and assessed for disease severity. The following clinical grading scale for SJL/J mice was employed: 0, asymptomatic; 1, mild to moderate waddling gait; 2, moderate to severe waddling gait with extensor spasms; 3, extreme difficulty walking, impaired righting reflex; 4, paralysis of 1 or more limbs inability to walk; 5, moribund. All mice were monitored daily and afforded easier access to food and water once their symptoms became severe.
2.4. TMEV Plaque Assay
Mice were anesthetized with 50 mg/kg Nembutal and perfused with 30–50 ml of PBS through the left ventricle. Spinal cords and brains were collected from perfused mice, weighed and homogenized on ice using a Polytron System PT1200C tissue homogenizer (Kinematica AG, Switzerland). Homogenates were serially diluted and added to culture plates (Nunc, Roskilde Denmark) of confluent BHK-21 cells for 1 hour at room temperature, with periodic gentle rocking. Cells were then covered with a media/agar solution containing 1% Noble Agar (BD, Sparks, MD) and 2X DMEM. Following a 5 day incubation at 34°C, the agar was removed and the BHK monolayer was fixed with formalin or methanol (Fisher Scientific, Fair Lawn, NJ). Plaques were visualized by staining with crystal violet. To determine PFU/ml homogenate, the number of plaques on each plate was multiplied by the dilution factor of the homogenate and divided by the amount of homogenate added per plate. The PFU/ml was divided by the weight of the tissue to calculate PFU/mg of tissue.
2.5. Isolation of CNS-Infiltrating Mononuclear Cells
Mice were anesthetized with 50 mg/kg Nembutal and perfused with 30–50 ml of PBS through the left ventricle. Brains and spinal cords were harvested, minced with scissors and treated with Collagenase IV (Invitrogen, Carlsbad, CA) and DNase I (Invitrogen, Carlsbad, Ca) for 45 minutes at 37°C. Samples were pushed through 40 mm Nytex (Sefar America, Kansas City, MO) to yield single cell suspensions. Mononuclear cells were isolated using a 30/70 Percoll (Amersham, Piscataway, NY) gradient centrifuged at 2500 × g for 20 minutes at room temperature [8,38].
2.6. Flow Cytometric Analysis
Cells to be analyzed by flow cytometry were blocked with anti-CD16/32 (BD Biosciences, San Jose, CA). For CD4, CD8, CD25, CD62L, IFNγ, and Foxp3 stains, cells were stained with 0.5 mg antibody per 106 cells using eBioscience antibodies (San Diego, CA). Cells were permeabilized and fixed using the Foxp3 staining buffer kit from eBioscience (San Diego, Ca). For BrdU labeling and staining, the FITC BrdU Flow Kit (BD Biosciences, San Jose, CA) was used according to manufacturers instructions. Briefly, animals were injected i.p. with 1 mg of BrdU at 24 and 12 hours prior to harvest. These cells were then fixed and permeabilized and DNAse treated prior to anti-BrdU staining. For intracellular cytokine staining cells were stimulated in vitro in media containing 10% FCS and 1:1000 β-mercaptoethanol 50ng/mL PMA (Sigma, St. Louis, MO) and 500ng/mL Ionomycin (Sigma, St. Louis, MO) for 6 hours, Brefeldin A (Sigma, St. Louis, MO) at 10μg/mL and Golgi Stop (BD Pharmingen, San Jose, CA). were added for the final two hours. After stimulation and surface staining. Cells were fixed and permeabilized using the BD Cytofix/Cytoperm Fixation Permeabilization Solution according to manufacturers instructions (BD Biosciences, San Jose, CA). Samples were run on the Canto II flow cytometer with FACS Diva Software (Becton-Dickinson, Mountain View, CA) and analyzed using Flowjo Software (Treestar Flowjo, Ashland, OR).
2.7. CD4+CD25+ Treg cell inactivation and GITR ligation
500 μg of anti-CD25 antibody (clone PC-61, Bioexpress Cell Culture Services, West Lebanon, NH) was injected i.p. on days -2 and 0 relative to TMEV infection. Previous work has shown that this treatment results in a functional inactivation of CD25+ cells for 7–10 days following the last treatment [35]. 1mg of anti-GITR (clone DTA-1, Bioexpress Cell Culture Services, West Lebanon, NH) was injected i.p. on day -1 relative to TMEV infection.
2.8. Enzyme-linked Immuno-SPOT (ELISPOT)
ELISPOT plates (Whatman Inc., Clifton, NJ) were coated with purified anti-IFN-γ antibody (BD Biosciences, San Jose, CA) one day prior to assay. Plates were blocked with DMEM (Sigma, St. Louis, MO) and bovine serum Albumin (BSA, Sigma) for 1 to 2 hours before plating. Cells isolated form the CNS, spleen or lymph nodes were plated in triplicate with antigen or anti-CD3 antibody and incubated at 37°C for 18–24 hours. Plates were washed, and biotinylated anti-IFN-γ (BD biosciences, San Jose, CA) was added. Following a 3–4 hour incubation, plates were washed and then anti-biotin alkaline phosphatase (Vector Laboratories, Burlingame, CA) was added for one hour. Following a final wash, cytokine producing cells were visualized with a developing kit (Bio-Rad Laboratories, Hercules, CA) per manufacturer’s instructions. Developed plates were read on an ImmunoSpot Analyzer and analyzed using ImmunoSpot software (Cellular Technology Ltd. Cleveland, OH). All ELISPOT data was presented as mean number of spots per well ± SEM.
2.9. In Vivo Cytolysis Assay
Splenocytes were collected from naïve animals, treated with NH4CL to remove red blood cells, and divided into two populations. Each population was pulsed with either cognate or irrelevant peptide, the two populations were labeled with differing concentrations of 5-carboxyfluorescein diacetate succinimidyl ester (CFSE). The two populations were injected in equal numbers into infected or naïve animals, at 6–10×106 total cells per mouse. After 5–6 hours, single cell suspensions of spleens of the recipients were analyzed by flow cytometry for the presence and relative numbers of cells in each CFSE peak. Cells loaded with cognate antigen were lysed by antigen-specific CD8+ T cells in infected animals and thus the corresponding peak was drastically reduced. Two equations were used to determine the percent lysis. The adjustment factor (A) was obtained from naïve controls, and for the percent lysis equation, the average of the A from 2 to 3 mice was used. The following equation was used: A = % cognate peptide/ % irrelevant peptide. % Lysis = (% irrelevant peptide × A) − (% cognate peptide/ % irrelevant peptide × A) [17].
2.10. Delayed Type Hypersensitivity (DTH)
For each mouse, baseline thickness of both ears was measured using a Mitutoyo model 7326 micrometer (Schlesinger’s Tools, Brooklyn, NY). Ears were then subcutaneously injected with 10 μg of either TMEV VP270–86 or VP3159–166 peptides in 10 μl PBS using a 100 μl Hamilton syringe fitted with a 30 gauge needle. 24 hours after injection of peptide, ear thickness was re-measured and mean ear swelling was determined by subtracting the baseline ear thickness.
2.11 Analysis of TMEV-specific Antibodies by ELISA
SJL/J mice were treated with 500μg anti-CD25 on days -2 and 0 relative to infection with 5×106 pfu/mouse of TMEV. On days 14, 21, and 28 post infection serum was collected through retro-orbital eye bleed from the same mice each week. Serum from animals was pooled. Plates were coated with 0.3 μg of UV-inactivated TMEV then blocked with 1% Blotto. Two-fold dilutions were plated in triplicate of pooled serum samples from 1:400 to 1:50. Polyclonal goat anti-mouse IgG was used to detect binding antibodies. The reaction was developed using p-nitrophenyl phosphate and measured colorimetrically by ELISA at 405nm.
2.12. In vitro Suppression Assay
GFP-Foxp3 mice on the SJL/J and C58Bl/6 backgrounds were i.p. injected with 1.5×107 PFU TMEV per mouse. Seven days later splenocytes were isolated and sorted for GFP+ Tregs by MoFlo (Dako Cytomation). Naïve splenocyte from (SJL/JxC57BL/6) F1 mice (Taconic Farms, Cambridge City, IN) were sorted for antigen presenting cells and naïve CD4+ T cells by AutoMACS according to manufacturers instructions. (Miltenyi Biotech, Auburn, CA). Cells were then plated at various Treg:Teffector cell ratios and stimulated with 0.2 μg/well anti-CD3 (clone 2C11). Incorporation of [3H]-thymidine at 72 hours was analyzed with a TopCount-NXT (Packard).
2.13. Statistical Analyses
For all in vitro and ex vivo assays, comparisons between groups were analyzed using unpaired Student’s t test. p values less than 0.05 were considered significant. All error bars are representative of standard deviation within the sample group.
3. Results
3.1. Functional Inactivation of CD25+ cells prior to infection with TMEV results in enhanced viral clearance and delayed disease in TMEV-susceptible SJL/mice
Previous data in our lab, as well as others, have shown that Tregs play an important protective role in autoimmune disease. For example, anti-CD25 mAb treatment of SJL/J mice resulted in exacerbated EAE [39] and anti-CD25 mAb treatment of normally EAE-resistant B10.S mice resulted in disease development [40]. Additionally, priming with H/K ATPase in incomplete Freund’s adjuvant (IFA) did not lead to in autoimmune gastritis, but lead to severe disease in anti-CD25 treated BALB/c mice [41]. Anti-CD25 can be used to functionally inactivate Tregs due to the high constitutive expression of CD25 on the cell surface, as T effector cells only express high surface levels of CD25 for a brief period after they are activated, they are not depleted by anti-CD25 treatment given prior to infection. Tregs have also been shown to be detrimental to anti-viral responses as Treg depletion results in increased viral clearance in a variety of infection models [23,26–30,32]. Based on the differential functional role of Tregs in autoimmune disease as compared to a viral infection, we wished to determine the role of Tregs in the TMEV-induced model of CNS autoimmunity. Firstly, we sought to determine if inactivation of Tregs in susceptible SJL/J mice prior to TMEV infection would enhance virus clearance and decrease the subsequent clinical demyelinating disease. SJL/J mice were treated with anti-CD25 mAb, which functionally inactivates Tregs by a combination of cell depletion and down-regulation of the IL-2 receptor [35], on days -2 and 0 prior to i.c. infection with 5×106 pfu of TMEV. Disease onset and progression in anti-CD25 treated SJL/J mice was significantly delayed. As expected, untreated C57BL/6 animals remained resistant to TMEV-IDD (Figure 1A&B).
Figure 1. Functional inactivation of CD25+ cells prior to infection with TMEV results in an ameliorated disease course, delayed progression of disease, and reduced viral load in SJL/J, but not C57BL/B6 mice.

SJL/J and C57Bl/6 (5/group) were treated with control Ig or 500 μg of anti-CD25 mAb (clone PC-61) i.p. on days -2 and 0 relative to i.c. TMEV infection with 5×106 PFU/mouse of TMEV. Mean clinical disease score (A) and percent of mice affected (B) were monitored for 100 days. Difference in clinical score and/or percent mice affected statistically different (*p<0.05). Virus load was assessed in brain and spinal cord tissue on day 8 in infected and PC-61-treated SJL/J mice (C) and B6 mice (F) and on days 12 (D) and 28 (E) post-infection in SJL/J mice. Virus load in SJL/J, but not B6 mice was significant reduced by PC-61 treatment (*p<0.05). Error bars are representative of standard deviation within a sample group. . All data is representative of 4–6 individual experiments.
We next asked if the lesser disease observed following Treg inactivation correlated with increased CNS virus clearance perhaps due to activation of elevated anti-viral responses in the absence of Tregs. Anti-CD25 treated SJL/J mice displayed a significantly lower viral load in the spinal cord at 8 days post-infection and a large decrease in viral load in the brain 12 days post infection (Figure 1C&D). As animals are infected i.c. it is not surprising there is no difference in viral load in the brain at day 8 due to the overwhelming amount of virus that was injected in that location (Figure 1C). By day 12 post-infection a significant decrease in viral load can be seen in the brain of anti-CD25 treated animals when compared to control treated animals (Figure 1D). As disease progresses, the virus spreads to the spinal cord where damage results in the onset of symptoms, anti-CD25 animals still develop symptoms therefore it is also not surprising that by day 12 post-infection they have only a slightly lower viral load in the spinal cord (Figure 1D). By day 28 post-infection the viral load in anti-CD25 treated and control treated animals is equivalent. In contrast, anti-CD25 treatment of TMEV-resistant C57BL/6 mice, which normally clear virus from the CNS within two weeks PI [9,42], did not affect viral titers in either the brain or spinal cord eight days PI (Figure 1F) and both groups had cleared the virus by 28 days post-infection (data not shown). Collectively, these results indicate that Tregs regulate early CNS virus clearance in susceptible SJL/J mice, but not in resistant C57BL/6 mice.
3.2. Functional inactivation of CD25+ cells prior to TMEV infection enhances anti-viral peptide-specific CD8+ T cell responses in susceptible SJL/J mice
Given the enhanced virus clearance observed in anti-CD25 mAb-treated SJL/J mice, we next wished to determine if the decreased CNS virus load was associated with enhanced anti-TMEV specific CD8+ T cell responses in the absence of Tregs. Previous work had shown that Treg inactivation resulted in increases in production of IFN-γ in response to viral antigens in several chronic viral infection models, including HCV, HSV, and influenza virus [27,28,43,44]. We thus determined the frequency of IFN-γ producing splenic CD8+ T cells from untreated and anti-CD25 mAb-treated SJL mice at days 7 and 26 PI in response to the dominant (VP3159–166) and subdominant (VP3173–181 and VP111–20) CD8+ T cell epitopes. These time points were chosen as representatives of the acute viral clearance phase of disease and the pre-clinical phase of demyelinating disease, respectively [45]. The frequencies of CD8+ IFN-γ-producing T cells specific for the dominant (VP3159–166) and subdominant (VP3173–181 and VP111–20) MHC class I H-2Ks-restricted epitopes were significantly elevated in anti-CD25 treated SJL/J mice at day 7, but only the response to the dominant VP3159–166 epitope was still elevated at day 26 PI (Figure 2A&C). Additionally, the in vivo lytic response [46] to both VP3159–166 and VP3173–181 peptide-loaded target cells was significantly enhanced at day 7 PI in mice infected in the absence of nTregs (Figure 2B), but the response to VP3159–166 was slightly, but not significantly elevated at day 26 PI compared to control Ig-treated SJL/J mice (Figure 2D). We also examined virus-specific CD8+ T cell responses in disease-resistant C57BL/6 mice who have been shown to mount an effective CD8+ T cell response resulting in TMEV clearance within 10–14 days PI [10,47]. Treg inactivation in C57BL/6 mice led to no significant differences in the frequencies of IFN-γ producing CD8+ T cells to the H-2Db MHC class I-restricted dominant and subdominant epitopes or to an increase of in vivo lysis of target cells loaded with the dominant VP2121–130 epitope at day 7 PI (Figure 2E&F). Collectively, these results indicate that Tregs exert significant suppressive control over the early anti-TMEV CD8 response in disease-susceptible SJL/J mice.
Figure 2. Functional inactivation of CD25+ cells significantly increases the magnitude of the TMEV-specific CD8+ T cell response in TMEV-infected SJL/J, but not C57BL/6 mice.

Groups of 4–5 SJL/J mice were treated with control Ig or 500 μg of anti-CD25 mAb (clone PC-61) i.p. on days -2 and 0 relative to i.c. infection with 5×106 pfu/mouse of TMEV on day 0. At 7 days (A) and 26 days (C) PI, splenic IFN-γ ELISPOT responses to the to the immunodominant (VP3159) and subdominant (VP3173 and VP111) SJL TMEV epitopes were assessed. In vivo lysis responses were also assessed on day 7 PI (B) to VP3159- and VP3173-pulsed targets and on day 26 PI (D) to VP3159-pulsed targets. C57BL/6 mice were also treated with control Ig or 500 μg of PC-61 i.p. on days -2 and 0 relative to TMEV infection and IFN-γ ELISPOT responses to the immunodominant (VP2121) and subdominant (VP2165 and VP3110) B6 epitopes (E) as well as in vivo lysis responses to VP3121-pulsed targets (F) were determined 7 days PI. The indicated ELISPOT and in vivo lytic responses in SJL mice were significantly enhanced in PC-61 treated mice (*p<0.05; **p<0.01). Data represent 2–3 individual experiments carried out at similar time points. Error bars are representative of the standard deviation within a sample group.
3.3 Treatment with anti-GITR mAb delays disease onset and enhances anti-viral clearance mechanisms
Glucocorticoid Induced TNF Receptor (GITR) is a type II transmembrane protein which is constitutively expressed on Tregs and is upregulated on CD4+ and CD8+ T effector cells after activation. GITR ligation on T regulatory cells inhibits their suppressive activity [48], while GITR ligation on effector cells often makes them impervious to Treg-mediated suppression [32,49]. It has been reported that treatment with anti-GITR mAb results in enhanced anti-viral and anti-tumor immunity by CD8+ T cells as shown by reduced viral load and tumor size as well as increased IFN-γ production by CD8+ and CD4+ T cells [23,30,50] Conversely, in EAE and collagen-induced arthritis, anti-GITR treatment significantly exacerbated clinical disease severity [51–53].
We next determined if anti-GITR treatment would also enhance the anti-viral response to TMEV. SJL/J mice were treated with 1mg of anti-GITR on day -1 relative to i.c. infection with 5×106 pfu of TMEV. Disease progression was significantly delayed in anti-GITR treated mice (Figure 3A & B) which was associated with enhanced virus clearance as shown by a decreased viral load in the brain and a significantly reduced viral load in the spinal cord at day 12 post-infection (Figure 3C). The anti-viral CD8+ in vivo lytic response to the dominant epitope VP3159–166 was significantly enhanced in anti-GITR treated animals at days 7 (Figure 3D) and 14 (Figure 3E) post-infection. Additionally, the IFN-γ response to the dominant and both subdominant epitopes was enhanced at day 7 post-infection in anti-GITR treated animals (Figure 3F). However by day 14 post-infection the IFN-γ response was only enhanced to VP3159–166 and VP3173–181 (Figure 3G). We also used anti-CD25 and anti-GITR treatment in combination treating animals with 0.5 μg of anti-CD25 at days -2 and 0 and 1mg of anti-GITR at day -1 relative to infection. These animals were assessed for disease score as well as anti-viral in vivo lytic response to VP3159–166. The combination of anti-CD25 and anti-GITR treatment did not further ameliorate disease progression compared to the individual treatments (Supplemental Figure 1A&B) and marginally, but not significantly, increased the in vivo lytic response (Supplemental Figure 1C). Thus inhibition of Treg-mediated suppression of CD8+ T cells by either anti-CD25 or anti-GITR treatment results in increased anti-viral responses and delayed disease progression, but that the treatments are not additive in their effect.
Figure 3. Anti-GITR antibody treatment results in an ameliorated disease course, delayed progression of disease, decreased viral load, and increased anti-TMEV specific CD8+ T cell responses.

SJL mice (4–5/group) were treated with 1mg of anti-GITR (clone DTA-1) at day -1 relative to i.c. infection with 5×106 pfu/mouse of TMEV. Mean clinical score (A) and disease incidence (B) were monitored for 100 days. Difference in clinical scores were statistically different (*p<0.05). At day 14 post-infection Brain and spinal cord tissue were taken for analysis of viral load by plaque assay (C). Viral load was significantly decreased in the spinal cord of anti-GITR treated animals and marginally decreased in the brain (C). Additionally, at 7 and 14 days post-infection the in vivo lytic response to VP3159–166-pulsed targets (D & E) and IFN-γ ELISPOT responses to VP3159–166, VP3173–181, and VP111–20 (F & G) were determined. In each case, anti-GITR treated animals had increased anti-TMEV specific responses compared to control Ig-treated animals (*p<0.01). Error bars are representative of standard deviation within the sample group. Data is representative of two individual experiments.
3.4. Functional inactivation of Treg cells enhances CD4+ Th1 and antibody responses in TMEV-infected SJL/J mice
The CD4+ T cell response is the primary mechanism mediating myelin damage and disease progression in TMEV-IDD (reviewed in [2]). Our previous studies have shown that myelin damage in TMEV-IDD is initiated by virus-specific CD4+ Th1 cells activated by virus epitopes presented by CNS-resident macrophages, microglia and dendritic cells (DCs) harboring persistent virus which cause myelin damage by bystander activation of CNS mononuclear cells. Epitope spreading to endogenous myelin epitopes is induced secondary to initial myelin destruction and this autoimmune response is primarily responsible for driving chronic progression of CNS demyelination [2,6,8,54,55]. CD4+ Th cells are also critical for antibody class switching and anti-viral antibody has been shown to play a role in resistance to TMEV-IDD [56,57]. SJL/J mice produce anti-TMEV antibodies which are found in both the CNS and serum of infected mice, furthermore SJL/J mice lacking B cells develop a more severe demyelinating disease. We thus determined if inactivation of Treg cells in anti-CD25 treated mice would affect the DTH (ear swelling) responses and/or CD4+ T cell-dependent anti-TMEV specific antibody responses. DTH responses to both the immunodominant CD4+ (VP270–86) and CD8+ T (VP3159–166) T cell epitopes were significantly enhanced in anti-CD25 treated SJL/J mice at time points corresponding to acute virus infection (day 7 PI), pre-clinical disease (day 21 PI), and disease onset (day 40 PI) (Figure 3A-C). The maximal response to the CD4+ T cell epitope was seen at disease onset (Figure 3C). Thus, the effect of Treg inactivation on development of the CD4+ T cell response to the immunodominant epitope lasted long after the recovery of Treg population in antibody-treated mice.
TMEV-specific antibody responses were also significantly increased in anti-CD25 mAb-treated mice when compared to control Ig-treated mice (Figure 3D-F). Serum pooled from 5 animals per group was analyzed on plates coated with 0.3μg of TMEV. Not surprisingly, on days 7 and 14 PI, antibody responses were low and the enhancement in anti-CD25-treated mice was minimal (Figure 3D and data not shown). However, by days 21 and 28 PI, enhanced antibody responses in anti-CD25-treated mice were considerably greater with the maximal difference noted at day 28 PI (Figure 3F). Considered together, the suppressed clinical disease, lower CNS virus titers, and enhanced TMEV-specific CD4+ T cell, CD8+ T cell and antibody responses observed following anti-CD25 treatment indicate that Tregs are a major factor in the failure to clear persistent CNS virus in disease-susceptible SJL/J mice.
3.5. SJL/J mice have a greater numbers of Tregs and an unfavorable ratio of Tregs to effector cells in the periphery and CNS after TMEV infection
The importance of Tregs in regulating susceptibility to autoimmunity and limiting autoimmune tissue damage has been documented in multiple models of autoimmune diseases including multiple sclerosis, type I diabetes, and rheumatoid arthritis [58–61]. Notably, in EAE, the balance of Tregs and effector CD4+ T cells has been reported to switch in favor of Tregs allowing for recovery from acute disease [62]. This indicates that Tregs can be activated in vivo to regulate ongoing autoreactive inflammatory tissue damage. Our data to this point indicate that Tregs differentially regulate TMEV immune responses in disease-susceptible SJL/J vs. resistant C57Bl/6 mice. To test the hypothesis that TMEV infection may directly or indirectly activate Tregs in TMEV-infected SJL/J, but not C57BL/6 mice, we quantitated the number of Tregs in both the periphery and CNS of these mouse strains during acute TMEV infection (days 3 and 7 PI). Significantly greater numbers of Tregs were observed in the CNS of TMEV-susceptible SJL/J as compared to resistant C57BL/6 mice at both time points (Figure 5A, Supplemental Figures 2–5). In fact, C57BL/6 mice had negligible amounts of Tregs in the CNS at days 3 and 7 post infection. The total number of CD4+Foxp3+ Tregs in the spleen of virus-infected SJL/J mice was also generally higher than in infected C57Bl/6 mice (Figure 5B, Supplemental Figures 2–5). This finding is consistent with the hypothesis that TMEV infection preferentially activates Treg expansion in TMEV-susceptible mouse strains which in turn suppresses anti-viral responses required for CNS virus clearance.
Figure 5. Accumulation of high numbers of Tregs in the CNS of SJL/J, but not C57BL/6 mice during acute TMEV infection.

Foxp3-GFP transgenic SJL/J and B6 mice were sham infected or infected i.c. with 5×106 PFU/mouse of TMEV on day 0. At days 3 and 7 PI, the numbers of CD3+CD4+Foxp3+ Treg cells were enumerated in the CNS (A) and spleen (B) by using the percentage of cells to back calculate the number of live cells as determined by trypan blue exclusion. The numbers of Tregs in the CNS of TMEV-infected SJL/J mice were significantly higher than those in the CNS of sham infected SJL/J mice and of TMEV-infected B6 mice (*p<0.01), (**p<0.03). This data is representative of 5–6 individual experiments.
It was recently reported in EAE that antigen-specific Tregs accumulate in the CNS but are not able to control the autoimmune inflammation due to an unfavorable ratio of antigen-specific Tregs to effector CD4+ T cells [63]. We thus compared the ratio of Tregs (CD4+Foxp3+) to CD8+ effector T cells (CD8+IFN-γ+) T cells in both CNS (Figure 6A & Supplemental Figure 6&8) and spleen (Figure 6B &Supplemental Figures 7&9) of TMEV-susceptible SJL/J vs. resistant C57BL/6 mice during the acute virus clearance phase (days 3 and 7 PI) of disease. The ratio of CD8+IFN-γ+ T cells to Tregs present within the CNS of SJL/J mice is 1:2.6 at day 3 and 1:4 at day 7 PI, while the activated CD8+ T cell to Tregs ratio present within the CNS of C57BL/6 mice was a much more favorable ratio (1:0.8 and 1:1.2, respectively) for virus clearance (Figure 6A). A similar trend was seen in the spleen with a 1:16 effector CD8:Treg ratio at day 3 and 1:4.2 at day 7 PI in SJL mice, but a ratio of 1:3 at day 3 post-infection in B6 mice. The ratio at day 7 PI in the spleen of C57BL/6 mice was also unfavorable at 1:4.7 (Figure 6B). While IFN-γ production is a hallmark of CD8+ T cell activation and effector function, CD8+ T cells are also known to kill via expression of FAS as well as production of Granzymes A and B and perforin. Additionally, not all cells are capable of producing both IFN-γ and lytic factors. Therefore we also assessed the ratio of Tregs (CD4+CD25+Foxp3+) to activated CD8+ T cells (CD8+CD25+CD62Llo) in the CNS of the two strains. SJL/J mice displayed a Treg to activated CD8+ T cell ratio of 1:9 and 1:6.2 at days 3 and 7 PI, respectively (Figure 6C), while C57BL/6 mice have a much more favorable ratio of 1:1 and 1:0.8 at days 3 and 7, respectively (Figure 6C). Thus following TMEV infection, SJL/J mice have an unfavorable CNS ratio of Tregs to activated CD8+ T cells during acute infection which would impede viral clearance. In contrast, the ratio in C57BL/6 mice favors rapid viral clearance.
Figure 6. The ratio of activated CD8+ T cells to Treg cells is less in the CNS of TMEV-infected SJL/J mice than C57BL/B6 mice.

Foxp3-GFP transgenic SJL/J and B6 mice were infected i.c. with 5×106 PFU/mouse of TMEV on day 0. At days 3 and 7 PI, the numbers of IFN-γ-producing CD8+ effector T cells (CD3+CD8+IFN-γ+) and Treg cells (CD3+CD4+Foxp3+) were enumerated in the CNS (A) and spleen (B). The ratio of Treg cells (CD3+CD4+Foxp3+) and activated effector cells (CD3+CD8+CD25+CD62Llo) were also enumerated in the CNS (C). The ratio of activated CD8+ T cells to Treg cells was then determined and is shown above each pair of bars. Data represents the mean ± SD of 4 mice per group on day 3 and 5 mice per group on day 7 post-infection. This data is representative of 2 individual experiments.
3.6. TMEV infection induces the proliferation of Tregs in the periphery and CNS of SJL/J mice
To determine if Tregs preferentially expand in SJL/J vs. C57BL/6 mice in response to TMEV infection, we measured Treg expansion using BrdU labeling. CD3+CD4+Foxp3+ Treg cells from the spleen and CNS were examined by flow cytometry at days 3 and 7 PI for uptake of BrdU (administered over the 24 hours prior to sacrifice). We found that SJL/J mice had approximately 2-fold greater numbers of BrdU+ cells in the CNS at days 3 and 7 than infected C57BL/6 mice (Figure 7A and Supplemental Figures 10 &12) and a similar increase was noted in the spleen (Figure 7D and Supplemental Figures 11&13). These data indicate that Tregs proliferate to a greater degree in the CNS of disease-susceptible SJL/J mice after TMEV infection.
Figure 7. TMEV infection promotes significant expansion of Treg cells in TMEV-infected SJL/J, but not in virus-infected C57BL/6B6 mice.

SJL/J and B6 mice (3/group) were sham infected or infected i.c. with 5×106 pfu/mouse of TMEV on day 0. At days 2 and 6 animals were injected i.p. with 1 mg of BrdU every 12 hours for 24 hours. On days 3 and 7, the number of CD4+CD25+Foxp3+BrdU+ cells in the CNS (A&B) and spleens (C&D) of sham-infected and TMEV-infected animals were enumerated by FACS analysis. Percentages were then used to back calculate the total number of viable cells as determined by trypan blue staining. The bars indicate the mean ± SEM of 3 mice per group and is representative of 3–4 separate experiments.
3.7. Tregs from SJL/J and B6 animals are equally able to suppress proliferation of CD4+ T cells
We next asked if Treg cells from TMEV-infected SJL/J mice had enhanced functional suppressive capacity in addition to their enhanced numbers and proliferative capacity in comparison to Tregs from C57BL/6 animals. We found that i.p. infection with TMEV results in a greater increase in activation of Treg cells and effector cells in the peripheral organs than i.c. infection at 7 days post infection (data not shown). We thus i.p. infected both B6 Foxp3-GFP and SJL/J Foxp3-GFP transgenic mice with 1.5×107 pfu TMEV/mouse. Seven days PI, spleens and lymph nodes were harvested and CD4+GFP+ Treg cells purified by MoFlow sorting. These cells were then cultured with CD4+Foxp3− cells plus T-cell depleted spleen APCs from (C57BL/6xSJL/J)F1 mice at various ratios (Figure 8). Treg cells from naïve and infected SJL/J and B6 animals were equally capable of suppressing proliferation of CD4+ T cells stimulated with anti-CD3 (Figure 8) indicating that enhanced regulation of TMEV-specific immune responses in SJL/J mice relates to their greater numbers and the unfavorable CNS ratio to effector T cells rather than enhanced suppressive capability.
Figure 8. Tregs from TMEV-infected SJL/J and C57BL/6 mice are equally able to suppress proliferation of CD4+ T cells.
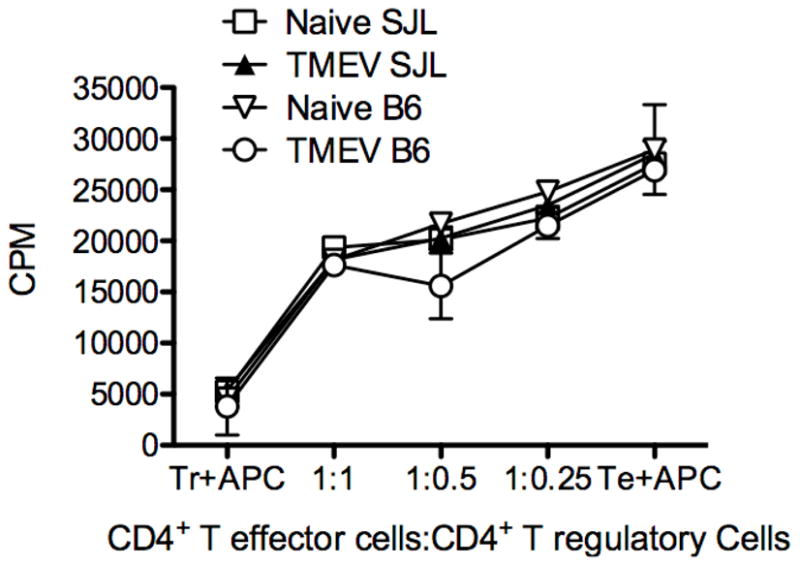
B6-GFP-Foxp3 and SJL/J-GFP-Foxp3 animals were i.p. infected with 1.5×107 pfu/ mouse TMEV. 7 days later splenocytes were isolated and CD4+GFP+ cells were sorted from naïve and infected SJL/J and B6 animals. These cells were cultured with CD4+CD25−Foxp3− cells from naïve (SJLxB6)F1 mice in the presence of 0.2 μg/well anti-CD3 (clone 2C11) using T cell-depleted F1 splenocytes as antigen presenting cells. Cultures were pulsed with 3H-TdR at the time of plating and cultured at 37° C for three days before harvesting. Data is representative of 2–3 individual experiments.
4. Discussion
Tregs have been shown to play contrasting roles in autoimmunity and chronic viral infection. They are protective against autoimmune disease [34,39,58–61,64]. Notably, in EAE, Tregs have been shown to mediate genetic resistance to disease induction in one model [40] and the balance of Tregs and effector T cells in the CNS has been reported to switch in favor of Tregs allowing for recovery from acute disease in another EAE model [62,63]. On the other hand, Treg cell function has been shown to impede virus clearance during chronic viral infections [23,26–30,32]. Since the majority of autoimmune diseases are thought to be induced in genetically susceptible individuals as a secondary consequence of viral or bacterial infection [1,2,5], Tregs present a potential conundrum in that they may interfere with efficient clearance of an infectious agent and, in turn, promote the development of autoimmunity via mechanisms such as molecular mimicry or epitope spreading [2,65].
Our laboratory has had a long-standing interest in studying the pathogenesis of Theiler’s murine encephalomyelitis virus-induced demyelinating disease (TMEV-IDD), a model of chronic MS, which provides an ideal system to study the interplay of genes, infection and immunoregulation in the induction and progression of a virus-induced CNS autoimmune demyelinating disease [1,2]. Susceptibility to TMEV-IDD is controlled by multiple genes, predominantly MHC class I, wherein resistant strains of mice [e.g., C57BL/6 (H-2b)] mount an efficient CTL response and rapidly clear the infection, while susceptible strains (H-2s,q,r,p, and f) of mice [e.g., SJL/J (H-2s)] fail to mount an effective CTL response [9–18] resulting in a life-long persistent CNS TMEV infection leading to induction of CNS autoimmunity via epitope spreading [6–8].
We thus asked if genetic susceptibility to development of a virus-induced autoimmune demyelinating disease, whose differential induction in SJL/J vs. C57BL/6 mice is dependent on the efficiency of the anti-TMEV CTL response, was regulated by virus-induced Treg activity. Our findings demonstrate that inactivation of Treg cells via anti-CD25 mAb treatment prior to infection of susceptible SJL/J mice results in delayed disease onset and progression, increased viral clearance, and increased adaptive anti-viral CD4, CD8 and antibody immune responses. To our knowledge, our results showing that TMEV infection of susceptible SJL/J mice rapidly induces the activation and expansion of Tregs, thereby inhibiting virus clearance by down-regulating anti-TMEV immune responses, is the first description of Tregs controlling genetic susceptibility to an infection-induced autoimmune disease. Furthermore, these findings provide a new putative mechanistic basis for explaining the induction of a variety of human autoimmune diseases secondary to virus or bacterial infection.
We showed that treatment with anti-CD25 mAb immediately prior to i.c. infection of susceptible SJL/J mice led to a significant delay in onset and severity of TMEV-IDD concomitant with reduced CNS virus loads (Figure 1) and significantly enhanced CD4+ and CD8+ T cell- and antibody-mediated anti-viral immune responses (Figures 2&4). Anti-CD25 mAb treatment has been previously successfully used to determine the role of Treg cells in autoimmunity, viral clearance, and tumor responses. While it has been suggested that anti-CD25 mAb treatment can deplete Tregs following in vivo treatment, we previously showed that anti-CD25 mAb causes functional inactivation of Tregs by downregulating CD25 surface expression without substantially reducing the numbers of Foxp3+ T cells [35]. Significant to the current study, inactivation of Tregs with anti-CD25 mAb is temporary and functional Tregs return to near normal frequencies within 7–10 days after the final mAb treatment. Additionally, we have shown using anti-GITR mAb making T effector cells impervious to Treg suppression also results in delayed disease progression as well as increased viral clearance and anti-viral responses by CD8+ T cells (Figure 3). This indicates that inactivation or inhibition of Tregs during acute TMEV infection promotes protection from TMEV-IDD by inducing more efficient virus clearance leading to less bystander damage to myelin. Tregs likely further regulate the induction of autoimmunity in anti-CD25-treated mice as their numbers have returned to normal levels by the time (days 35–45 PI) when myelin-specific autoimmune responses usually arise in TMEV-infected mice due to epitope spreading [6–8]. Interestingly, we did not see any enhancement of in vivo or in vitro CD8+ T cell (Figure 1F and 2E&F) or antibody (not shown) responses to TMEV epitopes in C57BL/6 mice indicating lack of functional Treg control of the anti-viral response in this disease-resistant strain.
Figure 4. Functional inactivation of CD25+ cells prior to TMEV infection of SJL/J mice results in enhanced TMEV-specific DTH (Th1) and antibody responses.

Groups of 5 SJL/J mice were treated with control Ig or 500 μg of anti-CD25 mAb (clone PC-61) i.p. on days -2 and 0 relative to i.c. infection with 5×106 pfu/mouse of TMEV on day 0. In vivo 24 hr ear swelling (DTH) responses upon ear challenge with the immunodominant MHC class II-restricted CD4 (VP270–86) and MHC class I-restricted CD8 (VP3159–166) TMEV epitopes were determined on days 7 (A), 21 (B) and 40 (C) PI (corresponding to the acute, pre-clinical, and disease onset stages, respectively). Error bars are representative of standard deviation within the sample group. Serum from 5 SJL/J mice was collected from the same animals once per week on days 14 (D), 21 (E), and 28 (F). Serum samples were then pooled before TMEV-specific IgG antibody responses were determined by ELISA on UV-inactivated TMEV-coated plates. Data is representative of three individual experiments.
It has been reported that inactivation of Tregs using anti-CD25 mAb can change the hierarchy of immunodominance in virus-specific CD8+ T cell responses during the course of respiratory syncytial virus infection [66]. However, we did not see any evidence of a change in immunodominance as a result of Treg inactivation in SJL/J mice. In fact, significant increases in anti-viral CD8 responses were noted to the immunodominant and both of the subdominant epitopes at 7 days PI and the order of immunodominance was retained. Previous work in the TMEV-IDD model has shown that CD8+ T cells are required for effective virus clearance. MHC class I-deficient mice on the resistant B6 background were shown to be susceptible to demyelination after TMEV infection [67]. In addition, we have shown that transfer of VP3159–166-specific CD8+ T cells to TMEV-infected SJL/J mice increased viral clearance thus decreasing development of clinical disease [18]. Paradoxically, transfer of exogenous TMEV-specific CD8+ T cells to resistant C57BL/6 mice has been reported to induce the development of a hemorrhagic fever [68]. Collectively these results indicate that the anti-viral CD8+ T response in SJL/J mice appears to be protective and not pathogenic, but that a CD8+ T cell response that is too robust can also be pathogenic causing CNS damage.
Our results also show that Treg cells are induced to proliferative preferentially in TMEV-susceptible SJL/J mice resulting in a CNS CD8+ T effector:Treg cell ratio unfavorable for virus clearance (Figs. 5–7). In contrast, the CD8+ T effector:Treg cell ratio is significantly greater in resistant C57BL/6 mice allowing virus clearance. Tregs have been reported to expand in response to other chronic virus infections. In HBV and HCV, chronically infected patients have more Treg cells than patients that recover from these infections and these Tregs are functionally superior at suppressing anti-viral CD8+ T cell responses [69,70]. In our model we show that TMEV-infected susceptible SJL/J mice have a greater absolute number of Treg cells than resistant C57BL/6 mice (Figure 5). It has been reported that the ratio of effector T cells to Tregs is an important factor for controlling an immune response within the target organ [63]. We show that the susceptible SJL/J strain of mice has an unfavorable ratio (1:4) of CD8 T effector:Treg cells during the acute phase of TMEV infection (day 7 PI), while the ratio in resistant C57BL/6 mice at this critical time point is approximately 1:1.2 (Figure 5). We speculate that the CD8+ T cells in the CNS of virus-infected SJL/J mice are largely TMEV-specific, but as tetramers are not available for the SJL/J H-2Ks haplotype, IFN-γ production or the activation markers CD25 and CD62L were therefore used to enumerate activated CD8+ T cells (Figure 6). Additionally, and of relevance to our findings, it has been reported that Tregs preferentially expand in the inflamed CNS in EAE, although they appear to be functionally ineffective in suppressing autoimmune effector cells [62]. We found that Tregs proliferated to a greater degree in the CNS of disease-susceptible SJL/J mice after TMEV infection resulting in about 2-fold greater numbers of BrdU+ cells in the CNS at days 3 and 7 than in infected C57BL/6 mice (Figure 7). In support of the idea that the suboptimal anti-TMEV CD8 response in susceptible mice was primarily due to the skewed CD8+ T effector:Treg cell ratio in the CNS, we also found that Tregs from SJL/J mice and C57BL/6 mice were equally capable of suppressing anti-CD3-induced proliferation of CD4+ T cells whether or not they were derived from naïve or TMEV-infected mice (Figure 8). It is significant that (SJL/JxB6)F1 CD4+ effector T cells and APCs were employed in this assay as it allowed us to directly compare the functional capacity of Treg cells from the two strains on identical target cells. Studies to determine the functional capacity and antigen-specificity (myelin vs. TMEV responses) of CNS Tregs during the autoimmune phase of disease in SJL/J mice that follows the acute viral phase are on-going, as are studies examining the molecular mechanisms by which TMEV-induced Tregs suppress virus clearance [71–73].
5. Conclusions
In summary, our findings strongly indicate that differences in the numbers and the ratio of CNS T effector:Treg cells during acute TMEV infection control the efficiency of the anti-viral immune response resulting in virus persistence and resulting long-lasting effects leading to the eventual development and progression of the autoimmune demyelinating disease. The preferential expansion of Tregs in TMEV-infected SJL/J mice, as compared to disease-resistant C57BL/6 mice, suggests the possibility that genetic susceptibility to certain autoimmune diseases may be in some instances due to ‘regulatory mimicry’. This would encompass a situation wherein a clonotype of nTregs expressing a self antigen-specific T cell receptor selected on particular MHC backgrounds could be activated to expand in response to a cross-reactive epitope expressed on an infectious agent. This expanded population of Tregs would then ameliorate or delay the clearance of the infectious agent in the target organ of the autoimmune disease setting the stage for promotion of the development of autoimmunity via mechanisms such as molecular mimicry or epitope spreading. These findings thus have important implications to our thinking about the potential mechanisms underlying induction of human autoimmune disease secondary to virus infection.
Supplementary Material
Supplemental Figure 1. Treatment with anti-CD25 and anti-GITR in combination does not increase TMEV-specific CD8+ T cell responses or ameliorate disease progression compared to either treatment alone. SJL/J mice were treated with control Ig or either 0.5 mg of anti-CD25 (clone PC-61) on days -2 and 0 relative to infection and/or 1 mg of anti-GITR (clone DTA-1 ) on day -1 relative to infection. Animals were followed for disease score (A) and incidence (B) for 100 days post-infection. Additionally, an in vivo lysis assay was performed at day 12 p.i. (C) to assess TMEV specific CD8+ T cell responses. Treatment with either antibody resulted in the same disease amelioration and increase in anti-viral responses as the combined treatment.
Supplemental Figure 2. FACS analysis of the T regulatory cell population in the CNS of TMEV-infected and Sham-infected SJL and B6 animals at day 3 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5 ×106 pfu/mouse. On day 3 post-infection brain and spinal cord tissue from sham and infected animals were analyzed for CD3+CD4+Foxp3+ cells.
Supplemental Figure 3. FACS analysis of the T regulatory cell population in the spleen of TMEV-infected and Sham-infected SJL and B6 animals at day 3 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5 ×106 pfu/mouse. On day 3 post-infection spleen tissue from sham and infected animals were analyzed for CD3+CD4+Foxp3+ cells.
Supplemental Figure 4. FACS analysis of the T regulatory cell population in the CNS of TMEV-infected and Sham-infected SJL and B6 animals at day 7 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5×106 pfu/mouse. On day 7 post-infection brain and spinal cord tissue from sham and infected animals were analyzed for CD3+CD4+Foxp3+ cells.
Supplemental Figure 5. FACS analysis of the T regulatory cell population in the spleen of TMEV-infected and Sham-infected SJL and B6 animals at day 7 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5×106 pfu/mouse. On day 7 post-infection spleen tissue from sham and infected animals were analyzed for CD3+CD4+Foxp3+ cells.
Supplemental Figure 6. FACS analysis of CD8+IFN-γ+ cells in the CNS of TMEV-infected and Sham-infected SJL and B6 mice at day 3 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5×106 pfu/mouse. On day 3 post-infection brain and spinal cord tissue from sham and infected animals were harvested. Cells were stimulated PMA and ionomycin for 6 hours and Brefeldin A and Monensin for the final two hours.
Supplemental Figure 7. FACS analysis of CD8+IFN-γ+ cells in the spleen of TMEV-infected and Sham-infected SJL and B6 mice at day 3 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5×106 pfu/mouse. On day 3 post-infection spleen tissue from sham and infected animals were harvested. Cells were stimulated PMA and ionomycin for 6 hours and Brefeldin A and Monensin for the final two hours.
Supplemental Figure 8. FACS analysis of CD8+IFN-γ+ cells in the CNS of TMEV-infected and Sham-infected SJL and B6 mice at day 7 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5×106 pfu/mouse. On day 3 post-infection brain and spinal cord tissue from sham and infected animals were harvested. Cells were stimulated PMA and ionomycin for 6 hours and Brefeldin A and Monensin for the final two hours.
Supplemental Figure 9. FACS analysis of CD8+IFN-γ+ cells in the spleen of TMEV-infected and Sham-infected SJL and B6 mice at day 7 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5×106 pfu/mouse. On day 3 post-infection spleen tissue from sham and infected animals were harvested. Cells were stimulated PMA and ionomycin for 6 hours and Brefeldin A and Monensin for the final two hours.
Supplemental Figure 10. FACS analysis of CD3+CD4+Foxp3+BrdU+ cells in the CNS of TMEV-infected and Sham-infected SJL and B6 mice at day 3 post-infection. SJL and B6 mice were sham-infected or TMEV-infected with 5×105 pfu/mouse. At 24 and 12 hours prior to harvest animals were treated i.p. with 1mg of BrdU. On day 3 brain and spinal cord samples were harvested and analyzed for CD3+CD4+Foxp3+BrdU+ cells.
Supplemental Figure 11. FACS analysis of CD3+CD4+Foxp3+BrdU+ cells in the spleen of TMEV-infected and Sham-infected SJL and B6 mice at day 3 post-infection. SJL and B6 mice were sham infected or TMEV infected with 5×105 pfu/mouse. At 24 and 12 hours prior to harvest animals were treated i.p. with 1mg of BrdU. On day 3 spleen samples were harvested and analyzed for CD3+CD4+Foxp3+BrdU+ cells.
Supplemental Figure 12. FACS analysis of CD3+CD4+Foxp3+BrdU+ cells in the CNS of TMEV-infected and Sham-infected SJL and B6 mice at day 7 post-infection. SJL and B6 mice were sham-infected or TMEV-infected with 5×105 pfu/mouse. At 24 and 12 hours prior to harvest animals were treated i.p. with 1mg of BrdU. On day 7 brain and spinal cord samples were harvested and analyzed for CD3+CD4+Foxp3+BrdU+ cells.
Supplemental Figure 13. FACS analysis of CD3+CD4+Foxp3+BrdU+ cells in the spleen of TMEV-infected and Sham-infected SJL and B6 mice at day 7 post-infection. SJL and B6 mice were sham-infected or TMEV-infected with 5×105 pfu/mouse. At 24 and 12 hours prior to harvest animals were treated i.p. with 1mg of BrdU. On day 7 spleen samples were harvested and analyzed for CD3+CD4+Foxp3+BrdU+ cells.
Acknowledgments
We thank members of the Miller lab for helpful comments in the preparation of this manuscript. This work was supported in part by United States Public Health Service, NIH Grants NS-023349, NS-062365, and National Multiple Sclerosis Society Grant RG4294A9/1. M.T.G. was supported by an NIH Individual Predoctoral Research Fellowship (F31 AI-060338) and M.H.R. by NIH Training Grant T32 AI-0007476.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Olson JK, Croxford JL, Miller SD. Virus-induced autoimmunity: Potential role of viruses in initiation, perpetuation, and progression of T cell-mediated autoimmune diseases. Viral Immunol. 2001;14:227–50. doi: 10.1089/088282401753266756. [DOI] [PubMed] [Google Scholar]
- 2.Munz C, Lunemann JD, Getts MT, Miller SD. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol. 2009;9:246–58. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–62. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 4.Oksenberg JR, Baranzini SE, Sawcer S, Hauser SL. The genetics of multiple sclerosis: SNPs to pathways to pathogenesis. Nat Rev Genet. 2008;9:516–26. doi: 10.1038/nrg2395. [DOI] [PubMed] [Google Scholar]
- 5.Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359:1221–31. doi: 10.1016/S0140-6736(02)08220-X. [DOI] [PubMed] [Google Scholar]
- 6.Miller SD, Vanderlugt CL, Begolka WS, Pao W, Yauch RL, Neville KL, et al. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat Med. 1997;3:1133–36. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- 7.Katz-Levy Y, Neville KL, Girvin AM, Vanderlugt CL, Pope JG, Tan LJ, et al. Endogenous presentation of self myelin epitopes by CNS-resident APCs in Theiler’s virus-infected mice. JClinInvest. 1999;104:599–610. doi: 10.1172/JCI7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. NatMed. 2005;11:335–39. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 9.Lipton HL, Melvold R. Genetic analysis of susceptibility to Theiler’s virus-induced demyelinating disease in mice. J Immunol. 1984;132:1821–25. [PubMed] [Google Scholar]
- 10.Lyman MA, Myoung J, Mohindru M, Kim BS. Quantitative, not qualitative, differences in CD8(+) T cell responses to Theiler’s murine encephalomyelitis virus between resistant C57BL/6 and susceptible SJL/J mice. Eur J Immunol. 2004;34:2730–39. doi: 10.1002/eji.200324811. [DOI] [PubMed] [Google Scholar]
- 11.Azoulay A, Brahic M, Bureau JF. FVB mice transgenic for the H-2Db gene become resistant to persistent infection by Theiler’s virus. J Virol. 1994;68:4049–52. doi: 10.1128/jvi.68.6.4049-4052.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clatch RJ, Melvold RW, Miller SD, Lipton HL. Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelinating disease in mice is influenced by the H-2D region: correlation with TMEV-specific delayed-type hypersensitivity. J Immunol. 1985;135:1408–14. [PubMed] [Google Scholar]
- 13.Brahic M, Bureau JF. Genetics of susceptibility to Theiler’s virus infection. Bioessays. 1998;20:627–33. doi: 10.1002/(SICI)1521-1878(199808)20:8<627::AID-BIES5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 14.Azoulay-Cayla A, Syan S, Brahic M, Bureau JF. Roles of the H-2D(b) and H-K(b) genes in resistance to persistent Theiler’s murine encephalomyelitis virus infection of the central nervous system. J Gen Virol. 2001;82:1043–47. doi: 10.1099/0022-1317-82-5-1043. [DOI] [PubMed] [Google Scholar]
- 15.Azoulay-Cayla A, Dethlefs S, Perarnau B, Larsson-Sciard EL, Lemonnier FA, Brahic M, et al. H-2D(b−/−) mice are susceptible to persistent infection by Theiler’s virus. J Virol. 2000;74:5470–76. doi: 10.1128/jvi.74.12.5470-5476.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lipton HL, Melvold R, Miller SD, Dal Canto MC, Jensen K. Mutation of a major histocompatibility class I locus, H-2D, leads to an increased virus burden and disease susceptibility in Theiler’s virus-induced demyelinating disease. J Neurovirol. 1995;1:138–44. doi: 10.3109/13550289509113960. [DOI] [PubMed] [Google Scholar]
- 17.Getts MT, Kim BS, Miller SD. Differential outcome of tolerance induction in naive versus activated Theiler’s virus epitope-specific CD8+ cytotoxic T cells. J Virol. 2007;81:6584–93. doi: 10.1128/JVI.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Getts MT, Richards MH, Miller SD. A critical role for virus-specific CD8(+) CTLs in protection from Theiler’s virus-induced demyelination in disease-susceptible SJL mice. Virology. 2010;402:102–11. doi: 10.1016/j.virol.2010.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyman MA, Lee HG, Kang BS, Kang HK, Kim BS. Capsid-specific cytotoxic T lymphocytes recognize three distinct H-2D(b)-restricted regions of the BeAn strain of Theiler’s virus and exhibit different cytokine profiles. J Virol. 2002;76:3125–34. doi: 10.1128/JVI.76.7.3125-3134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang BS, Lyman MA, Kim BS. Differences in avidity and epitope recognition of CD8(+) T cells infiltrating the central nervous systems of SJL/J mice infected with BeAn and DA strains of Theiler’s murine encephalomyelitis virus. J Virol. 2002;76:11780–84. doi: 10.1128/JVI.76.22.11780-11784.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang BS, Lyman MA, Kim BS. The majority of infiltrating CD8+ T cells in the central nervous system of susceptible SJL/J mice infected with Theiler’s virus are virus specific and fully functional. J Virol. 2002;76:6577–85. doi: 10.1128/JVI.76.13.6577-6585.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–62. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 23.Dittmer U, He H, Messer RJ, Schimmer S, Olbrich AR, Ohlen C, et al. Functional impairment of CD8(+) T cells by regulatory T cells during persistent retroviral infection. Immunity. 2004;20:293–303. doi: 10.1016/s1074-7613(04)00054-8. [DOI] [PubMed] [Google Scholar]
- 24.Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–42. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 25.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–41. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 26.Eggena MP, Barugahare B, Jones N, Okello M, Mutalya S, Kityo C, et al. Depletion of regulatory T cells in HIV infection is associated with immune activation. J Immunol. 2005;174:4407–14. doi: 10.4049/jimmunol.174.7.4407. [DOI] [PubMed] [Google Scholar]
- 27.Boettler T, Spangenberg HC, Neumann-Haefelin C, Panther E, Urbani S, Ferrari C, et al. T cells with a CD4+CD25+ regulatory phenotype suppress in vitro proliferation of virus-specific CD8+ T cells during chronic hepatitis C virus infection. J Virol. 2005;79:7860–7. doi: 10.1128/JVI.79.12.7860-7867.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sehrawat S, Suvas S, Sarangi PP, Suryawanshi A, Rouse BT. In vitro-generated antigen-specific CD4+ CD25+ Foxp3+ regulatory T cells control the severity of herpes simplex virus-induced ocular immunoinflammatory lesions. J Virol. 2008;82:6838–51. doi: 10.1128/JVI.00697-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zelinskyy G, Kraft AR, Schimmer S, Arndt T, Dittmer U. Kinetics of CD8+ effector T cell responses and induced CD4+ regulatory T cell responses during Friend retrovirus infection. Eur J Immunol. 2006;36:2658–70. doi: 10.1002/eji.200636059. [DOI] [PubMed] [Google Scholar]
- 30.He H, Messer RJ, Sakaguchi S, Yang G, Robertson SJ, Hasenkrug KJ. Reduction of retrovirus-induced immunosuppression by in vivo modulation of T cells during acute infection. J Virol. 2004;78:11641–7. doi: 10.1128/JVI.78.21.11641-11647.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hasenkrug KJ, Dittmer U. Immune control and prevention of chronic Friend retrovirus infection. Front Biosci. 2007;12:1544–51. doi: 10.2741/2167. [DOI] [PubMed] [Google Scholar]
- 32.Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, et al. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol. 2004;173:5008–20. doi: 10.4049/jimmunol.173.8.5008. [DOI] [PubMed] [Google Scholar]
- 33.Muriglan SJ, Ramirez-Montagut T, Alpdogan O, Van Huystee TW, Eng JM, Hubbard VM, et al. GITR activation induces an opposite effect on alloreactive CD4(+) and CD8(+) T cells in graft-versus-host disease. J Exp Med. 2004;200:149–57. doi: 10.1084/jem.20040116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McHugh RS, Shevach EM. Cutting edge: depletion of CD4+CD25+ regulatory T cells is necessary, but not sufficient, for induction of organ-specific autoimmune disease. J Immunol. 2002;168:5979–83. doi: 10.4049/jimmunol.168.12.5979. [DOI] [PubMed] [Google Scholar]
- 35.Kohm AP, McMahon JS, Podojil JR, Smith Begolka W, DeGutes M, Kasprowicz DJ, et al. Cutting Edge: Anti-CD25 mAb injection results in the functional inactivation, not depletion of CD4+CD25+ Treg cells. J Immunol. 2006;176:3301–05. doi: 10.4049/jimmunol.176.6.3301. [DOI] [PubMed] [Google Scholar]
- 36.Suvas S, Kumaraguru U, Pack CD, Lee S, Rouse BT. CD4+CD25+ T cells regulate virus-specific primary and memory CD8+ T cell responses. J Exp Med. 2003;198:889–901. doi: 10.1084/jem.20030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Couper KN, Blount DG, de Souza JB, Suffia I, Belkaid Y, Riley EM. Incomplete depletion and rapid regeneration of Foxp3+ regulatory T cells following anti-CD25 treatment in malaria-infected mice. J Immunol. 2007;178:4136–46. doi: 10.4049/jimmunol.178.7.4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4(+) T(H)-17 cells in relapsing EAE. Nat Immunol. 2007;8:172–80. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- 39.Kohm AP, Carpentier PA, Miller SD. Regulation of experimental autoimmune encephalomyelitis (EAE) by CD4+CD25+ regulatory T cells. NovartisFoundSymp. 2003;252:45–52. [PubMed] [Google Scholar]
- 40.Reddy J, Illes Z, Zhang X, Encinas J, Pyrdol J, Nicholson L, et al. Myelin proteolipid protein-specific CD4+CD25+ regulatory cells mediate genetic resistance to experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2004;101:15434–9. doi: 10.1073/pnas.0404444101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McHugh RS, Shevach EM. Cutting edge: depletion of CD4+CD25+ regulatory T cells is necessary, but not sufficient, for induction of organ-specific autoimmune disease. J Immunol. 2002;168:5979–83. doi: 10.4049/jimmunol.168.12.5979. [DOI] [PubMed] [Google Scholar]
- 42.Oleszak EL, Chang JR, Friedman H, Katsetos CD, Platsoucas CD. Theiler’s virus infection: a model for multiple sclerosis. Clin Microbiol Rev. 2004;17:174–207. doi: 10.1128/CMR.17.1.174-207.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Godkin A, Ng WF, Gallagher K, Betts G, Thomas HC, Lechler RI. Expansion of hepatitis C-specific CD4+CD25+ regulatory T cells after viral clearance: a mechanism to limit collateral damage? J Allergy Clin Immunol. 2008;121:1277–84. e3. doi: 10.1016/j.jaci.2008.01.070. [DOI] [PubMed] [Google Scholar]
- 44.Haeryfar SM, DiPaolo RJ, Tscharke DC, Bennink JR, Yewdell JW. Regulatory T cells suppress CD8+ T cell responses induced by direct priming and cross-priming and moderate immunodominance disparities. J Immunol. 2005;174:3344–51. doi: 10.4049/jimmunol.174.6.3344. [DOI] [PubMed] [Google Scholar]
- 45.Miller SD, Clatch RJ, Pevear DC, Trotter JL, Lipton HL. Class II-restricted T cell responses in Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelinating disease. I. Cross-specificity among TMEV substrains and related picornaviruses, but not myelin proteins. J Immunol. 1987;138:3776–84. [PubMed] [Google Scholar]
- 46.Oehen S, Brduscha-Riem K. Differentiation of naive CTL to effector and memory CTL: correlation of effector function with phenotype and cell division. J Immunol. 1998;161:5338–46. [PubMed] [Google Scholar]
- 47.Begolka WS, Haynes LM, Olson JK, Padilla J, Neville KL, Canto MD, et al. CD8-deficient SJL mice display enhanced susceptibility to Theiler’s virus infection and increased demyelinating pathology. J Neurovirol. 2001;7:409–20. doi: 10.1080/135502801753170264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nocentini G, Riccardi C. GITR: a multifaceted regulator of immunity belonging to the tumor necrosis factor receptor superfamily. Eur J Immunol. 2005;35:1016–22. doi: 10.1002/eji.200425818. [DOI] [PubMed] [Google Scholar]
- 49.Shevach EM, Stephens GL. The GITR-GITRL interaction: co-stimulation or contrasuppression of regulatory activity? Nat Rev Immunol. 2006;6:613–8. doi: 10.1038/nri1867. [DOI] [PubMed] [Google Scholar]
- 50.Suvas S, Kim B, Sarangi PP, Tone M, Waldmann H, Rouse BT. In vivo kinetics of GITR and GITR ligand expression and their functional significance in regulating viral immunopathology. J Virol. 2005;79:11935–42. doi: 10.1128/JVI.79.18.11935-11942.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kohm AP, Williams JS, Miller SD. Cutting Edge: Ligation of the glucocorticoid-induced TNF receptor enhances autoreactive CD4+ T cell activation and experimental autoimmune encephalomyelitis. J Immunol. 2004;172:4686–90. doi: 10.4049/jimmunol.172.8.4686. [DOI] [PubMed] [Google Scholar]
- 52.Cuzzocrea S, Ayroldi E, Di Paola R, Agostini M, Mazzon E, Bruscoli S, et al. Role of glucocorticoid-induced TNF receptor family gene (GITR) in collagen-induced arthritis. FASEB J. 2005;19:1253–65. doi: 10.1096/fj.04-3556com. [DOI] [PubMed] [Google Scholar]
- 53.Patel M, Xu D, Kewin P, Choo-Kang B, McSharry C, Thomson NC, et al. Glucocorticoid-induced TNFR family-related protein (GITR) activation exacerbates murine asthma and collagen-induced arthritis. Eur J Immunol. 2005;35:3581–90. doi: 10.1002/eji.200535421. [DOI] [PubMed] [Google Scholar]
- 54.Katz-Levy Y, Neville KL, Padilla J, Rahbe SM, Begolka WS, Girvin AM, et al. Temporal development of autoreactive Th1 responses and endogenous antigen presentation of self myelin epitopes by CNS-resident APCs in Theiler’s virus-infected mice. J Immunol. 2000;165:5304–14. doi: 10.4049/jimmunol.165.9.5304. [DOI] [PubMed] [Google Scholar]
- 55.Neville KL, Padilla J, Miller SD. Myelin-specific tolerance attenuates the progression of a virus-induced demyelinating disease: Implications for the treatment of MS. J Neuroimmunol. 2002;123:18–29. doi: 10.1016/s0165-5728(01)00479-9. [DOI] [PubMed] [Google Scholar]
- 56.Kang BS, Palma JP, Lyman MA, Dal Canto M, Kim BS. Antibody response is required for protection from Theiler’s virus-induced encephalitis in C57BL/6 mice in the absence of CD8+ T cells. Virology. 2005;340:84–94. doi: 10.1016/j.virol.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 57.Rossi CP, Cash E, Aubert C, Coutinho A. Role of the humoral immune response in resistance to Theiler’s virus infection. J Virol. 1991;65:3895–99. doi: 10.1128/jvi.65.7.3895-3899.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting Edge: CD4(+)CD25(+) regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169:4712–16. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- 59.Herman AE, Freeman GJ, Mathis D, Benoist C. CD4+CD25+ T regulatory cells dependent on ICOS promote regulation of effector cells in the prediabetic lesion. J Exp Med. 2004;199:1479–89. doi: 10.1084/jem.20040179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Piccirillo CA, Tritt M, Sgouroudis E, Albanese A, Pyzik M, Hay V. Control of type 1 autoimmune diabetes by naturally occurring CD4+CD25+ regulatory T lymphocytes in neonatal NOD mice. Ann N Y Acad Sci. 2005;1051:72–87. doi: 10.1196/annals.1361.048. [DOI] [PubMed] [Google Scholar]
- 61.Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–65. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Connor RA, Anderton SM. Foxp3+ regulatory T cells in the control of experimental CNS autoimmune disease. J Neuroimmunol. 2008;193:1–11. doi: 10.1016/j.jneuroim.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 63.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–38. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 64.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. NatImmunol. 2003;4:330–36. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 65.Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nature RevImmunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- 66.Ruckwardt TJ, Bonaparte KL, Nason MC, Graham BS. Regulatory T cells promote early influx of CD8+ T cells in the lungs of respiratory syncytial virus-infected mice and diminish immunodominance disparities. J Virol. 2009;83:3019–28. doi: 10.1128/JVI.00036-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pullen LC, Miller SD, Dal Canto MC, Kim BS. Class I-deficient resistant mice intracerebrally inoculated with Theiler’s virus show an increased T cell response to viral antigens and susceptibility to demyelination. Eur J Immunol. 1993;23:2287–93. doi: 10.1002/eji.1830230935. [DOI] [PubMed] [Google Scholar]
- 68.Pirko I, Suidan GL, Rodriguez M, Johnson AJ. Acute hemorrhagic demyelination in a murine model of multiple sclerosis. J Neuroinflammation. 2008;5:31. doi: 10.1186/1742-2094-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ebinuma H, Nakamoto N, Li Y, Price DA, Gostick E, Levine BL, et al. Identification and in vitro expansion of functional antigen-specific CD25+ FoxP3+ regulatory T cells in hepatitis C virus infection. J Virol. 2008;82:5043–53. doi: 10.1128/JVI.01548-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barboza L, Salmen S, Goncalves L, Colmenares M, Peterson D, Montes H, et al. Antigen-induced regulatory T cells in HBV chronically infected patients. Virology. 2007;368:41–9. doi: 10.1016/j.virol.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 71.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–96. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174:1783–6. doi: 10.4049/jimmunol.174.4.1783. [DOI] [PubMed] [Google Scholar]
- 73.Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589–601. doi: 10.1016/j.immuni.2004.09.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Treatment with anti-CD25 and anti-GITR in combination does not increase TMEV-specific CD8+ T cell responses or ameliorate disease progression compared to either treatment alone. SJL/J mice were treated with control Ig or either 0.5 mg of anti-CD25 (clone PC-61) on days -2 and 0 relative to infection and/or 1 mg of anti-GITR (clone DTA-1 ) on day -1 relative to infection. Animals were followed for disease score (A) and incidence (B) for 100 days post-infection. Additionally, an in vivo lysis assay was performed at day 12 p.i. (C) to assess TMEV specific CD8+ T cell responses. Treatment with either antibody resulted in the same disease amelioration and increase in anti-viral responses as the combined treatment.
Supplemental Figure 2. FACS analysis of the T regulatory cell population in the CNS of TMEV-infected and Sham-infected SJL and B6 animals at day 3 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5 ×106 pfu/mouse. On day 3 post-infection brain and spinal cord tissue from sham and infected animals were analyzed for CD3+CD4+Foxp3+ cells.
Supplemental Figure 3. FACS analysis of the T regulatory cell population in the spleen of TMEV-infected and Sham-infected SJL and B6 animals at day 3 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5 ×106 pfu/mouse. On day 3 post-infection spleen tissue from sham and infected animals were analyzed for CD3+CD4+Foxp3+ cells.
Supplemental Figure 4. FACS analysis of the T regulatory cell population in the CNS of TMEV-infected and Sham-infected SJL and B6 animals at day 7 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5×106 pfu/mouse. On day 7 post-infection brain and spinal cord tissue from sham and infected animals were analyzed for CD3+CD4+Foxp3+ cells.
Supplemental Figure 5. FACS analysis of the T regulatory cell population in the spleen of TMEV-infected and Sham-infected SJL and B6 animals at day 7 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5×106 pfu/mouse. On day 7 post-infection spleen tissue from sham and infected animals were analyzed for CD3+CD4+Foxp3+ cells.
Supplemental Figure 6. FACS analysis of CD8+IFN-γ+ cells in the CNS of TMEV-infected and Sham-infected SJL and B6 mice at day 3 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5×106 pfu/mouse. On day 3 post-infection brain and spinal cord tissue from sham and infected animals were harvested. Cells were stimulated PMA and ionomycin for 6 hours and Brefeldin A and Monensin for the final two hours.
Supplemental Figure 7. FACS analysis of CD8+IFN-γ+ cells in the spleen of TMEV-infected and Sham-infected SJL and B6 mice at day 3 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5×106 pfu/mouse. On day 3 post-infection spleen tissue from sham and infected animals were harvested. Cells were stimulated PMA and ionomycin for 6 hours and Brefeldin A and Monensin for the final two hours.
Supplemental Figure 8. FACS analysis of CD8+IFN-γ+ cells in the CNS of TMEV-infected and Sham-infected SJL and B6 mice at day 7 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5×106 pfu/mouse. On day 3 post-infection brain and spinal cord tissue from sham and infected animals were harvested. Cells were stimulated PMA and ionomycin for 6 hours and Brefeldin A and Monensin for the final two hours.
Supplemental Figure 9. FACS analysis of CD8+IFN-γ+ cells in the spleen of TMEV-infected and Sham-infected SJL and B6 mice at day 7 post-infection. SJL and B6 GFP-Foxp3 transgenic mice were sham-infected or TMEV-infected i.c. with 5×106 pfu/mouse. On day 3 post-infection spleen tissue from sham and infected animals were harvested. Cells were stimulated PMA and ionomycin for 6 hours and Brefeldin A and Monensin for the final two hours.
Supplemental Figure 10. FACS analysis of CD3+CD4+Foxp3+BrdU+ cells in the CNS of TMEV-infected and Sham-infected SJL and B6 mice at day 3 post-infection. SJL and B6 mice were sham-infected or TMEV-infected with 5×105 pfu/mouse. At 24 and 12 hours prior to harvest animals were treated i.p. with 1mg of BrdU. On day 3 brain and spinal cord samples were harvested and analyzed for CD3+CD4+Foxp3+BrdU+ cells.
Supplemental Figure 11. FACS analysis of CD3+CD4+Foxp3+BrdU+ cells in the spleen of TMEV-infected and Sham-infected SJL and B6 mice at day 3 post-infection. SJL and B6 mice were sham infected or TMEV infected with 5×105 pfu/mouse. At 24 and 12 hours prior to harvest animals were treated i.p. with 1mg of BrdU. On day 3 spleen samples were harvested and analyzed for CD3+CD4+Foxp3+BrdU+ cells.
Supplemental Figure 12. FACS analysis of CD3+CD4+Foxp3+BrdU+ cells in the CNS of TMEV-infected and Sham-infected SJL and B6 mice at day 7 post-infection. SJL and B6 mice were sham-infected or TMEV-infected with 5×105 pfu/mouse. At 24 and 12 hours prior to harvest animals were treated i.p. with 1mg of BrdU. On day 7 brain and spinal cord samples were harvested and analyzed for CD3+CD4+Foxp3+BrdU+ cells.
Supplemental Figure 13. FACS analysis of CD3+CD4+Foxp3+BrdU+ cells in the spleen of TMEV-infected and Sham-infected SJL and B6 mice at day 7 post-infection. SJL and B6 mice were sham-infected or TMEV-infected with 5×105 pfu/mouse. At 24 and 12 hours prior to harvest animals were treated i.p. with 1mg of BrdU. On day 7 spleen samples were harvested and analyzed for CD3+CD4+Foxp3+BrdU+ cells.