

Abstract
Chronic allograft rejection (CR) is the leading cause of late graft failure following organ transplantation. CR is a progressive disease, characterized by deteriorating graft function, interstitial fibrosis, cardiac hypertrophy and occlusive neointima development. TGFβ, known for its immunosuppressive qualities, plays a beneficial role in the transplant setting by maintaining alloreactive T cells in a hyporesponsive state, but has also been implicated in promoting graft fibrosis and CR. In the mouse vascularized cardiac allograft model, transient depletion of CD4+ cells promotes graft survival but leads to CR, which is associated with intragraft TGFβ expression. Decorin, an extracellular matrix protein, inhibits both TGFβ bioactivity and gene expression. In this study, gene transfer of decorin into cardiac allografts was employed to assess the impact of intragraft TGFβ neutralization on CR, systemic donor-reactive T cell responses, and allograft acceptance. Decorin gene transfer and neutralization of TGFβ in cardiac allografts significantly attenuated interstitial fibrosis, cardiac hypertrophy and improved graft function, but did not result in systemic donor-reactive T cell responses. Thus, donor-reactive T and B cells remained in a hyporesponsive state. These findings indicate that neutralizing intragraft TGFβ inhibits the cytokine's fibrotic activities, but does not reverse its beneficial systemic immunosuppressive qualities.
Keywords: Gene therapy, suppression, T cells, Th1/Th2, transplantation
Introduction
The accepted treatment for end-stage heart failure failure is transplantation (1). Immunosuppressive therapies diminish the incidence of graft loss due to acute graft rejection, leaving chronic rejection (CR)3 as the main impediment to long-term transplant survival (2). CR in cardiac allografts is characterized by interstitial fibrosis, vascular occlusion, cardiac hypertrophy and progressive dysfunction of the graft (3-6). The cellular mechanisms and cascade of events that lead to CR remain poorly defined and no effective therapies exist except retransplantation.
Transforming growth factor β (TGFβ) is a widely expressed cytokine that exerts pleiotropic effects on cell proliferation, migration, differentiation and survival (reviewed in (7)). TGFβ contributes to multiple biological processes, including tumorigenesis, development, wound healing, fibrosis and suppression of immune responses (7). The importance of TGFβ as an immune regulator was demonstrated in TGFβ deficient mice, which manifest a severe autoimmune phenotype that results in death at 3-4 weeks of age (8, 9). TGFβ controls T cell proliferation and survival and acts to inhibit Th1/Th2 differentiation and effector function (7, 10). B cells respond to TGFβ with decreased proliferation, survival and activation (11). TGFβ is also a critical cytokine in T regulatory cell (Treg) development and function (10, 12, 13). The Treg lineage-specific transcription factor, FoxP3, is induced by TGFβ, and results in the conversion of CD4+CD25- T cells into Treg (14). Furthermore, TGFβ signaling in Treg is essential for peripheral maintenance of this cell subset (7, 15).
TGFβ also contributes to wound healing and tissue repair (16). During normal wound healing, transient upregulation of TGFβ stimulates the production of factors that act in concert to increase extracellular matrix (ECM) deposition, decrease matrix degradation, and restore normal tissue composition (17). While TGFβ is critical in wound healing and tissue repair, enhanced and prolonged TGFβ production is detrimental and observed in a number of fibrotic diseases, including pulmonary fibrosis (18), glomerulonephrtitis (19), scleroderma (20), and CR (21, 22).
Decorin, an ECM protein and member of the small leucine rich proteoglycan family, plays a role in TGFβ regulation (23). The core protein of decorin binds the active form of TGFβ, thereby inhibiting TGFβ's interaction with its receptor and sequestering the cytokine to the ECM (24, 25). In addition to inhibiting TGFβ's bioactivity, decorin negatively impacts TGFβ gene expression (26, 27). Decorin gene transfer ameliorated TGFβ-mediated fibrosis in a glomerulonephritis model (26) and in a pulmonary fibrosis model (28).
TGFβ mediates many beneficial anti-inflammatory effects in the transplant setting (29, 30) and we have previously reported an association with TGFβ and CR using the mouse vascularized cardiac allograft model (21). Intragraft TGFβ levels are readily detectable in the CR grafts from recipients transiently depleted of CD4+ T cells, but not in the grafts of anti-CD40L treated recipients, which remain free of CR. Adenoviral-mediated gene transfer of the active form of TGFβ into allografts induces fibrosis and results in CR in recipients treated with anti-CD40L that do not normally exhibit CR (21). This supports a critical role for TGFβ in the progression of CR.
In recipients transiently depleted of CD4+ T cells, CD4+ T cells begin to repopulate the periphery 3-4 weeks post-transplantation (31-33). Donor-reactive T cells in these animals are functionally distinct from naïve cells in that repopulating CD4+ are hyporesponsive toward the graft but mount Th2 recall responses (34). The importance of TGFβ in allograft acceptance and suppression of graft-reactive T and B cells was revealed in transplant experiments employing mice with a dominant negative TGFβRII transgene (CD4-DNR) (Faust et al., submitted), which render the animals unresponsive to T cell TGFβ signaling (35). Transient CD4+ T cell depletion of CD4-DNR recipients resulted in both alloreactive cellular and humoral responses, which remained hyporesponsive in wild type (WT) recipients indicating that TGFβ is critical to suppression of T and B cell responses in this system. Graft rejection in these recipients correlated with CD4+ T cell repopulation of the periphery (Faust et al., submitted). These studies revealed that inductive anti-CD4 mAb treatment is a TGFβ dependent model of allograft acceptance and that IL-17 is a critical element in TGFβ driven fibrosis (Faust et al., submitted).
In this study, we employed decorin gene transfer into cardiac allografts to assess the impact of intragraft TGFβ neutralization on CR, graft function, donor-reactive T and B cell responses, and allograft acceptance. We demonstrate that neutralizing intragraft TGFβ inhibits the cytokine's fibrotic activities, but does not reverse its beneficial systemic immunosuppressive qualities.
Materials and Methods
Mice
C57BL/6 WT (H-2b) and BALB/c (H-2d) mice were purchased from the Jackson Laboratory. All mice were housed under specific pathogen-free conditions in the Unit for Laboratory Animal Medicine at the University of Michigan. These experiments were approved by the University Committee on Use and Care of Animals at the University of Michigan.
Culture medium
Culture medium consisted of the following: RPMI 1640 supplemented with 2% FCS, 1 mM sodium pyruvate, 100 U/mL penicillin, 100 μg/mL streptomyin, 1.6 mM L-glutamine, 10 mM HEPES buffer (all from Invitrogen), 0.27 mM L-asparagine, 1.4 mM L-arginine HCl, 14 μM folic acid, and 50 μM 2-ME (all from Sigma-Aldrich).
Vascularized cardiac transplantation
C57BL/6 mice were transplanted with intact BALB/c cardiac allografts, as described (36). Briefly, the aorta and pulmonary artery of the donor heart were anastomosed end-to-side to the recipients abdominal aorta and inferior vena cava, respectively. Upon perfusion with the recipient's blood, the transplanted heart resumes contraction. Graft function is monitored by abdominal palpation.
Adenoviral-mediated transduction of cardiac allografts
As described (37, 38), cardiac allografts were transduced by perfusion via the aorta with E1/E3 deleted adenoviral vectors (5 × 108 pfu) encoding rat decorin (AdDec) or beta-galactosidase (Adβgal). AdDec was constructed in Dr. Elizabeth Nabel's lab using a cDNA kindly provided by Dr. Wayne Border (26). Following perfusion, grafts were recovered and placed in iced Ringer's for approximately 1 h prior to transplantation. Reporter gene studies with Adβgal have revealed that the distribution of transgene expression within the cardiac allograft is patchy, and that both cardiac myocytes and cells of the vasculature express the transgene product (37).
In vivo mAb treatment
The hybridoma secreting anti-CD4 mAb (clone GK1.5) was obtained from American Type Culture Collection. Anti-CD4 was purified and resusupended in PBS by Bio X Cell (West Lebanon, NH). Mice received 1 mg i.p. of anti-CD4 mAb on days -1, 0, 7 (21, 31, 32). All doses are relative to day of transplant.
Histology
Allografts were recovered at the times indicated post-transplantation, fixed in formalin, and embedded in paraffin. Sections were stained with hematoxylin-eosin (H&E) to assess myocyte viability (presence of cross striation and myocyte nuclei), and the nature and intensity of graft infiltrating cells.
Morphometric analysis of cardiac allograft fibrosis and hypertrophy
Graft fibrosis was quantified by morphometric analysis of Masson's trichrome stained tissues using iPLab software (Scanalytics Inc., Fairfax, VA) (3). Masson's trichrome stains fibrotic tissue blue. Mean fibrotic areas were calculated from 10 to 12 areas per heart section analyzed at 200× magnification. Nine individual hearts were analyzed per group. To quantify cardiomyocyte area as a measure of hypertrophy, digital outlines were drawn around 100 cardiomyocytes from views of H&E stained grafts at 200× magnification. Areas within outlines were quantified using SCION IMAGE Beta 4.0.2 software (Scion Corporation, Frederick, MD) to measure cardiomyocyte cell size (3). Five individual hearts were analyzed per group.
ELISPOT assays for cytokine-producing cells
ELISPOT assays were performed as previously described (39). Capture and detection mAbs specific for IFNγ (R4-6A2, XMG1.2), IL-4 (11B11, BVD6-24G2) and IL-17 (TC11-18H10.1, TC11-8H4.1) were purchased from Pharmingen (San Diego, CA). PVDF-backed microtiter plates (Millipore, Bedford, MA) were coated with unlabeled mAb and blocked with 1% BSA in PBS. Irradiated (1000 rad) donor splenocytes (4×105) and 1×106 recipient splenocytes were added to the plates. After washing, a 1:1000 dilution of anti-biotin alkaline phosphatase (AP) conjugate (Vector Laboratories, Burlingame, CA) was added to IFNγ and IL-17 plates, and a 1:2000 dilution of horseradish peroxidase-conjugated streptavidin (SA-HRP; Dako, Carpinteria, CA) was added to IL-4 plates. Plates were washed and spots visualized by addition of nitroblue tetrazolium (NBT; Biorad, Hercules, CA)/3-bromo-4-chloro-inolyl-phosphate (BCIP; Sigma) to IFNγ and IL-17 plates, or 3-amino-9-ethylcarbazole (AEC; Pierce, Rockford, IL) to IL-4 plates. Color development continued until spots were visible and stopped by adding H2O. Plates were dried and spots quantified with an Immunospot Series 1 ELISPOT analyzer (Cellular Technology Ltd., Cleveland, OH).
RNA isolation and Quantitative RT-PCR
Cardiac allografts were homogenized in 1 mL TRIzol® (Invitrogen Life Technologies, Carlsbad, CA) and RNA was isolated as per manufacturers protocol. 5 μg of total RNA were reverse transcribed using 10× PCR buffer (Roche), 10 mM dNTPs, Oligo (dt), M-MLV-RT (all from Invitrogen), and RNAsin (Promega). Products were then cleaned with 1:1 phenol/chloroform/isoamyl (25:24:1) and re-precipitated with 7.5 M NH4OAC in pure EtOH overnight at -80°C.
Quantitative RT-PCR was performed on cDNA using a Rotor-Gene 3000 TM (Corbett Life Science, CA). Primer binding to DNA was detected by SYBR Green ITM dye (Roche, Indianapolis, IN). Relative expression of the gene of interest was calculated by accompanying Rotor-Gene software as the concentration of the gene product compared to GAPDH. Significance was determined with an unpaired Student t-test.
Primer sequences:
Rat Decorin sense: 5′ AGCATAAATATGTCCAGGTCGTC
Rat Decorin anti-sense: 5′GAAGTCTTCCTAGTCTGGTATGAAGG
TGFβ sense: 5′CCTGAGTGGCTGTCTTTTGAC
TGFβ anti-sense: 5′CCTGTATTCCGTCTCCTTGGT
Collagen (pro-collagen 1a) sense: 5′ TCCCTACTCAGCCGTCTGTGCC
Collagen anti-sense: 5′ AGCCCTCGCTTCCGTACTCG
IL-17 sense: 5′ GGACTCTCCACCGCAATGA
IL-17 anti-sense: 5′GACCAGGATCTCTTGCTGGA
FoxP3 sense: 5′CCAAGGTGAGCGAGTGTC
FoxP3 anti-sense: 5′AAGGCAGAGTCAGGAGAAGT
GAPDH sense: 5′CTGGTGCTGAGTATGTCGTG
GAPDH anti-sense: 5′CAGTCTTCTGAGTGGCAGTG
Donor-reactive Ab determination
P815 cells (H-2d) were stained for flow cytometric analysis using diluted (1:50) sera obtained from mice as the primary antibody, followed by FITC-conjugated isotype specific anti-mouse IgG and IgM secondary antibodies (The Binding Site, San Diego, CA, USA) used at a 1:50 dilution (33). Data are reported as the mean channel fluorescence determined on a Becton Dickinson FACSCaliber (San Jose, CA, USA).
Statistical analysis
Data were analyzed with GraphPad Prism 4.0c software using unpaired Student t-tests. p values of ≤ 0.05 were considered statistically significant.
Results
Rationale
Prolonged allograft survival can be accomplished in the mouse cardiac allograft model by depleting CD4+ T cells transiently at the time of transplant. However, allografts in anti-CD4 mAb treated recipients develop interstitial fibrosis and CR, which is associated with intragraft TGFβ expression (21). TGFβ is beneficial in the transplant setting, and has been associated with the progression of donor-reactive T and B cells to a hyporesponsive state in recipients treated inductively with anti-CD4 mAb ((34), Faust et al., submitted). Conversely, T cell responsiveness to TGFβ can be deleterious for the graft by inducing fibrosis (Faust et al., submitted). Therefore, we explored the impact of local TGFβ neutralization employing decorin gene transfer into the allografts. We assessed the impact of intragraft TGFβ neutralization on allograft acceptance, graft function, T and B cell hyporesponsiveness and CR.
Decorin gene transfer into cardiac allografts
Prior studies utilizing adenoviral transduction of allografts revealed long-term transgene expression and negligible off-target tissue effects (21, 37). To evaluate efficacy of decorin gene transfer, allografts were transduced with adenoviral vectors that encode rat decorin (AdDec) or βgal (Adβgal) and transplanted into recipients treated with inductive anti-CD4 mAb. Functioning allografts were harvested on day 7, day 14 and day 50 post-transplant (Figure 1). Employing rat decorin specific primers and quantitative RT-PCR, we verified that over-expression of rat decorin was detected predominantly within the cardiac allografts at day 7 and day 14 and not within non-target tissue, such as the spleen (Figure 1A). In addition, long-term rat decorin gene expression was detected in the AdDec infected allografts and not in the βgal controls at day 50 post-transplant (Figure 1B). These results demonstrate the efficacy and tissue localization of decorin gene transfer into allografts using adenoviral vectors, and the persistence of transgene expression.
Figure 1. Decorin gene transfer and gene expression.
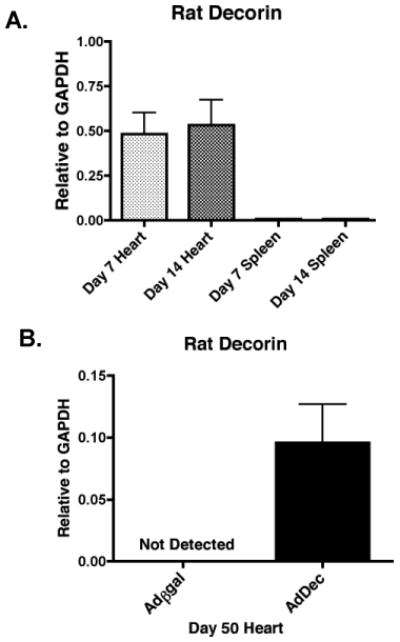
BALB/c allografts were transduced with AdDec or Adβgal and transplanted into C57BL/6 mice that were given inductive anti-CD4 therapy on days -1, 0, and 7 relative to transplant. Spleens and allografts were recovered on day 7 (light shaded bars) and day 14 (dark shaded bars) for AdDec transduced allografts (A). Functioning allografts were recovered on day 50 post-transplant for Adβgal (open bars) and AdDec (shaded bars) transduced allografts (B). Rat decorin expression relative to GAPDH was assessed by real-time RT-PCR. Bars represent the mean of RNA expression from a minimum of 4 Adβgal and 4 AdDec transduced allografts (+/- S.E.M.).
Intragraft TGFβ neutralization by decorin gene transfer does not reverse T and B cell hyporesponsiveness in recipients treated with anti-CD4 mAb
While TGFβ is a known pro-fibrotic cytokine (40) it also has beneficial anti-inflammatory effects in the transplant setting and is frequently observed within accepted grafts (29, 30, 41, 42). Since TGFβ is critical in controlling donor-reactive responses following transient CD4+ T cell depletion (Faust et al., submitted), it was possible that intragraft neutralization of TGFβ might reverse immune hyporesponsiveness if decorin acted beyond the local confines of the allograft and inhibited TGFβ systemically (26). To determine if localized TGFβ neutralization within the allografts affected systemic donor-reactive immune responses, ELISPOT was employed to quantify the number of in vivo primed donor-reactive Th1 (IFNγ), Th2 (IL-4) and Th17 (IL-17) responses (Figure 2A). Gene transfer of βgal or decorin to allografts resulted in negligible T cell responses compared to untreated transplant recipients, indicating that TGFβ neutralization within cardiac allografts did not reverse graft-reactive T cell hyporesponsiveness in recipients depleted of CD4+ T cells (Figure 2A). In addition, cardiac allograft contractions were noticeably stronger in AdDec transduced allografts when compared to βgal controls. These data demonstrate that intragraft inhibition of TGFβ had a beneficial effect on graft function and did not reverse systemic donor-reactive T cell hyporesponsiveness normally observed in recipients transiently depleted of CD4+ T cells.
Figure 2. Intragraft TGFβ neutralization does not reverse donor-reactive T and B cell hyporesponsiveness.
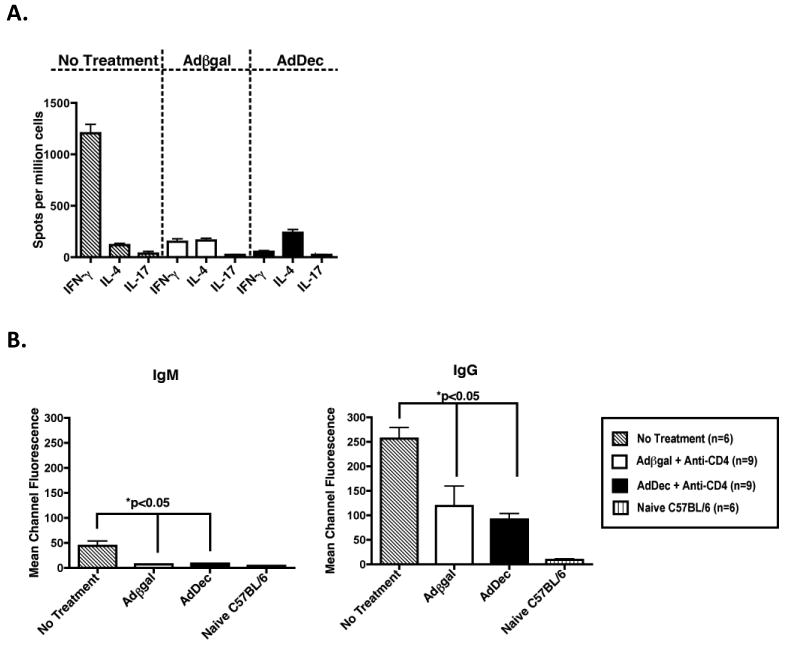
(A) On day 50 post-transplantation, splenocytes from Adβgal or AdDec transduced recipients were processed for ELISPOT assays to quantify primed, donor-reactive IFNγ, IL-4 or IL-17-producing cells. C57BL/6 recipients of BALB/c allografts that received no treatment (striped bars) served as positive controls and splenocytes were harvested at the time of rejection. Bars represent the average number of cytokine producing cells (+/- S.E.M.). Numbers in parentheses represent the number of recipients in each group. (B) Fifty days post-transplant, sera were obtained from recipients transduced with Adβgal (open bars) or AdDec (shaded bars) and treated with inductive anti-CD4 mAb. P815 (H-2d) cells were incubated with of sera and bound donor-reactive Ab was detected by incubation with FITC-tagged anti-IgM or anti-IgG Abs. The mean channel fluorescence is indicative of the relative amount of donor-reactive Ab. Bars represent the average mean channel fluorescence of 9 Adβgal or 9 AdDec transduced recipient samples (+/- S.E.M.).
TGFβ also inhibits B cell responses by affecting B cell proliferation, survival signals, activation, and IgG class switching (7). In CD4-DNR recipients treated with anti-CD4 mAb, T cells differentiate into effector cells and provide help to B cells, which produce donor-reactive IgG (Faust et al., submitted). To examine the effect of intragraft decorin over-expression on donor-reactive antibody, we quantified donor-reactive IgM and IgG production by flow cytometry (Figure 2B). No difference in alloantibody production was observed in recipients whose allografts expressed decorin or βgal. This demonstrates that B cell hyporesponsiveness is also not reversed in recipients that overexpress decorin within their grafts. This further indicates that there are no systemic effects on the alloreactive immune responses by local TGFβ neutralization.
Effect of intragraft decorin gene transfer on TGFβ-induced gene expression
TGFβ is a pleiotropic cytokine that exerts a variety of effects on many different cell types. A reciprocal developmental pathway exists for the generation of pathogenic effector Th17 cells and Treg in response to TGFβ, with IL-6 being the co-factor required for Th17 induction (43-45). In addition, TGFβ induces cardiac fibroblasts to differentiate into myofibroblasts, which produce significant amounts of collagen and contribute to fibrosis (40). To assay for the effect of decorin on TGFβ-induced genes such as TGFβ, collagen A1, FoxP3 and IL-17, RNA was isolated from the allografts and quantitative RT-PCR was performed (Figure 3). In recipients transduced with AdDec, intragraft TGFβ (p<0.05), collagen A1 (p<0.05) and IL-17 (p<0.05) transcript levels were significantly reduced compared to control allografts (Figure 3). In contrast, intragraft FoxP3 expression was comparable between AdDec and Adβgal transduced allografts. These observations indicate that localized TGFβ neutralization by decorin can significantly decrease gene expression associated with fibrosis, but does not affect FoxP3 expression. These data suggest Th17 polarization occurs within the site of inflammation--the graft--while Treg induction and maintenance occurs systemically in the secondary lymphoid tissues (46).
Figure 3. Effects of TGFβ neutralization on intragraft gene expression.
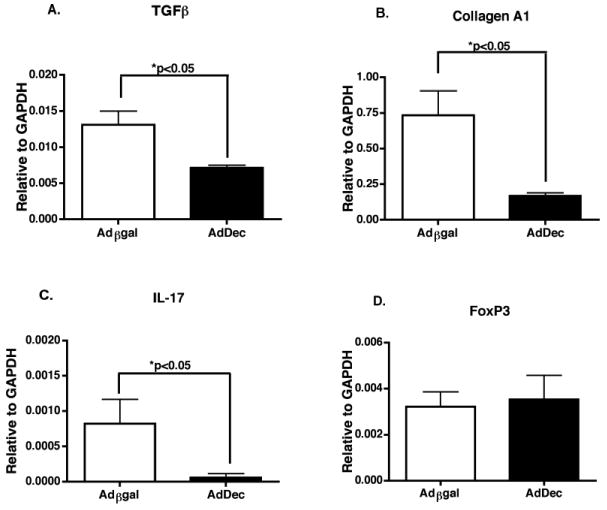
RNA samples from grafts of recipients transiently depleted of CD4+ T cells and transduced with Adβgal or AdDec were recovered 50 days post-transplant. Intragraft expression of (A) TGFβ, (B) collagen A1, (C) IL-17 and (D) FoxP3 relative to GAPDH was assessed by real-time RT-PCR. Bars depict the means of RNA expression from 9 Adβgal or 8 AdDec transduced allografts (+/- S.E.M.).
Intragraft TGFβ neutralization significantly attenuates graft fibrosis and hypertrophy
To investigate the effect of localized TGFβ neutralization on graft fibrosis, quantitative morphometric trichrome analysis was performed (Figure 4). Assessment of allograft fibrosis revealed that intragraft decorin expression resulted in a significant reduction of collagen deposition compared to control grafts (p<0.01) (Figure 4A and 4B). These data indicate that TGFβ promotes cardiac fibrosis and that localized neutralization of TGFβ can significantly attenuate CR.
Figure 4. Decorin gene transfer and intragraft TGFβ neutralization attenuates fibrosis and hypertrophy.
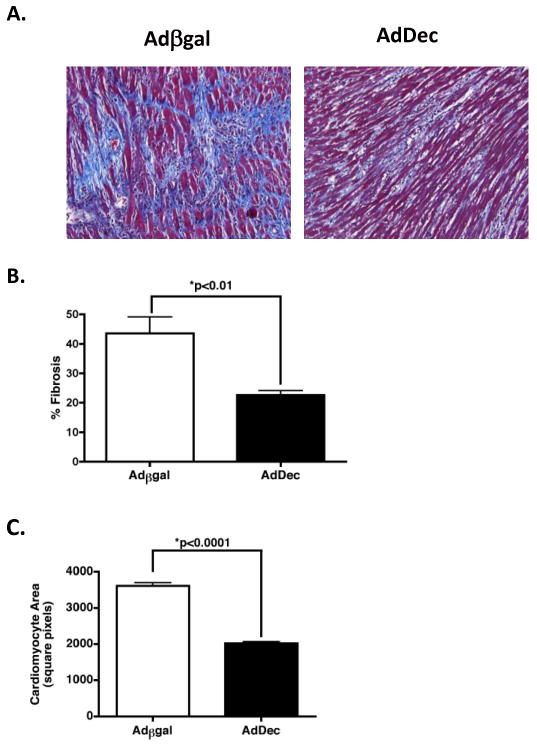
(A) Sections of grafts from recipients treated with anti-CD4 and transduced with Adβgal or AdDec were stained with Masson's trichrome stain on day 50 post-transplant. Fibrotic tissue appears blue. Magnification, 200×. (B) Morphometric analysis of trichrome staining. Bars represent the average percentage of (+/- S.E.M.) of graft area positive for collagen in 9 Adβgal (open bars) or 9 AdDec (shaded bars) transduced allografts. (C) Cardiomyocyte area quantification of groups described in (A). Bars represent mean (+/- S.E.M.) of area measurements from 100 cardiomyocytes per allograft at 200× magnification. Nine individual hearts were analyzed per group.
Cardiac hypertrophy is defined as an increase in the heart mass (47). An increase in the size of the cardiac myocytes, as opposed to the number, is the primary basis of cardiac hypertrophy (47). TGFβ is critical in driving this process (reviewed in (48)). An upregulation of TGFβ in cardiac tissue increases cardiomyocyte size and leads to cardiac dysfunction (48-50). To evaluate the effect of intragraft TGFβ neutralization on cardiac hypertrophy, cardiomyocyte cell size was measured employing histologic analysis (3). Reduced cardiac hypertrophy was observed in AdDec transduced grafts (Figure 4C). These findings indicate that intragraft TGFβ correlates with both fibrosis and hypertrophy in CR allografts and that decorin gene transfer can attenuate both pathologies.
Discussion
CR is an intractable disease characterized by interstitial fibrosis, occlusive neointima development, and graft dysfunction (4-6). The etiology of CR is poorly understood and no therapies exist to block its progression. TGFβ plays a beneficial role in the transplant setting because of its immunsuppressive qualities (29, 30), but has also been implicated in promoting graft fibrosis and CR (51, 52). In the mouse vascularized cardiac model, we have previously reported an association between TGFβ and CR (21). Intragraft TGFβ transcript levels were readily detected in the CR grafts from recipients transiently depleted of CD4+ T cells, but not in the grafts of anti-CD40L treated recipients, which remain free of CR (21), suggesting the importance of TGFβ in this pathology. In this study, we employed decorin gene transfer and local neutralization of TGFβ in cardiac allografts to assess the impact of intragraft TGFβ neutralization on allograft acceptance, T and B cell hyporesponsiveness and CR. We demonstrate that local neutralization of TGFβ in cardiac allografts significantly attenuated interstitial fibrosis and improved graft function, but did not reverse the hyporeactive state of donor-reactive T or B cells.
Intragraft transcript levels of TGFβ are frequently detected in accepted grafts, including the cardiac allografts from recipients transiently depleted of CD4+ T cells. TGFβ expression is believed to promote graft survival through the induction of Treg which control graft-reactive Th1 and Th2 responses (29, 30, 41). Previous studies in recipients treated with inductive anti-CD4 mAb have revealed that repopulating CD4+ T cells are hyporesponsive toward donor antigen and mount Th2 responses upon re-challenge, while naïve T cells mount a dominant Th1 response (34). In this CR model, alloreactive T cells only progress to a hyporesponsive state in response to TGFβ (Faust et al., submitted). The critical role for TGFβ in this CR model was revealed when CD4-DNR mice were used as recipients and transiently depleted of CD4+ T cells. T cell TGFβ signaling was requisite for both long-term graft acceptance and suppression of graft-reactive T and B cell responses as well as graft fibrosis (Faust et al., submitted). Therefore, systemic strategies targeting TGFβ are not feasible since this could alter the hyporesponsiveness of graft-reactive T and B cells. In contrast, intragraft TGFβ inhibition would be beneficial in attenuating fibrosis. TGFβ neutralization within allografts, however, did not result in a reversal of T or B cell hyporesponsiveness (Figure 2). These findings demonstrate that localized TGFβ inhibition does not alter the systemic regulation of graft-reactive cells or lead to graft loss but was effective at reducing fibrosis associated with CR.
In transplantation, FoxP3+ Treg have been shown to play a central role in suppression of alloreactive T cells and in long-term allograft acceptance (reviewed in (53)). In both human and animal transplant, allograft acceptance strongly correlates with Treg infiltration into the graft as detected by enhanced intragraft FoxP3 transcript levels (53-56). Treg can be divided into two populations: natural Treg (nTreg), which arise in the thymus and do not require TGFβ to develop, and induced peripheral Treg (iTreg), which do require TGFβ to differentiate from naïve CD4+FoxP3- T cells into Treg (reviewed in (57)). In the current study, local neutralization of TGFβ did not affect intragraft FoxP3 expression, indicating that Treg migration and/or generation within the graft was unchanged between decorin and βgal transduced recipients (Figure 3). However, these data argue against Treg induction within the grafts since intragraft TGFβ was neutralized by decorin gene transfer. Indeed, Treg have been reported to be generated in lymph nodes and subsequently migrate to the graft (46). While thymic-derived Treg do not require TGFβ for their generation, they do depend on TGFβ for their persistence in the periphery (58). Given that decorin gene transfer spares the systemic effects of TGFβ, it is also possible that nTreg may contribute in regulating alloreactive responses within the graft (59).
Interstitial fibrosis represents a hallmark of CR and results in pathogenic cardiac remodeling and graft dysfunction (22). The effector cells in this process are cardiac fibroblasts, which respond to TGFβ by inducing the expression of pro-fibrotic mediators that upregulate extracellular matrix synthesis and down-regulate matrix degradation (22). During remodeling, cardiac fibroblasts located within the interstitium proliferate and produce proteins such as collagen (60), resulting in a significant increase in interstitial fibrosis (61). Cardiac fibrosis impairs contractility and reduces cardiac function.
Previous studies demonstrate that decorin gene transfer ameliorates TGFβ-induced fibrosis of multiple organs (26, 28). Decorin inhibits TGFβ bioactivity by sequestering TGFβ to the ECM (24, 25). In addition, decorin negatively impacts TGFβ gene expression by interrupting TGFβ/Smad-dependent transcriptional events (24, 25, 27). One mechanism by which decorin inhibits fibrosis is through the reduction of TGFβ-induced collagen transcript levels in cultured human cardiac fibroblasts (62). Gene transfer of decorin into allografts transplanted into recipients transiently depleted of CD4+ T cells significantly attenuated collagen deposition and fibrosis compared to control allografts (Figure 4). Decorin over-expression also inhibited cardiac hypertrophy, demonstrating amelioration of an additional TGFβ-induced parameter correlated with CR (Figure 4).
In addition to reduced TGFβ gene expression, decorin reduced intragraft transcript levels IL-17 (Figure 3). IL-17 amplifies inflammatory responses (reviewed in (63, 64)) and has recently been identified as a cytokine with pro-fibrotic activities (65-70). Prior studies in IL-17-/- recipients treated inductively with anti-CD4 mAb revealed that allografts from the deficient mice exhibited a significant reduction in fibrosis compared to WT (Faust et al., submitted). IL-17 may induce fibrosis though multiple mechanisms. Upregulation of collagen gene expression in direct response to IL-17 has been observed in mouse cardiac fibroblasts (66). IL-17 may also induce endothelial cells and fibroblasts to secrete pro-inflammatory cytokines and chemokines (63, 64) that result in the recruitment of APC and alloreactive T cells into allograft. These inflammatory cells may secrete factors that lead to myocardial damage and tissue remodeling that favors fibrosis. The reduction of intragraft IL-17 expression in AdDec transduced grafts compared to WT counterparts further implicates this pro-inflammatory cytokine in CR.
Decorin has multiple molecular targets in cell growth in addition to its interaction with TGFβ (23). Decorin negatively impacts cell proliferation, an effect mediated through the induction of p21 (23). Decorin-induced cell cycle arrest might reduce fibrosis by suppressing cardiac fibroblasts from proliferating and differentiating into myofibroblasts. Decorin also interacts with complement C1q, inhibiting activation of the classical complement pathway (71). Hence, under inflammatory tissue damage and ECM remodeling, decorin may suppress complement activation and prevent further cardiomyocyte injury. Decorin may further inhibit the production of inflammatory chemokines and cytokines, including MCP-1 and IL-8 by preventing C1q from binding graft endothelial cells (71, 72). Therefore, in addition to decorin's inhibitory effects on TGFβ, suppression of complement activation may help to reduce fibrosis by limiting the damage inflicted on the allograft.
In summary, TGFβ is a critical cytokine in fibroproliferative disorders following inflammatory responses (reviewed in (16)). TGFβ can have both exacerbating and ameliorating effects in immune-mediated fibrotic diseases, making global inhibition undesirable and local neutralization of TGFβ an attractive therapy. As evidenced in this model of CR, systemic TGFβ production is requisite for T cell hyporesponsiveness (Faust et. al., submitted), while local TGFβ production at the site of inflammation induces graft fibrosis. We demonstrate that neutralizing intragraft TGFβ inhibits the cytokine's fibrotic activities, but does not reverse its beneficial immunosuppressive qualities. These data provide insight into the underlying causes of CR, and identify intragraft TGFβ as a therapeutic target for treatment of this disease.
Footnotes
Disclosures: The authors declare no conflict of interest.
This work was supported by R01 HL070613 (DKB) and by R01 AI061469 (DKB) and by T90 DK070071 (SMF).
Abbreviations used in this paper: adenovirus, Ad; AdDec, adenovirus that encodes rat decorin; Adβgal, adenovirus that encodes beta-galactosidase; CD4-DNR, transgenic mice with a dominant negative TGFβ receptor; CR, chronic rejection; ECM, extracellular matrix; iTreg, induced peripheral T regulatory cells; nTreg, thymic-derived natural T regulatory cells; TGFβ, transforming growth factor beta; Treg, T regulatory cell; WT, wild type.
References
- 1.Al-khaldi A, Robbins RC. New directions in cardiac transplantation. Annu Rev Med. 2006;57:455–471. doi: 10.1146/annurev.med.57.082704.130518. [DOI] [PubMed] [Google Scholar]
- 2.Hayry P, Isoniemi H, Yilmaz S, Mennander A, Lemstrom K, Raisanen-Sokolowski A, Koskinen P, Ustinov J, Lautenschlager I, Taskinen E, et al. Chronic allograft rejection. Immunol Rev. 1993;134:33–81. doi: 10.1111/j.1600-065x.1993.tb00639.x. [DOI] [PubMed] [Google Scholar]
- 3.Diaz JA, Booth AJ, Lu G, Wood SC, Pinsky DJ, Bishop DK. Critical role for IL-6 in hypertrophy and fibrosis in chronic cardiac allograft rejection. Am J Transplant. 2009;9:1773–1783. doi: 10.1111/j.1600-6143.2009.02706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Libby P, Pober JS. Chronic rejection. Immunity. 2001;14:387–397. doi: 10.1016/s1074-7613(01)00119-4. [DOI] [PubMed] [Google Scholar]
- 5.Orosz CG, Pelletier RP. Chronic remodeling pathology in grafts. Curr Opin Immunol. 1997;9:676–680. doi: 10.1016/s0952-7915(97)80048-9. [DOI] [PubMed] [Google Scholar]
- 6.Weis M, von Scheidt W. Cardiac allograft vasculopathy: a review. Circulation. 1997;96:2069–2077. doi: 10.1161/01.cir.96.6.2069. [DOI] [PubMed] [Google Scholar]
- 7.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 8.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kulkarni AB, Ward JM, Yaswen L, Mackall CL, Bauer SR, Huh CG, Gress RE, Karlsson S. Transforming growth factor-beta 1 null mice. An animal model for inflammatory disorders. Am J Pathol. 1995;146:264–275. [PMC free article] [PubMed] [Google Scholar]
- 10.Rubtsov YP, Rudensky AY. TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat Rev Immunol. 2007;7:443–453. doi: 10.1038/nri2095. [DOI] [PubMed] [Google Scholar]
- 11.Lebman DA, Edmiston JS. The role of TGF-beta in growth, differentiation, and maturation of B lymphocytes. Microbes Infect. 1999;1:1297–1304. doi: 10.1016/s1286-4579(99)00254-3. [DOI] [PubMed] [Google Scholar]
- 12.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levings MK, Bacchetta R, Schulz U, Roncarolo MG. The role of IL-10 and TGF-beta in the differentiation and effector function of T regulatory cells. Int Arch Allergy Immunol. 2002;129:263–276. doi: 10.1159/000067596. [DOI] [PubMed] [Google Scholar]
- 14.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 16.Branton MH, Kopp JB. TGF-beta and fibrosis. Microbes Infect. 1999;1:1349–1365. doi: 10.1016/s1286-4579(99)00250-6. [DOI] [PubMed] [Google Scholar]
- 17.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 18.Kang HR, Cho SJ, Lee CG, Homer RJ, Elias JA. Transforming growth factor (TGF)-beta1 stimulates pulmonary fibrosis and inflammation via a Bax-dependent, bid-activated pathway that involves matrix metalloproteinase-12. J Biol Chem. 2007;282:7723–7732. doi: 10.1074/jbc.M610764200. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto T, Noble NA, Cohen AH, Nast CC, Hishida A, Gold LI, Border WA. Expression of transforming growth factor-beta isoforms in human glomerular diseases. Kidney Int. 1996;49:461–469. doi: 10.1038/ki.1996.65. [DOI] [PubMed] [Google Scholar]
- 20.Denton CP, Abraham DJ. Transforming growth factor-beta and connective tissue growth factor: key cytokines in scleroderma pathogenesis. Curr Opin Rheumatol. 2001;13:505–511. doi: 10.1097/00002281-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 21.Csencsits K, Wood SC, Lu G, Faust SM, Brigstock D, Eichwald EJ, Orosz CG, Bishop DK. Transforming growth factor beta-induced connective tissue growth factor and chronic allograft rejection. Am J Transplant. 2006;6:959–966. doi: 10.1111/j.1600-6143.2006.01292.x. [DOI] [PubMed] [Google Scholar]
- 22.Leask A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res. 2007;74:207–212. doi: 10.1016/j.cardiores.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 23.Stander M, Naumann U, Wick W, Weller M. Transforming growth factor-beta and p-21: multiple molecular targets of decorin-mediated suppression of neoplastic growth. Cell Tissue Res. 1999;296:221–227. doi: 10.1007/s004410051283. [DOI] [PubMed] [Google Scholar]
- 24.Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature. 1990;346:281–284. doi: 10.1038/346281a0. [DOI] [PubMed] [Google Scholar]
- 25.Border WA, Noble NA, Yamamoto T, Harper JR, Yamaguchi Y, Pierschbacher MD, Ruoslahti E. Natural inhibitor of transforming growth factor-beta protects against scarring in experimental kidney disease. Nature. 1992;360:361–364. doi: 10.1038/360361a0. [DOI] [PubMed] [Google Scholar]
- 26.Isaka Y, Brees DK, Ikegaya K, Kaneda Y, Imai E, Noble NA, Border WA. Gene therapy by skeletal muscle expression of decorin prevents fibrotic disease in rat kidney. Nat Med. 1996;2:418–423. doi: 10.1038/nm0496-418. [DOI] [PubMed] [Google Scholar]
- 27.Abdel-Wahab N, Wicks SJ, Mason RM, Chantry A. Decorin suppresses transforming growth factor-beta-induced expression of plasminogen activator inhibitor-1 in human mesangial cells through a mechanism that involves Ca2+-dependent phosphorylation of Smad2 at serine-240. Biochem J. 2002;362:643–649. doi: 10.1042/0264-6021:3620643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolb M, Margetts PJ, Sime PJ, Gauldie J. Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1327–1334. doi: 10.1152/ajplung.2001.280.6.L1327. [DOI] [PubMed] [Google Scholar]
- 29.Josien R, Douillard P, Guillot C, Muschen M, Anegon I, Chetritt J, Menoret S, Vignes C, Soulillou JP, Cuturi MC. A critical role for transforming growth factor-beta in donor transfusion-induced allograft tolerance. J Clin Invest. 1998;102:1920–1926. doi: 10.1172/JCI4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daley SR, Ma J, Adams E, Cobbold SP, Waldmann H. A key role for TGF-beta signaling to T cells in the long-term acceptance of allografts. J Immunol. 2007;179:3648–3654. doi: 10.4049/jimmunol.179.6.3648. [DOI] [PubMed] [Google Scholar]
- 31.Bishop DK, Li W, Chan SY, Ensley RD, Shelby J, Eichwald EJ. Helper T lymphocyte unresponsiveness to cardiac allografts following transient depletion of CD4-positive cells. Implications for cellular and humoral responses. Transplantation. 1994;58:576–584. doi: 10.1097/00007890-199409150-00009. [DOI] [PubMed] [Google Scholar]
- 32.Piccotti JR, Li K, Chan SY, Eichwald EJ, Bishop DK. Cytokine regulation of chronic cardiac allograft rejection: evidence against a role for Th1 in the disease process. Transplantation. 1999;67:1548–1555. doi: 10.1097/00007890-199906270-00008. [DOI] [PubMed] [Google Scholar]
- 33.Csencsits K, Burrell BE, Lu G, Eichwald EJ, Stahl GL, Bishop DK. The classical complement pathway in transplantation: unanticipated protective effects of C1q and role in inductive antibody therapy. Am J Transplant. 2008;8:1622–1630. doi: 10.1111/j.1600-6143.2008.02295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wood SC, Lu G, Burrell BE, Bishop DK. Transplant acceptance following anti-CD4 versus anti-CD40L therapy: evidence for differential maintenance of graft-reactive T cells. Am J Transplant. 2008;8:2037–2048. doi: 10.1111/j.1600-6143.2008.02372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 36.Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation. 1973;16:343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- 37.Chan SY, Li K, Piccotti JR, Louie MC, Judge TA, Turka LA, Eichwald EJ, Bishop DK. Tissue-specific consequences of the anti-adenoviral immune response: implications for cardiac transplants. Nat Med. 1999;5:1143–1149. doi: 10.1038/13467. [DOI] [PubMed] [Google Scholar]
- 38.Chan SY, Goodman RE, Szmuszkovicz JR, Roessler B, Eichwald EJ, Bishop DK. DNA-liposome versus adenoviral mediated gene transfer of transforming growth factor beta1 in vascularized cardiac allografts: differential sensitivity of CD4+ and CD8+ T cells to transforming growth factor beta1. Transplantation. 2000;70:1292–1301. doi: 10.1097/00007890-200011150-00006. [DOI] [PubMed] [Google Scholar]
- 39.Matesic D, Lehmann PV, Heeger PS. High-resolution characterization of cytokine-producing alloreactivity in naive and allograft-primed mice. Transplantation. 1998;65:906–914. doi: 10.1097/00007890-199804150-00008. [DOI] [PubMed] [Google Scholar]
- 40.Cutroneo KR. TGF-beta-induced fibrosis and SMAD signaling: oligo decoys as natural therapeutics for inhibition of tissue fibrosis and scarring. Wound Repair Regen. 2007;15 1:S54–60. doi: 10.1111/j.1524-475X.2007.00226.x. [DOI] [PubMed] [Google Scholar]
- 41.Walsh PT, Taylor DK, Turka LA. Tregs and transplantation tolerance. J Clin Invest. 2004;114:1398–1403. doi: 10.1172/JCI23238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yong Z, Chang L, Mei YX, Yi L. Role and mechanisms of CD4+CD25+ regulatory T cells in the induction and maintenance of transplantation tolerance. Transpl Immunol. 2007;17:120–129. doi: 10.1016/j.trim.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 43.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 44.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 45.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen D, Bromberg JS. T regulatory cells and migration. Am J Transplant. 2006;6:1518–1523. doi: 10.1111/j.1600-6143.2006.01372.x. [DOI] [PubMed] [Google Scholar]
- 47.Marian AJ. Genetic determinants of cardiac hypertrophy. Curr Opin Cardiol. 2008;23:199–205. doi: 10.1097/HCO.0b013e3282fc27d9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider MD. Serial killer: angiotensin drives cardiac hypertrophy via TGF-beta1. J Clin Invest. 2002;109:715–716. doi: 10.1172/JCI15292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raichlin E, Villarraga HR, Chandrasekaran K, Clavell AL, Frantz RP, Kushwaha SS, Rodeheffer RJ, McGregor CG, Daly RC, Park SJ, Kremers WK, Edwards BS, Pereira NL. Cardiac allograft remodeling after heart transplantation is associated with increased graft vasculopathy and mortality. Am J Transplant. 2009;9:132–139. doi: 10.1111/j.1600-6143.2008.02474.x. [DOI] [PubMed] [Google Scholar]
- 50.Torre-Amione G. Cardiac allograft hypertrophy: a new target for therapy, a surrogate marker for survival? Am J Transplant. 2009;9:7–8. doi: 10.1111/j.1600-6143.2008.02503.x. [DOI] [PubMed] [Google Scholar]
- 51.Mannon RB. Therapeutic targets in the treatment of allograft fibrosis. Am J Transplant. 2006;6:867–875. doi: 10.1111/j.1600-6143.2006.01261.x. [DOI] [PubMed] [Google Scholar]
- 52.Jain S, Furness PN, Nicholson ML. The role of transforming growth factor beta in chronic renal allograft nephropathy. Transplantation. 2000;69:1759–1766. doi: 10.1097/00007890-200005150-00001. [DOI] [PubMed] [Google Scholar]
- 53.Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol. 2003;3:199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 54.Cobbold SP, Castejon R, Adams E, Zelenika D, Graca L, Humm S, Waldmann H. Induction of foxP3+ regulatory T cells in the periphery of T cell receptor transgenic mice tolerized to transplants. J Immunol. 2004;172:6003–6010. doi: 10.4049/jimmunol.172.10.6003. [DOI] [PubMed] [Google Scholar]
- 55.Bestard O, Cruzado JM, Mestre M, Caldes A, Bas J, Carrera M, Torras J, Rama I, Moreso F, Seron D, Grinyo JM. Achieving donor-specific hyporesponsiveness is associated with FOXP3+ regulatory T cell recruitment in human renal allograft infiltrates. J Immunol. 2007;179:4901–4909. doi: 10.4049/jimmunol.179.7.4901. [DOI] [PubMed] [Google Scholar]
- 56.Lee I, Wang L, Wells AD, Dorf ME, Ozkaynak E, Hancock WW. Recruitment of Foxp3+ T regulatory cells mediating allograft tolerance depends on the CCR4 chemokine receptor. J Exp Med. 2005;201:1037–1044. doi: 10.1084/jem.20041709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity. 2009;30:616–625. doi: 10.1016/j.immuni.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liston A, Rudensky AY. Thymic development and peripheral homeostasis of regulatory T cells. Curr Opin Immunol. 2007;19:176–185. doi: 10.1016/j.coi.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 59.Zhang N, Schroppel B, Lal G, Jakubzick C, Mao X, Chen D, Yin N, Jessberger R, Ochando JC, Ding Y, Bromberg JS. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity. 2009;30:458–469. doi: 10.1016/j.immuni.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petrov VV, Fagard RH, Lijnen PJ. Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension. 2002;39:258–263. doi: 10.1161/hy0202.103268. [DOI] [PubMed] [Google Scholar]
- 61.Weber KT, Brilla CG, Campbell SE, Zhou G, Matsubara L, Guarda E. Pathologic hypertrophy with fibrosis: the structural basis for myocardial failure. Blood Press. 1992;1:75–85. doi: 10.3109/08037059209077497. [DOI] [PubMed] [Google Scholar]
- 62.Jahanyar J, Joyce DL, Southard RE, Loebe M, Noon GP, Koerner MM, Torre-Amione G, Youker KA. Decorin-mediated transforming growth factor-beta inhibition ameliorates adverse cardiac remodeling. J Heart Lung Transplant. 2007;26:34–40. doi: 10.1016/j.healun.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 63.Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051–1057. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cortez DM, Feldman MD, Mummidi S, Valente AJ, Steffensen B, Vincenti M, Barnes JL, Chandrasekar B. IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK- and ERK1/2-dependent C/EBP-beta, NF-kappaB, and AP-1 activation. Am J Physiol Heart Circ Physiol. 2007;293:H3356–3365. doi: 10.1152/ajpheart.00928.2007. [DOI] [PubMed] [Google Scholar]
- 66.Venkatachalam K, Mummidi S, Cortez DM, Prabhu SD, Valente AJ, Chandrasekar B. Resveratrol inhibits high glucose-induced PI3K/Akt/ERK-dependent interleukin-17 expression in primary mouse cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2008;294:H2078–2087. doi: 10.1152/ajpheart.01363.2007. [DOI] [PubMed] [Google Scholar]
- 67.Burlingham WJ, Love RB, Jankowska-Gan E, Haynes LD, Xu Q, Bobadilla JL, Meyer KC, Hayney MS, Braun RK, Greenspan DS, Gopalakrishnan B, Cai J, Brand DD, Yoshida S, Cummings OW, Wilkes DS. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest. 2007;117:3498–3506. doi: 10.1172/JCI28031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Page N, Olivenstein R, Elias J, Chakir J. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol. 2001;108:430–438. doi: 10.1067/mai.2001.117929. [DOI] [PubMed] [Google Scholar]
- 69.Dubin PJ, McAllister F, Kolls JK. Is cystic fibrosis a TH17 disease? Inflamm Res. 2007;56:221–227. doi: 10.1007/s00011-007-6187-2. [DOI] [PubMed] [Google Scholar]
- 70.Simonian PL, Roark CL, Wehrmann F, Lanham AK, Diaz del Valle F, Born WK, O'Brien RL, Fontenot AP. Th17-polarized immune response in a murine model of hypersensitivity pneumonitis and lung fibrosis. J Immunol. 2009;182:657–665. [PMC free article] [PubMed] [Google Scholar]
- 71.Groeneveld TW, Oroszlan M, Owens RT, Faber-Krol MC, Bakker AC, Arlaud GJ, McQuillan DJ, Kishore U, Daha MR, Roos A. Interactions of the extracellular matrix proteoglycans decorin and biglycan with C1q and collectins. J Immunol. 2005;175:4715–4723. doi: 10.4049/jimmunol.175.7.4715. [DOI] [PubMed] [Google Scholar]
- 72.Wehner J, Morrell CN, Reynolds T, Rodriguez ER, Baldwin WM., 3rd Antibody and complement in transplant vasculopathy. Circ Res. 2007;100:191–203. doi: 10.1161/01.RES.0000255032.33661.88. [DOI] [PubMed] [Google Scholar]