

Abstract
The kinesin superfamily of motor proteins are known to be ATP-dependent transporters of various types of cargoes. In neurons, KIF17 is found to transport vesicles containing the N-methyl-D-aspartate receptor NR2B subunit from the cell body specifically to the dendrites. These subunits are intimately associated with glutamatergic neurotransmission as well as with learning and memory. Glutamatergic synapses are highly energy-dependent, and recently we found that the same transcription factor, nuclear respiratory factor 1 (NRF-1), co-regulates energy metabolism (via its regulation of cytochrome c oxidase and other mitochondrial enzymes) and neurochemicals of glutamatergic transmission (NR1, NR2B, GluR2, and nNOS). The present study tested our hypothesis that NRF-1 also transcriptionally regulates KIF17. By means of in silico analysis, electrophoretic mobility shift and supershift assays, in vivo chromatin immunoprecipitation assays, promoter mutations, and real-time quantitative PCR, we found that NRF-1 (but not NRF-2) functionally regulates Kif17, but not Kif1a, gene. NRF-1 binding sites on Kif17 gene is highly conserved among mice, rats, and humans. Silencing of NRF-1 with small interference RNA blocked the up-regulation of Kif17 mRNA and proteins (and of Grin1 and Grin2b) induced by KCl-mediated depolarization, whereas over-expressing NRF-1 rescued these transcripts and proteins from being suppressed by TTX. Thus, NRF-1 co-regulates oxidative enzymes that generate energy and neurochemicals that consume energy related to glutamatergic neurotransmission, such as KIF17, NR1, and NR2B, thereby ensuring that energy production matches energy utilization at the molecular and cellular levels.
Keywords: excitatory neurotransmission, gene regulation, KCl depolarization, NRF-1 over-expression, NMDA receptor, TTX
1. Introduction
The kinesin superfamily proteins (KIFs) transport membranous organelles, protein complexes, and mRNAs to specific destinations in a microtubule- and ATP- dependent manner [1, 2]. They play a significant role in neuronal development, synaptic functioning, intraflagellar transport, and retinal outer segment renewal [2–5]. Kif17, a homodimeric motor protein, binds specifically through its tail domain to the postsynaptic density-95/disc large/zona occludens-1 (PDZ) domain of Mint 1 (mLin10) within a large scaffolding protein complex that transports vesicles containing the NR2B subunit of the NMDA receptors (NMDARs) from the cell body exclusively to the dendrites [6]. Over-expressing KIF17 enhances learning and memory [7], which is dependent on NMDA receptors [8–10]. Likewise, up-regulating NR2B increases the expression of KIF17, whereas down-regulating KIF17 reduces NR2B expression [11]. The positive relationship between KIF17 and NR2B raises the possibility that the two may be co-regulated [11]. Our laboratory has recently found that neuronal NR2B and the obligatory NR1 subunit of NMDA receptors are transcriptionally regulated by nuclear respiratory factor 1 (NRF-1) [12], which also regulates all subunits of cytochrome c oxidase (COX; [13]), a marker of energy metabolism and neuronal activity [14]. The goal of the present proposal was to test our hypothesis that NRF-1 also transcriptionally regulates KIF17. By means of in silico analysis, electrophoretic mobility shift and supershift assays, in vivo chromatin immunoprecipitation assays, promoter mutations, and real-time quantitative PCR, we found that NRF-1 indeed functionally regulates KIF17 in neurons.
2. Materials and methods
All experiments were carried out in accordance with the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Medical College of Wisconsin regulations. All efforts were made to minimize the number of animals and their suffering.
2.1. Cell culture
Murine neuroblastoma N2a cells were obtained from the American Type Culture Collection (ATCC, CCL-131). Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, 50 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA) at 37°C in a humidified atmosphere with 5% CO2.
Rat primary cortical neurons were cultured as described previously [15]. Briefly, 1-day-old neonatal rat pups were anesthetized with CO2 and killed by decapitation. Brains were removed from the skull and the meninges were removed. Visual cortical tissue was dissected, trypsinized, and triturated to release individual neurons. These primary cortical neurons were then plated in 35 mm poly-L-lysine-coated dishes at a density of 50,000 cells/dish. Cells were maintained in Neurobasal-A media supplemented with B27 (Invitrogen). Ara-C (Sigma, St. Louis, MO) was added to the media to suppress the proliferation of glial cells.
2.2. In silico analysis of murine Kif17 promoter
DNA sequences surrounding the transcription start points (TSPs) of Kif17 gene was derived from the mouse genome database in GenBank™, as previously described [7]. These promoter sequences encompassed 1 kb upstream and up to 200 bps downstream (excluding protein-coding sequence) of the TSP of each gene analyzed. Computer-assisted search for putative NRF-1 core binding sequences “GCGCAT/CGC” or “GCGCAG/CGC” and for NRF-2α (GGAA or TTCC) were conducted on Kif17 and Kif1a promoters. Kif1a was chosen because it has a putative NRF-1 binding site, and NRF-2 was chosen because it was found previously to regulate all subunits of cytochrome c oxidase genes[15, 16], which NRF-1 also regulates [13]. Promoters of neuronal Kif3a and Kif5b were also analyzed for putative NRF-1 binding sites. Alignment of human, mouse, and rat promoter sequences was done as previously described, using the Genome VISTA genome alignment tool [15]. Murine Kif17 promoter sequence was compared with rat and human genomic sequences using a 5-bp calculation window. Regions of high homology and/or that contained known NRF-1 binding sites were compared for the conservation of NRF-1 binding.
2.3. Electrophoretic mobility shift and supershift assays
In vitro NRF-1 and NRF-2α interactions with Kif17 were assayed with EMSA using protocols as described previously [15]. Oligonucleotide probes with putative NRF-1 (sites A & B) and NRF-2α binding sites on the Kif17 promoter region were synthesized (Table 2A), annealed, and labeled by a Klenow fragment fill-in reaction with [α-32P]dATP (50 μCi/200 ng). Each of the labeled probes was incubated with 2 μg of calf thymus DNA and 5 μg of HeLa nuclear extract (Promega, Madison, WI) and was processed for EMSA. For the supershift assays, 1–1.5 μg of specific antibodies for the appropriate reactions were added to the probe/nuclear extract mixture and the reaction was incubated for 20 min at room temperature. For the competition reactions, 100-fold excess of unlabeled oligonucleotide was incubated with the nuclear extract before adding the labeled oligonucleotide. All reactions were loaded onto a 4% polyacrylamide gel and ran at 200 V for 2.5 h in 0.25X TBE buffer. The results were visualized with autoradiography. Rat cytochrome c with a NRF-1 binding site at position −172/−147 was used as a positive control for NRF-1 and was designed based on previous report [17]. Similarly, COX4i1 was used as a positive control for NRF-2α [15]. Mutants of NRF-1 and NRF-2α sequences as shown in Table 2A were used as negative controls.
Table 2A.
EMSA PROBES
| Gene Promoter | Position | Sequence |
|---|---|---|
| Kif17 NRF-1A | +3/+28 | F 5′ TTTTGCGGCGGCGCCTGCAGCGCGGATCC 3′ R 3′ CGCCGCCGCGGACGTCGCGCCTAGGTTTT 5′ |
| Kif17 NRF-1B | −53/−25 | F 5′ TTTTGCCGAGACGGCGTGCACGAGCGCGAGCG 3′ R 3′ CGGCTCTGCCGCACGTGCTCGCGCTCGCTTTT 5′ |
| Kif17 NRF-2α | −106/−81 | F 5′ TTTTGCACTCGCCATTCCGTCCTGTGACG 3′ R 3′ CGTGAGCGGTAAGGCAGGACACTGCTTTT 5′ |
| Kif17 NRF-1A mutant | +3/+28 | F 5′ TTTTGCGGCGGCGCCTTTTGCTTTGATCC 3′ R 3′ CGCCGCCGCGGAAAACGAAACTAGGTTTT 5′ |
| Kif17 NRF-1B mutant | −53/−25 | F 5′ TTTTGCCGAGACGGCGTTTTCGAGTTTGAGCG 3′ R 3′ CGGCTCTGCCGCAAAAGCTCAAACTCGCTTTT 5′ |
| Kif17 NRF-2α mutant | −106/−81 | F 5′ TTTTGCACTCGCCATAGAGTCCTGTGACG 3′ R 3′ CGTGAGCGGTATCTCAGGACACTGCTTTT 5′ |
| Rat Cyt C | −172/−147 | F 5′TTTTCTGCTAGCCCGCATGCGCGCGCACCTTA3′ R 3′GACGATCGGGCGTACGCGCGCGTGGAATTTTT5′ |
| COX4i1 | +13/+36 | F 5′TTTTCGGGACCCGCTCTTCCGGTCGCGA 3′ R 3′GCCCTGGGCGAGAAGGCCAGCGCTTTTT 5′ |
Positions of probes are given relative to TSP. Putative NRF-1 and NRF-2α binding sites are in boldface. Mutated nucleotide sequences are underlined.
2.4. Chromatin immunoprecipitation assays
In vivo ChIP assays were performed using protocols as described previously [12, 18]. For each immunoprecipitation, ~750,000 primary neurons were used and fixed with 1% formaldehyde for 10 min at room temperature. A ChIP assay kit (Upstate, Charlottesville, VA) was used with minor modifications. The cells were resuspended in a swelling buffer (5 mM PIPES, pH 8.0, 85 mM KCl, and 1% Nonidet P-40, and protease inhibitors added right before use). The nuclei were isolated by centrifugation and then sonicated. The sonicated lysate was immunoprecipated with 0.2 μg of the appropriate (NRF-1, NRF-2α) polyclonal rabbit antibodies or 2 μg of anti-nerve growth factor receptor (NGFR) p75 polyclonal goat antibodies (C20 from Santa Cruz Biotechnology, Santa Cruz, CA). NGFR was used as an antibody control, as it should not immunoprecipitate any transcription factor.
Semi-quantitative PCR was performed with 1/20th of the precipitated chromatin, and primers targeting NRF-1 and NRF-2α sequences were designed near the TSP of Kif17 and Kif1a genes (Table 2B) using approaches as described previously [15]. Transcription factor B2 of mitochondria (TFB2M) promoter, previously found to be activated by NRF-1 in neurons [13], was used as a positive control, and exon 5 of β-actin gene was used as a negative control (Table 2B). COX6a1 promoter, previously found to be activated by NRF-2α in neurons [15], was used as a positive control. PCR reactions were carried out with the EX Taq hot-start polymerase (Takara Mirus Bio, Madison, WI) with the following cycling parameters: 30 s denaturation at 94°C, 30 s annealing at 59.5°C, and 20 s extension at 72°C (32–36 cycles per reaction). All reactions were hot-started by heating to 94°C for 120 s. Use of hot-start polymerase and PCR additives (magnesium and/or 0.5 M betaine (Sigma)) significantly improved the quality and reproducibility of ChIP. PCR products were visualized on 2% agarose gels stained with ethidium bromide.
Table 2B.
ChIP assay primers
| Gene Promoter | Position | Sequence | Amplicon length |
|---|---|---|---|
| Kif17 NRF-1A&B | −87 to +112 | F 5′ TGACGTCACGGAGGTTGC 3′ R 5′ AAGTGTGCGGGCTGGAAC 3′ |
199 bps |
| Kif17 NRF-2α | −238 to −64 | F 5′ CTTACCCTGCCTACCTCTGC 3′ R 5′ CTGGGCAACCTCCGTGAC 3′ |
174 bps |
| Kif1a | −154 to +43 | F 5′ AACCTCGTCTCTTCTGTGCCGCTTT3′ R 5′ GGAACTGATGCTTCTTACTGCCTG 3′ |
197 bps |
| COX6a1 | −163 to +96 | F 5′ATGGTACCTCACCCAGCAGCAGAGGAG 3′ R 5′CCAGAGACTCGAGACGCACTC 3′ |
259 bps |
| TFB2M promoter | −64 to +115 | F 5′GAAGCGAGTGAGCAAAGGAC 3′ R 5′GGTCCCCTCATCCTCCTCTA 3′ |
179 bps |
| β-Actin exon5 | −134 to +53 | F 5′GCTCTTTTCCAGCCTTCCTT 3′ R 5′CGGATGTCAACGTCACACTT 3′ |
187 bps |
Positions of amplicons are given relative to TSP.
2.5. Promoter mutagenesis study
A luciferase reporter construct of Kif17 was created by PCR, and the proximal promoter sequences were cloned using genomic DNA prepared from mouse N2a cells as template, digested with KpnI and HindIII, and the product was ligated directionally into pGL3 (Promega). Site-directed mutagenesis of both NRF-1 binding sites on each promoter was produced using QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Sequencing was used to verify all constructs.
The Kif17 promoter construct was transfected into N2a cells in a 24-well plate using Lipofectamine 2000 (Invitrogen). Each well received 0.6 μg of reporter construct and 0.03 μg of pCMVβgal, which constitutively expressed β-galactosidase. Transfected neurons were stimulated with KCl at a final concentration of 20 mM in the culture media for 5 h as previously described [19]. After five hours of treatment, cell lysates were harvested and measured for luciferase activity as described previously [13]. Data from six independent transfections were averaged for each promoter construct.
2.6. Plasmid construction of NRF-1 shRNA
NRF-1 silencing was carried out using small hairpin RNAs (shRNA) against murine NRF-1 (GenBank™ accession no. for NRF-1: NM_010938) cloned into pLVTHM vector with H1 promoter and green fluorescent protein reporter (gift of Dr. P. Aebischer, Swiss Federal Institute of Technology). Four shRNA sequences were selected: 5′-GAAAGCTGCAAGCCTATCT-3′; 5′GCCACAGGAGGTTAATTCA-3′; 5′-GCATTACGGACCATAGTTA-3′; and 5′-AGAGCATGATCCTGGAAGA-3′, with a linker sequence (5′-TTCAAGAGA- 3′) and complementary sequence for each to form the NRF-1-shRNA cassette. The pLL3.7/U6 promoter vector with puromycin resistance (Addgene, Cambridge, MA) vector was used concurrently for puromycin selection. The empty vector pLVTHM or scrambled shRNA in pLVTHM served as negative controls. The basic gene cloning method was followed as described previously [16]. Primary neurons were plated in 35-mm dishes at a density of 5 to 8 × 106 cells/dish. They were co-transfected 3 days post-plating with NRF-1 shRNA expression vectors (four sequences at equal amounts; 1 μg total for primary neurons) and the pLL3.7/U6 vector for puromycin resistance (1 μg for primary neurons) via NeuroFect™ (Genlantis, San Diego, CA). Empty vectors or scrambled shRNA vectors alone were used at the same concentrations as vectors with shRNA against NRF-1. Puromycin at a final concentration of 0.5 μg/ml was added to the culture medium on the second day after transfection to select for purely transfected cells. Green fluorescence was used to monitor transfection efficiency. Transfection for primary cortical neurons was from 40% to 60%. However, puromycin selection effectively yielded 100% of transfected cells. Cells were harvested after 48 h of silencing and lysed for either protein or total RNA preparation.
To check for the effect of silencing NRF-2 on the expression of Kif17, the same protocols as above was used with shRNA sequences against NRF-2α as described previously [16].
To determine the effect of KCl stimulation, neurons transfected with shRNA against NRF-1 were exposed to KCl at a final concentration of 20 mM in the culture media for 5 h as described previously [19]. Cells were then harvested for RNA isolation.
2.7. RNA isolation and cDNA synthesis
Total RNA was isolated by RNeasy kits (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Three micrograms of total RNA was treated with DNase I and purified by phenol-chloroform. cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions.
2.8. Real-time quantitative PCR
Real-time quantitative PCRs were carried out in a Cepheid Smart Cycler Detection system (Cepheid, Sunnyvale, CA). SyBr Green (BioWhittaker Molecular Application, Rockland, ME) and EX Taq real-time quantitative PCR hotstart polymerase were used following the manufacturer’s protocols and as described previously [13]. Primer sequences are shown in Table 3. PCR runs: hot start 2 min at 95°C, denaturation 10 s at 95°C, annealing 15 s according to the Tm of each primer, and extension 10 s at 72°C for 15–30 cycles. Melt curve analyses verified the formation of single desired PCR product. Rat 18S was used as an internal control, and the 2−ΔΔCT method [20] was carried out for the relative amount of transcripts.
Table 3.
Primers for real-time PCR
| Gene | Sequence | Amplicon length | Tm |
|---|---|---|---|
| Kif17 | F 5′ AAGTGTGGGTGTAGAAGTGGCTGT 3′ R 5′ TGCTTCTCCCTCAGGTCCTTGTTT 3′ |
144 | 60º |
| Kif1a | F 5′CATGAGCGCATCTTGTTTGCTCCA 3′ R 5′ACACACCCAAGGTACCACCATCTT 3′ |
196 | 60º |
| Grin2b | F 5′TCATGGTATCTCGCAGCAATGGGA 3′ R 5′ACCGCAGAAACAATGAGCAGCATC 3′ |
108 | 60º |
| Grin1 | F 5′TGGCTTCTGCATAGACCTGCTCAT 3′ R 5′TTGTTGCTGTTGTTTACCCGCTCC 3′ |
117 | 60º |
| NRF-1 | F 5′ AAAAGGCCTCATGTGTTTGAGT 3′ R 5′ AGGGTGAGATGCAGAGAACAAT 3′ |
139 | 59.5º |
| NRF-2α | F 5′CTCCCGCTACACCGACTAC 3′ R 5′TCTGACCATTGTTTCCTGTTCTG 3′ |
145 | 59.5º |
| 18S | F 5′CGCGGTTCTATTTTGTTGGT 3′ R 5′AGTCGGCATCGTTTATGGTC 3′ |
219 | 59.5º |
2.9. Western blot assays
Control and NRF-1 shRNA samples were loaded onto 10% SDS-PAGE gel and electrophoretically transferred onto polyvinylidene difluoride membranes (Bio-Rad). Subsequent to blocking, blots were incubated in primary antibodies (polyclonal antibodies against KIF17 (H-280, SC-50455, 1:200) and KIF1A (E-20, SC-19106, 1:100; Santa Cruz Biotechnology). Monoclonal antibodies against β-actin (Sigma) at 1:3,000 dilution were used as loading controls. Blots were then incubated in secondary antibodies (goat-anti-rabbit; Bio-Rad, rabbit-anti-goat; Millipore), reacted with ECL, and exposed to autoradiographic film (Santa Cruz Biotechnology). Quantitative analyses of relative changes were done with a Phospho Imager (Bio-Rad).
2.10. NRF-1 over-expression and TTX treatment
pSG5NRF-1 plasmid (gift of Dr. Richard Scarpulla) was used for NRF-1 over-expression. Primary neurons were each plated in 35 mm dishes at a density of 2 to 5 × 105 cells/dish. Neurons were co-transfected 3 days post-plating with either 2 μg of the pSG5NRF-1 plasmid or an empty vector plus 0.5 μg of pLL3.7 vector, using NeuroFect™ (Genlantis) at 1:3 ratio. Puromycin at a final concentration of 0.5μg/ml was added on the second day after transfection to select for purely transfected cells. After one day of over-expression, TTX at a final concentration of 0.4 μM was added to the culture media for 4 days. Primary neurons were harvested on the 5th day for RNA isolation.
2.11. Statistical Analysis
Significance among group means was determined by analysis of variance (ANOVA). Significance between two groups was analyzed by Student’s t test. P-values of 0.05 or less were considered significant.
3. Results
3.1. In silico promoter analysis of Kif17 gene
Proximal promoters of murine Kif17 gene with DNA sequence 1 kb 5′ upstream and 100 bps beyond 3′ of transcription start points (TSP) were analyzed in silico for potential NRF-1 and NRF-2α binding sites (Table 2A). Promoter showed two atypical sequence for NRF-1 binding (A and B sites, Table 1) and both had the invariant GCA core described previously for COX subunit genes [13]. There were two NRF-2α binding sites but they were not tandem arrays. NRF-1 binding sites bore a high (85–95%) homology with both the rat and human Kif17 genes (Table 1). Kif1a promoter also had atypical NRF-1 binding sites but without the invariant GCA core. It was found not to functionally bind to NRF-1 and so was used as a negative control in subsequent studies. Promoters of neuronal Kif3a and Kif5b were also analyzed, and they did not contain either typical or atypical NRF-1 binding sites.
Table 1.
Aligned partial sequences of Kif17 promoter from rat (R), mouse (M), and human (H) genomes indicate conservation of atypical NRF-1 (A–B) binding sites. Conserved binding site sequences are in boldface. Solid boxes highlight NRF-1 sites that are highly conserved in all three or at least two species.
 |
3.2. In vitro binding of Kif17 with NRF-1 and NRF-2α
In vitro electrophoretic mobility shift assays (EMSAs) were carried out using 32P-labeled probes (Table 2A) to determine the specificity of the two NRF-1 binding sites (A and B) in the promoters of murine Kif17 (Fig. 1A–B). Rat cytochrome c promoter with a NRF-1 binding site at positions −172/−147 served as a positive control [13], and it formed specific DNA/NRF-1 shift and supershift complexes (Fig. 1A, lanes 1 and 3, respectively). When an excess of unlabeled probe was added as a competitor, no shift band was formed (Fig. 1A, lane 2). To rule out any non-specific antibody-oligonucleotide interactions, labeled oligonucleotides were incubated with NRF-1 antibody without HeLa nuclear extract, and no shift bands were observed (Fig. 1A, lane 8; Fig. 1B, lane 4). As shown in Fig. 1A-B, Kif17 promoter with both NRF-1A and B sites formed specific DNA-protein shift complexes when incubated with purified HeLa nuclear extract (Fig. 1A, lane 4; Fig. 1B, lane 1, respectively). Competition with excess unlabeled probes eliminated these complexes (Fig. 1A, lane 6; Fig. 1B, lane 2, respectively), whereas the addition of mutant NRF-1 probes had no effect (Fig. 1A, lanes 5 and Fig. 1B, lane 3). A super-shift band was produced when anti-NRF-1 antibodies were added to shift assays of Kif17 (Fig. 1A, lane 7 and Fig. 1B, lane 5, respectively). Kif17 mutant NRF-1 probes (A and B sites) yielded no band or complexes (Fig. 1A, lanes 9–11 and Fig. 1B, lanes 6–8).
Fig. 1. NRF-1 and NRF-2α interactions in vitro with Kif17 gene promoter.

(A–B) EMSAs for NRF-1. 32P- labeled oligonucleotides, excess unlabeled oligos as competitors, excess unlabeled mutant NRF-1 as competitors, HeLa extract, and NRF-1 antibodies are indicated by a + or a – sign. Arrowheads indicate NRF-1 shift and supershift complexes. The positive control, cytochrome c, shows a shift (A, lane 1) and a supershift (A, lane 3) band. When excess unlabeled competitor was added, it did not yield any band (A, lane 2). Kif17 promoter containing two putative NRF-1 binding sites (NRF-1A and NRF-1B) showed specific shift and supershift bands that were eliminated by excess unlabeled competitors (A, lanes 4, 7, and 6; B, lanes 1, 5, and 2, respectively). Labeled mutated NRF-1 A and B sites on Kif17 were used as negative controls, and they did not yield any band (A, lanes 9–11; B, lanes 6–8, respectively). Excess unlabeled but mutated NRF-1 (containing both NRF-1A and B sites) could not compete (A, lane 5; B, lane 3, respectively). Labeled oligos with NRF-1 antibodies alone with no HeLa extract did not yield any band (A, lane 8; B, lane 4, respectively). (C) EMSA for NRF-2α. The positive control, COX4i1, shows shift (C, lane 1) and supershift (C, lane 3) bands, while excess unlabeled competitors did not yield any band (C, lane 2). Kif17 promoter had no shift or supershift bands for NRF-2α (C, lanes 4 & 7). Labeled mutated NRF-2α site did not yield any bands (C, lanes 9–11). Excess unlabeled and mutated NRF-2α sites did not show any bands (C, lane 5–6). Labeled oligos with NRF-2α antibodies alone did not yield any band (C, lane 8).
In vitro binding of Kif17 promoter to NRF-2α were carried out using 32P-labeled probes (Table 1A). As shown in Fig. 1C, the rat COX4i1 promoter with NRF-2α site served as a positive control [15], and it formed specific DNA/NRF-2α shift and supershift complexes (Fig. 1C, lanes 1 and 3, respectively). Kif17 with putative NRF-2α sites yielded no band or complexes (Fig. 1C, lanes 4–8). When an excess of unlabeled probe was added as a competitor, no shift band was formed (Fig. 1C, lane 2). Mutated NRF-2α sites did not show any bands (Fig. 1C, lanes 9–11).
3.3. In vivo NRF-1 and NRF-2α interactions with Kif17 promoter
Chromatin immunoprecipitation assays (ChIP) were performed to verify possible NRF-1 and NRF-2α interactions with Kif17 promoter in vivo. β-actin exon 5 served as a negative control, whereas transcription factor B2 of mitochondria (TFB2M) with a known NRF-1 binding site [13] and COX6a1 with known NRF-2α sites [15] served as a positive controls. Parallel immunoprecipitation assays were carried out with the same neuronal cell lysates and NRF-1 and NRF-2α antibodies. Anti-nerve growth factor receptor p75 antibodies (NGFR) served as a negative immunoprecipitation control. Polymerase chain reactions (PCRs) using primers shown in Table 2B were done to determine which promoters interacted with NRF-1 and NRF-2α in vivo. Individual 0.5 and 0.1% dilutions of input chromatin were used as standards to indicate the efficiency of the PCRs (Fig. 2A–2B). Kif17 and TFB2M each produced a band from DNA co-immunoprecipitated with anti-NRF-1 antibodies at a position identical to that from the genomic DNA control (input) (Fig. 2A). On the other hand, Kif1a and β-actin yielded no bands (Fig. 2A). COX6a1 produced a band from DNA co-immunoprecipitated with anti-NRF-2α antibodies at a position identical to that from the genomic DNA control (input) (Fig. 2B), whereas Kif17 and β-actin yielded no band (Fig. 2B). In all cases, co-immunoprecipitation with NGFR antibodies did not yield any PCR product.
Fig. 2.

(A–B) ChIP assays. Input lanes represent 0.5% and 0.1% of chromatin. TFB2M promoter was the positive control for NRF-1, whereas COX6a1 promoter was the positive control for NRF-2α. β-actin was the negative control for both NRF-1 and NRF-2α. Kif17 promoter immunoprecipitated with NRF-1, whereas Kif1a did not. Anti-nerve growth factor receptor p75 antibodies (NGFR) represent a negative control. Kif17 promoter did not immunoprecipitate with NRF-2α.
3.4. Mutational analysis of NRF-1 binding
Based on EMSA probes (Table 2A) that formed NRF-1 specific complexes (Fig. 1A, B), site-directed mutations of these same putative NRF-1 binding sites on Kif17 and previously studied Grin1 promoters [12] were constructed (Table 2C), generated in luciferase reporter plasmids, and analyzed by gene transfection. As shown in Fig. 3, mutation of either NRF-1 A or B binding site on Kif17 as well as on Grin1 led to ~ 35 – 60% reduction in promoter activity of these genes (P < 0.05 – 0.01). Grin1 subunit promoter served as a positive control and confirmed our previous report [12].
Table 2C.
PCR cloning primers
| Cloning Primers | Primer sequence |
|---|---|
| Grin1 | F 5′AAGGTACCCGGCCGTCACACCTATTCT 3′ R 5′AAAAGCTTGAAAAGGCGAAAAAGACAGC 3′ |
| Kif17 | F 5′AAGGTACCGCGTGTGAACCTGCTTACAA 3′ R 5′AAAAGCTTCCATCGAAGGTGAACTGCTT 3′ |
| Mutagenesis Primers | |
| Grin1Mut | F 5′CAAGCATTTACGCCAACTTTGGCTTTTGTCAGGAGGCGCGCGCT 3′ R 5′AGCGCGCGCCTCCTGACAAAAGCCAAAGTTGGCGTAAATGCTTG 3′ |
| Kif17 NRF-1 Mut1 | F 5′ GTCGTGCGGCGGCGCCTTTTGCTTTGATCCCCCGTT 3′ R 5′ AACGGGGGATCAAAGCAAAAGGCGCCGCCGCACGAC 3′ |
| Kif17 NRF-1 Mut2 | F 5′ AACGGCGAACGCCGAGACGGCGTAAACGAAAACGAGCGTGA 3′ R 5′TCACGCTCGTTTTCGTTTACGCCGTCTCGGCGTTCGCCGTT 3′ |
Fig. 3. Site-directed mutational analysis of both NRF-1 binding sites (A and B) on Kif17 and Grin1 promoters.
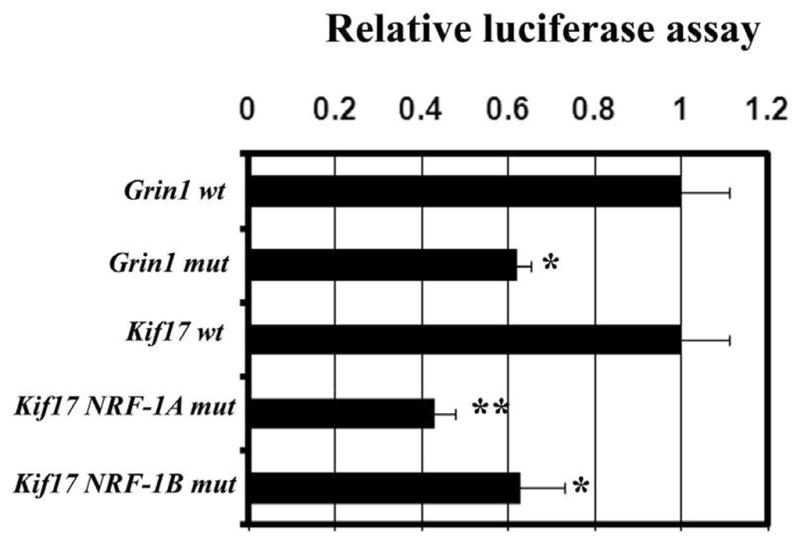
Mutated NRF-1 binding sites (NRF-1A mut and NRF-1B mut) on Kif17 and Grin1 subunit resulted in significant reductions in luciferase activity as compared to wild type (wt). (N = 6 for each construct). *, P < 0.05, **, P < 0.01.
3.5. NRF-1 silencing by RNA interference
To determine the effect of silencing NRF-1 transcript on the expression of NMDA receptors and Kif17, plasmid vectors expressing small hairpin RNA (shRNA) against four target sequences of NRF-1 mRNA were used. These vectors were previously found to silence NRF-1 expression in neurons [12]. Transfection of neurons with shRNA vectors resulted in ~ 70– 78 % decrease in levels of KIF17 and NRF-1 protein as measured by western blots (P < 0.05) (Fig. 4A). However, KIF1A showed no appreciable alterations in protein levels (Fig. 4A). To determine that this effect was not limited to N2a cells, cDNAs from primary cortical neurons (Fig. 4B) transfected with NRF-1 shRNA vectors, scrambled shRNA vectors, or empty vectors were analyzed with quantitative real-time PCRs. As shown in Fig. 4B, mRNA levels of NRF-1, Kif17, Grin1, and Grin2b were significantly reduced in primary neurons transfected with shRNA as compared to those transfected with empty vectors. The extent of reduction ranged between 62 to 78% (P < 0.05). On the other hand, the expressions of Kif1a and nuclear respiratory factor 2a (NRF-2α, a negative control) remained unchanged. The scrambled shRNA also did not have any effect on any mRNA level tested (Fig. 4B).
Fig. 4. RNA interference-mediated silencing of NRF-1 suppresses mRNAs of Kif17 and NMDAR subunit genes.

(A) Western blot reveals a down-regulation of KIF17 and NRF-1 protein levels in shRNA-transfected neurons, whereas KIF1A protein levels were not affected. β-Actin served as a loading control. (B) Primary neurons were transfected with shRNA against NRF-1 (dotted bars) or with empty vectors (black bars) or with scrambled shRNA (hatched bars). NRF-2α served as a negative control. NRF-1, Kif17, Grin1 and Grin2b subunit mRNAs were significantly reduced in shRNA-treated samples as compared to those with empty vectors, whereas Kif1a and NRF-2α mRNAs remained unchanged. N = 6 for each data point. * P < 0.05.
Silencing of NRF-2 with shRNA sequences as described previously [16] did not yield any statistically significant difference in the expression of Kif17 (data not shown), consistent with the negative EMSA and ChIP data.
3.6. Response of Kif17 promoter to KCl depolarizing stimulation
To determine if depolarizing stimulation altered the expression of Kif17 gene in N2a cells, 20 mM of potassium chloride was added to the culture media for 5 hours, a mild regimen previously found to activate NRF-1 and COX gene expression in primary neurons [19, 21, 22]. As shown in Figure 5A for N2a cells, depolarizing stimulation resulted in a significant increase (~109%) in the activity of Kif17 promoter as monitored by luciferase assays (P < 0.05). This increase was abolished by mutating either the NRF-1 A or B site, confirming a link between KCl–induced depolarization and the activation of Kif17 via NRF-1 binding.
Fig. 5. Depolarization-induced up-regulation of promoter gene expressions and mRNA levels of Kif17 and NMDAR subunit genes in neurons and the effects of NRF-1 binding site mutation and NRF-1 silencing.

(A) Site-directed mutations of NRF-1 binding sites on Kif17 promoter resulted in a significant reduction in luciferase activity as compared to the wild type (wt). KCl depolarization increased promoter activity in the wild type but not in the mutated Kif17 (NRF-1A mut and NRF-1B mut sites). (Group means were analyzed for overall statistical significance using the Student’s t-test and ANOVA, N = 6 for each construct). *, P < 0.05, as compared to Kif17 wild type. # = P < 0.05 as compared to Kif17 wt with KCl depolarization. (B) Data from real time quantitative PCR indicate that NRF-1, Kif17, Kif1a, Grin1, and Grin2b gene expression in neurons were increased by KCl depolarization as compared to controls. NRF-1 silencing with shRNA prevented the up-regulation of NRF-1, Kif17, Grin1, and Grin2b mRNAs by KCl, whereas Kif1a was not affected. Values represent mean ± S.E.M of combined data from 3 independent experiments. * P < 0.05, ** P < 0.01 versus controls. All # P values were compared to 20 mM KCl-treated samples (# P < 0.05). (C) Western blot reveals increased protein levels for both KIF17 and KIF1A following KCl depolarization as compared to controls. NRF-1 silencing with shRNA prevented the KCl-induced up-regulation of KIF17 protein levels, whereas those of KIF1A were not affected. * P < 0.01 versus controls. β-Actin served as a loading control.
3.7. Effect of NRF-1 silencing on Kif17, Grin1, and Grin2b mRNAs and protein in the presence of KCl stimulation
To ensure that NRF-1 has a functional effect on primary neurons, these neurons were transfected with shRNA against NRF-1 and were then subjected to 20 mM KCl for five hours. As shown in Fig. 5B, depolarizing stimulation without gene silencing resulted in a significant increase in the expressions of NRF-1, Kif17, Kif1a, Grin1, and Grin2b genes, as monitored by real-time quantitative PCR (P < 0.05, P < 0.01). The increase ranged from 180% to 470%, indicating that KCl has a general stimulatory effect on transcription (at least of the genes tested). However, in the presence of NRF-1 silencing, KCl was no longer able to up-regulate mRNA levels of NRF-1, Kif17, Grin1, and Grin2b. On the other hand, NRF-1 silencing did not prevent Kif1a from being up-regulated by KCl, and the levels remained higher than those of controls but not different statistically from those with KCl alone (Fig. 5B).
As shown in Fig. 5C, depolarizing stimulation without gene silencing resulted in a significant increase in the protein level of KIF17 (132%, P < 0.01), whereas that of KIF1A was increased but not significantly different from controls. Transfection of neurons with NRF-1 shRNA vectors in the presence of KCl stimulation resulted in ~ 80% decrease in the level of KIF17 protein as measured by western blots (P < 0.01) (Fig. 5C). However, KIF1A showed no statistically significant alterations in protein levels under the same treatment (Fig. 5C). These results confirmed the specificity of NRF-1 in the regulation of Kif17, Grin1, and Grin2b subunits in response to increased neuronal activity.
3.8. NRF-1 over-expression increased Grin1 and Kif17 mRNA levels and rescued neurons from tetrodotoxin-induced transcript reduction
A low concentration of TTX (0.4 μM) has been shown to decrease the level of COX subunit mRNAs as well as COX enzyme activity in vivo and in primary neurons [21, 23]. To determine if over-expression of NRF-1 could rescue not only COX but also Kif17 transcript, a pSG5NRF-1 construct (gift of Dr. Richard Scarpulla) for NRF-1 over-expression was transfected into primary neurons that were then exposed to TTX (0.4 μM) for 4 days. When neurons were transfected with empty vectors, exposure to TTX led to a 66% reduction in Grin1 and a 73 – 75% reduction in Kif17 and Kif1a mRNA levels (Figs. 6A–C), indicating an overall suppressive effect of TTX on gene expression in neurons. Neurons transfected with the pSG5NRF-1 construct had a 338% increase in mRNA levels of Grin1 (P < 0.05) (Fig. 6A) and a 219% increase in Kif17 mRNA levels (P < 0.05) (Fig. 6B) as compared to empty vector controls, but no statistically significant change in Kif1a level was evident (Fig. 6C). When exposed to TTX, neurons transfected with pSG5NRF-1 expressed 145 – 157% more Grin1 and Kif17 transcripts as compared to those with empty vectors (P < 0.05) (Fig. 6A–B). No statistically significant increase was found for Kif1a (Fig. 6C). These results confirmed that NRF-1 could rescue Grin1 and Kif17 mRNAs but not that of Kif1a in the presence of TTX.
Fig. 6. NRF-1 over-expression in primary neurons significantly increased mRNA levels for Kif17 and Grin1 genes and rescued them from TTX-induced suppression. Grin1, Kif17, and Kif1a mRNA levels.
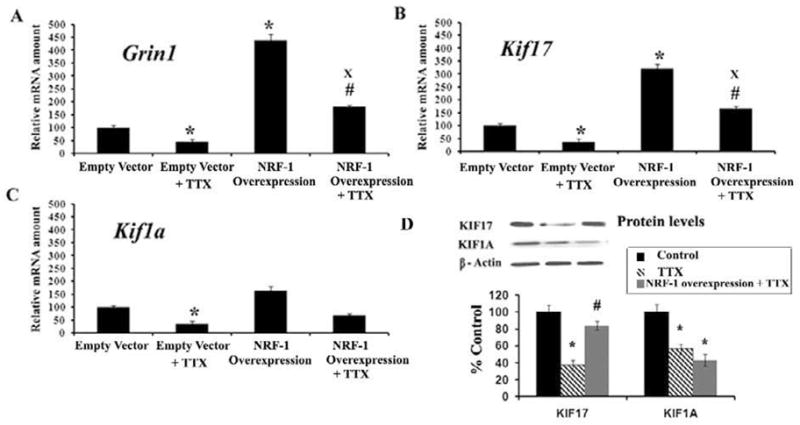
(A–C) were all reduced by TTX as compared to controls. Over-expression of NRF-1 significantly increased transcript levels of Grin1 and Kif17, but not of Kif1a. Over-expression of NRF-1 was able to rescue Grin1 and Kif17, but not Kif1a from TTX-induced down-regulation. Group means were analyzed for overall statistical significance using the Student’s t-test (N = 6 for each group). All * P values were compared to empty vectors (* P < 0.05). All # P values were compared to empty vector plus TTX (# P < 0.05), and all X P values were compared to NRF-1 over-expression (X P < 0.05). (D) Protein levels for KIF17 and KIF1A were all reduced with TTX impulse blockade as compared to controls in neurons. However, NRF-1 over-expression rescued protein levels of KIF17, but not of KIF1A, from being down-regulated by TTX. All * P values were compared to controls (* P < 0.01). All #P values were compared to TTX (#P < 0.01).
Neurons exposed to TTX had ~ 40 – 70% decrease in levels of KIF17 and KIF1A proteins as compared to controls in western blots (P < 0.01) (Fig. 6D). Neurons transfected with NRF-1 and exposed to TTX expressed 50% more KIF17 protein as compared to TTX alone (P < 0.01). However, NRF-1 over-expression could not rescue KIF1A protein levels after TTX exposure, confirming the specificity of NRF-1 on KIF17 (Fig. 6D).
4. Discussion
The present study documents for the first time that KIF17 is regulated by the same transcription factor, NRF-1, as its cargo NR2B of NMDA receptors, thereby linking transport motor and synaptic cargo associated with glutamatergic neurotransmission at the transcriptional level of regulation in neurons. The high homology of NRF-1 binding sites among mice, rats, and humans for Kif17 (Table 1), Grin2b, and Grin1 [12] genes underscores the conservation of such co-regulation through evolution. The findings are specific in that NRF-1 (but not NRF-2) regulates Kif17 (but not Kif1a) (the present study) and Grin2b (but not Grin2a) [12].
The kinesin superfamily of motor proteins are encoded by 45 genes, classified into 15 families (numbered kinesin 1 to 14B), and grouped into 3 types (N-, M-, and C-) depending on the location of the motor domain in the protein [2]. KIF17 (also known as OSM3) is a member of the N4 kinesin-2 family, with its motor domain in its N-terminus, and it is neuron-specific [6, 24]. Unlike its family member KIF3, which transports synaptic vesicles down the axons, KIF17 moves vesicles containing the NR2B and NR1 subunits of the NMDA receptors along microtubules from the cell body strictly to dendrites in a plus-end-directed, ATP-dependent manner. The head domain of KIF17 interacts with microtubules, while its tail domain interacts with the PDZ domain of the scaffolding protein MINT1 (LIN10), which forms a complex with LIN2 and LIN7 to support the NR2B-containing cargo vesicles [2, 6]. Over-expression of KIF17 in transgenic mice increased both the mRNA and protein levels of NR2B and enhanced spatial and working memories of mice [7]. Likewise, an up-regulation of NR2B in hippocampal neurons with the NMDA receptor antagonist APV led to an increased level of KIF17 [11]. On the other hand, a knockdown of KIF17 expression with antisense oligonucleotides reduced NR2B expression and its synaptic localization [11]. These parallel changes strongly suggest co-regulation of KIF17 and NR2B [11], and the present study provides solid evidence for such a co-regulation at the transcriptional level. NRF-1 regulates the expression of not only Grin2b (NR2B) and the obligatory Grin1 (NR1) subunits of the NMDA receptors [12], but also their transporter motor Kif17 in neurons (the present study). Significantly, the NR2A subunit of the NMDA receptors responds to perturbations of KIF17 in a direction opposite to that of NR2B [11], and it is neither transported by KIF17 [11] nor regulated by NRF-1 [12].
The NMDA receptors are important for glutamatergic synaptic transmission, synaptic plasticity, circuit development, learning, and memory [8–10]. Functional NMDA receptors contain heteromeric combinations of NR1 subunit and one or more of NR2A-D subunits [25]. In isolation, NR1 (Grin1) gene expresses a functional receptor with a weak response activated by glutamate and glycine, whereas none of the NR2 (Grin2) subunits is functional when expressed alone [26]. In the adult cortex, 40% of NMDA receptors are composed of NR1/NR2B subunits, 37% are made of NR1/NR2A, and only 6% are a combination of all threes subunits [27, 28]. However, the NR2B subunit appears to be critical for a number of basic structural and functional attributes associated with the NMDA receptor. This subunit predominates in the forebrain and hippocampus, and is essential for the synaptic localization of the NMDAR channel [29, 30]. Moreover, the cytoplasmic tail of NR2B mediates protein interaction independent of its channel participation, and it is necessary for long-term potentiation even when its role in channel formation is decreasing with development, whereas NR2A is not necessary for LTP [31]. The importance of NR2B in learning and memory is likely to be the basis for its continual transportation into the dendrites via KIF17 motor through postnatal development and into adulthood [6], as well as for their co-regulation by the same transcription factor NRF-1 (the present study).
NRF-1 is known to be a key transcriptional activator of nuclear genes encoding a number of mitochondrial respiratory enzymes, including subunits of the five respiratory chain complexes [32–34]. Recently, we found that NRF-1 regulates all 13 subunits of cytochrome c oxidase derived from the nuclear and mitochondrial genomes [13]. Thus, NRF-1 plays a significant role in coordinating the two genomes in critically regulating the expression of an enzyme vital for energy generation. Energy in neurons is used primarily to repolarize membrane potentials subsequent to glutamate-induced depolarization, and neuronal activity and energy metabolism are tightly coupled processes [14]. Under conditions of altered neuronal activity, such as afferent impulse blockade by TTX or depolarizing treatment with KCl, neuronal COX activity is adjusted to match the new energy demand [14, 23], and the expressions of glutamate and NMDA receptors are adjusted in parallel [12, 23, 35]. Likewise, NRF-1 protein and mRNA levels are up-regulated by depolarizing neuronal activity [19] and down-regulated by TTX-induced impulse blockade in vitro or afferent removal in vivo [22]. These changes preceded those of NRF-1′s target genes, such as cytochraome c oxidase [19, 22]. Thus, NRF-1 itself is directly regulated by neuronal activity. Remarkably, NRF-1 also directly regulates the expression of critical components of glutamatergic synapses, including NR1 and NR2B of the NMDA receptors, subunit GluR2 of the AMPA receptors, and neuronal nitric oxide synthase [12, 36, 37]. Thus, the tight coupling of neuronal activity and energy metabolism is extended to the molecular level of regulation. The present study adds a third dimension to the coupling by documenting that NRF-1 also regulates the expression of the NR2B transporter protein KIF17 that is highly ATP-dependent. A knockdown of NRF-1 expression with shRNA reduced both message and protein levels of Kif17 (KIF17), Grin1 (NR1), and Grin2b (NR2B) and prevented them from being up-regulated by KCl depolarization, whereas an over-expression of NRF-1 up-regulates the transcripts of all three genes and rescued them from being suppressed by TTX [12; the present study]. On the other hand, perturbations of NRF-1 do not affect the expression of Kif1a, whose protein is not known to be involved in NMDA receptor transportation. Of significance is the fact that dendrites, to which KIF17 exclusively targets and where NR1 and NR2B subunits of NMDA receptors are enriched, are also the major energy consumers of the brain [14]. Dendrites represent the major receptive sites for excitatory synapses, and their membranes have to be constantly repolarized after depolarization, a highly energy-demanding process fueled by ATP generated from oxidative metabolism. For this reason, the level of COX is also the highest in dendrites [14]. Thus, the tightly coupled regulatory mechanism via NRF-1 is exquisitely coordinated, efficient, and precise.
5. Conclusions
In conclusion, the present study documents that the same transcriptional regulator NRF-1 coordinately regulates the expressions of Kif17, Grin1 and Grin2b subunit genes of NMDA receptors, as well as previously reported COX subunit genes in neurons. This ensures that the production and transportation of critical receptors of glutamatergic neurotransmission is tightly coupled to energy generation via the oxidative pathway (Fig. 7). No doubt, NRF-1 may not act alone. CREB, Sp1, and AP1 binding sites have been reported on the Kif17 promoter that may play additional regulatory roles in the transcription of the Kif17 gene, but they have not been functionally characterized in neurons [7]. Future studies can be directed at elucidating these issues.
Fig. 7. Schematic representation of transcriptional co-regulation by a common transcription factor, NRF-1.
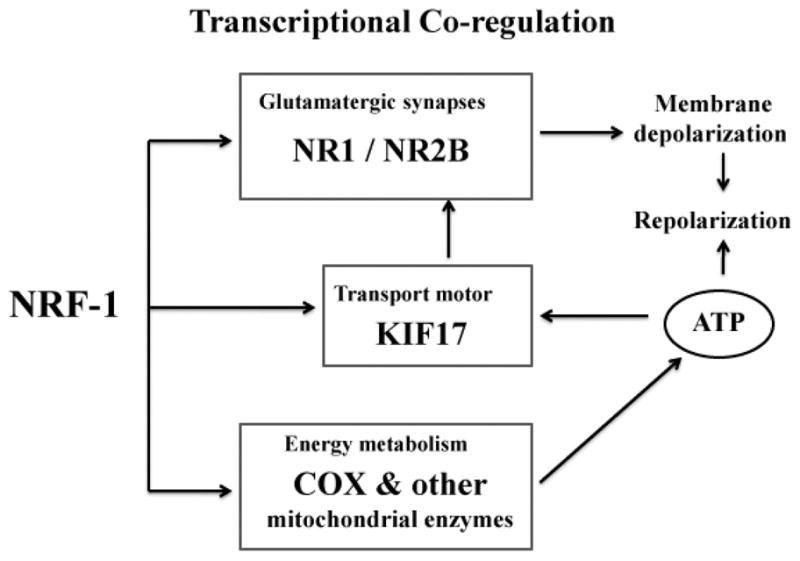
Genes for KIF17, NMDA receptor subunits NR1 and NR2B, as well as COX subunits are all regulated by NRF-1. Thus, NRF-1 coordinates the tight coupling between glutamatergic synaptic transmission and energy metabolism at the molecular level in neurons.
Acknowledgments
It gives us great pleasure to thank Dr. Richard Scarpulla for his generous gift of NRF-1 antibodies and pSG5NRF-1 plasmid, and Dr. P. Aebischer for his gift of PLVTHM. We thank Drs. H. Liang and H. Meng for assisting in the construction of shRNA vectors. Supported by NIH Grant R01 EY018441.
Abbreviations
- ChIP
chromatin immunoprecipitation
- CMV
cytomegalovirus
- COX
cytochrome c oxidase
- EMSA
electrophoretic mobility shift assay
- LTP
long term potentiation
- NGFR
nerve growth factor receptor
- NRF-1
nuclear respiratory factor-1
- NRF-2
nuclear respiratory factor 2
- shRNA
short hairpin RNA
- Sp1
specificity protein 1
- TFB2M
transcription factor B2 of mitochondria
- TSP
transcription start point
- TTX
tetrodotoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- 2.Hirokawa N, Nitta R, Okada Y. The mechanisms of kinesin motor motility: lessons from the monomeric motor KIF1A. Nat Rev Mol Cell Biol. 2009;10:877–884. doi: 10.1038/nrm2807. [DOI] [PubMed] [Google Scholar]
- 3.Hirokawa N, Takemura R. Kinesin superfamily proteins and their various functions and dynamics. Exp Cell Res. 2004;301:50–59. doi: 10.1016/j.yexcr.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 4.Insinna C, Humby M, Sedmak T, Wolfrum U, Besharse JC. Different roles for KIF17 and kinesin II in photoreceptor development and maintenance. Dev Dyn. 2009;238:2211–2222. doi: 10.1002/dvdy.21956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silverman MA, Leroux MR. Intraflagellar transport and the generation of dynamic, structurally and functionally diverse cilia. Trends Cell Biol. 2009;19:306–316. doi: 10.1016/j.tcb.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Setou M, Nakagawa T, Seog DH, Hirokawa N. Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science. 2000;288:1796–1802. doi: 10.1126/science.288.5472.1796. [DOI] [PubMed] [Google Scholar]
- 7.Wong RW, Setou M, Teng J, Takei Y, Hirokawa N. Overexpression of motor protein KIF17 enhances spatial and working memory in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:14500–14505. doi: 10.1073/pnas.222371099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 9.Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H, Mishina M. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature. 1995;373:151–155. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- 10.Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- 11.Guillaud L, Setou M, Hirokawa N. KIF17 dynamics and regulation of NR2B trafficking in hippocampal neurons. J Neurosci. 2003;23:131–140. doi: 10.1523/JNEUROSCI.23-01-00131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhar SS, Wong-Riley MTT. Coupling of energy metabolism and synaptic transmission at the transcriptional level: role of nuclear respiratory factor 1 in regulating both cytochrome c oxidase and NMDA glutamate receptor subunit genes. J Neurosci. 2009;29:483–492. doi: 10.1523/JNEUROSCI.3704-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dhar SS, Ongwijitwat S, Wong-Riley MTT. Nuclear respiratory factor 1 regulates all ten nuclear-encoded subunits of cytochrome c oxidase in neurons. J Biol Chem. 2008;283:3120–3129. doi: 10.1074/jbc.M707587200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong-Riley MTT. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]
- 15.Ongwijitwat S, Wong-Riley MTT. Is nuclear respiratory factor 2 a master transcriptional coordinator for all ten nuclear-encoded cytochrome c oxidase subunits in neurons? Gene. 2005;360:65–77. doi: 10.1016/j.gene.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 16.Ongwijitwat S, Liang HL, Graboyes EM, Wong-Riley MTT. Nuclear respiratory factor 2 senses changing cellular energy demands and its silencing down-regulates cytochrome oxidase and other target gene mRNAs. Gene. 2006;374:39–49. doi: 10.1016/j.gene.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 17.Evans MJ, Scarpulla RC. NRF-1: a trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev. 1990;4:1023–1034. doi: 10.1101/gad.4.6.1023. [DOI] [PubMed] [Google Scholar]
- 18.Ongwijitwat S, Wong-Riley MTT. Functional analysis of the rat cytochrome c oxidase subunit 6A1 promoter in primary neurons. Gene. 2004;337:163–171. doi: 10.1016/j.gene.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 19.Yang SJ, Liang HL, Wong-Riley MTT. Ultrastructural study of depolarization-induced translocation of NRF-2 transcription factor in cultured rat visual cortical neurons. Neurosci. 2006;141:1181–1192. doi: 10.1111/j.1460-9568.2004.03250.x. [DOI] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.Liang HL, Ongwijitwat S, Wong-Riley MTT. Bigenomic functional regulation of all 13 cytochrome c oxidase subunit transcripts in rat neurons in vitro and in vivo. Neurosci. 2006;140:177–190. doi: 10.1016/j.neuroscience.2006.01.056. [DOI] [PubMed] [Google Scholar]
- 22.Liang HL, Wong-Riley MTT. Activity-dependent regulation of nuclear respiratory factor-1, nuclear respiratory factor-2, and peroxisome proliferator-activated receptor gamma coactivator-1 in neurons. NeuroReport. 2006;17:401–405. doi: 10.1097/01.wnr.0000204980.98876.11. [DOI] [PubMed] [Google Scholar]
- 23.Wong-Riley MT, Huang Z, Liebl W, Nie F, Xu H, Zhang C. Neurochemical organization of the macaque retina: effect of TTX on levels and gene expression of cytochrome oxidase and nitric oxide synthase and on the immunoreactivity of Na+ K+ ATPase and NMDA receptor subunit I. Vision Res. 1998;38:1455–1477. doi: 10.1016/s0042-6989(98)00001-7. [DOI] [PubMed] [Google Scholar]
- 24.Miki H, Setou M, Kaneshiro K, Hirokawa N. All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci U S A. 2001;98:7004–7011. doi: 10.1073/pnas.111145398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- 26.Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S. Molecular cloning and characterization of the rat NMDA receptor. Nature. 1991;354:31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- 27.Chazot PL, Stephenson FA. Biochemical evidence for the existence of a pool of unassembled C2 exon-containing NR1 subunits of the mammalian forebrain NMDA receptor. J Neurochem. 1997;68:507–516. doi: 10.1046/j.1471-4159.1997.68020507.x. [DOI] [PubMed] [Google Scholar]
- 28.Luo J, Wang Y, Yasuda RP, Dunah AW, Wolfe BB. The majority of N-methyl-D-aspartate receptor complexes in adult rat cerebral cortex contain at least three different subunits (NR1/NR2A/NR2B) Mol Pharmacol. 1997;51:79–86. doi: 10.1124/mol.51.1.79. [DOI] [PubMed] [Google Scholar]
- 29.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 30.Mori H, Manabe T, Watanabe M, Satoh Y, Suzuki N, Toki S, Nakamura K, Yagi T, Kushiya E, Takahashi T, Inoue Y, Sakimura K, Mishina M. Role of the carboxy-terminal region of the GluR epsilon2 subunit in synaptic localization of the NMDA receptor channel. Neuron. 1998;21:571–580. doi: 10.1016/s0896-6273(00)80567-x. [DOI] [PubMed] [Google Scholar]
- 31.Foster KA, McLaughlin N, Edbauer D, Phillips M, Bolton A, Constantine-Paton M, Sheng M. Distinct roles of NR2A and NR2B cytoplasmic tails in long-term potentiation. J Neurosci. 2010;30:2676–2685. doi: 10.1523/JNEUROSCI.4022-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scarpulla RC. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene. 2002;286:81–89. doi: 10.1016/s0378-1119(01)00809-5. [DOI] [PubMed] [Google Scholar]
- 33.Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006;97:673–683. doi: 10.1002/jcb.20743. [DOI] [PubMed] [Google Scholar]
- 34.Scarpulla RC. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann N Y Acad Sci. 2008;1147:321–334. doi: 10.1196/annals.1427.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong-Riley M, Anderson B, Liebl W, Huang Z. Neurochemical organization of the macaque striate cortex: correlation of cytochrome oxidase with Na+K+ATPase, NADPH-diaphorase, nitric oxide synthase, and N-methyl-D-aspartate receptor subunit 1. Neurosci. 1998;83:1025–1045. doi: 10.1016/s0306-4522(97)00432-6. [DOI] [PubMed] [Google Scholar]
- 36.Dhar SS, Liang HL, Wong-Riley MTT. Nuclear respiratory factor 1 co-regulates AMPA glutamate receptor subunit 2 and cytochrome c oxidase: tight coupling of glutamatergic transmission and energy metabolism in neurons. J Neurochem. 2009;108:1595–1606. doi: 10.1111/j.1471-4159.2009.05929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dhar SS, Liang HL, Wong-Riley MTT. Transcriptional coupling of synaptic transmission and energy metabolism: role of nuclear respiratory factor 1 in co-regulating neuronal nitric oxide synthase and cytochrome c oxidase genes in neurons. Biochim Biophys Acta. 2009;1793:1604–1613. doi: 10.1016/j.bbamcr.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]