

Abstract
Transgenic expression of the α7 integrin can ameliorate muscle pathology in a mouse model of Duchenne muscular dystrophy (mdx/utr−/−) and thus can compensate for the loss of dystrophin in diseased mice. In spite of the beneficial effects of the α7 integrin in protecting mice from dystrophy, identification of molecular signaling events responsible for these changes remains to be established. The purpose of this study was to determine a role for signaling in the amelioration of muscular dystrophy by α7 integrin. Activation of PI3K, ILK, AKT, mTOR, p70S6K, BAD, ERK, and p38 was measured in the muscle from wild type (WT), mdx/utr−/− and α7BX2-mdx/utr−/− mice using in vitro activity assays or phosphospecific antibodies and western blotting. Significant increases in PI3K activity (47%), ILK activity (2.0-fold), mTOR (Ser2448) (57%), p70S6K (Thr389) (11.7-fold), and ERK (Thr202/Tyr204) (66%) were demonstrated in dystrophic mdx/utr−/− muscle compared to WT. A significant decrease in p38 phosphorylation (2.9-fold) was also observed. Although most of these signaling events were similar in dystrophic mdx/utr−/− mice overexpressing the α7 integrin, the AKT (Ser473): AKT ratio (2-fold vs. WT) and p70S6K phosphorylation (18-fold vs. WT) were higher in α7BX2-mdx/utr−/− compared to mdx/utr−/− mice. In addition, increased phosphorylation of BAD Serine 112 may contribute to the significant reduction in TUNEL+ cells observed in α7BX2-mdx/utr−/− mice. We conclude that the α7β1 integrin confers a protective effect in dystrophic muscle through the activation of the ILK, AKT, p70S6K and BAD signaling to promote muscle cell survival.
Keywords: α7 Integrin, Muscular dystrophy, Integrin linked kinase (ILK), AKT, Cell signaling, Apoptosis
1. Introduction
Duchenne muscular dystrophy (DMD) is caused by mutations in the dystrophin gene resulting in the absence of the dystrophin protein [1,2]. Dystrophin is a key component of a macromolecular complex that links the cell cytoskeleton to laminin in the extracellular matrix [3-5]. The lack of dystrophin in DMD patients results in severe progressive muscle wasting. Patients are usually confined to a wheelchair in their early teens and death from cardiopulmonary failure often occurs in their early twenties. In contrast, mdx mice, a mouse model for DMD, show mild muscle pathology and live a normal life span [2,6]. The muscles of mdx mice show increased expression of utrophin, a homologue of dystrophin normally found at neuromuscular and myotendinous junctions in the adult. In mdx mice, utrophin is localized around the sarcolemma suggesting that it may partially compensate for the absence of dystrophin [7]. Mdx/utr−/− mice, which lack both dystrophin and utrophin, develop severe pathology that closely resembles that seen in DMD and die at 4–20 weeks of age [8,9].
Both mdx mice and DMD patients have elevated levels of α7β1 integrin, a heterodimeric transmembrane receptor found in skeletal and cardiac muscle that also links laminin in the extracellular matrix to the cell cytoskeleton [10]. This led us to hypothesize that the dystrophin and integrin linkage systems may be complementary, such that in the absence of dystrophin, the level of α7β1 integrin is increased. The results led us to question whether further increasing integrin levels in the absence of the dystrophin linkage system could prevent the development of muscle disease. This hypothesis was confirmed in transgenic mdx/utr−/− mice in which expression of the α7BX2 integrin chain was further increased in skeletal muscle. An additional two-fold increase in α7β1 integrin protein reduced the development of muscular dystrophy and increased longevity of the α7BX2-mdx/utr−/− transgenic mice three-fold [11].
The mechanism by which the mdx/utr−/− mice were rescued by increased α7β1 integrin is unclear. Additional integrin may provide increased mechanical stability to muscle fibers by means of attachment to the basal lamina. This could be accomplished by increased integrin-mediated adhesion and/or stabilization of components of the dystrophin complex that are expressed but markedly reduced in the plasma membrane in the absence of dystrophin. The preservation of membrane structure and reduction in inflammation observed in α7BX2-mdx/utr−/− mice [11] may slow the rapid onset of degeneration–regeneration cycles observed during the first few weeks of development, allowing differentiation and growth of fibers to dominate. Alternatively, the α7β1 integrin may initiate an orchestrated series of signaling events inside the fiber that prevent apoptosis [12,13], a more controlled method for reducing fiber number which has been demonstrated in mdx mice [14]. ILK, the integrin-associated kinase, and its downstream substrate, the anti-apoptotic protein kinase B (PKB/AKT) appear to be important for the preservation of the myotendinous junction, such that the absence of ILK decreases AKT phosphorylation and leads to a milder form of muscular dystrophy in mice [15] and decreased cardiomyocyte defects in mice and zebrafish [16,17]. A comparative evaluation of the signaling events taking place in α7BX2-mdx/utr−/− versus mdx/utr−/− skeletal muscle provides an important step in understanding the underlying mechanism of α7 integrin-driven protection during dystrophy.
The purpose of this study was to evaluate the activation and expression of integrin-associated proteins that mediate cell signaling in mdx, mdx/utr−/−, and α7BX2-mdx/utr−/− mouse skeletal muscle. Elevations in ILK activity, ILK protein, and the AKT phosphorylation: AKT ratio were observed in α7BX2-mdx/utr−/− mice compared to mdx and mdx/utr−/− mice. The activation of this signaling pathway was confirmed through the parallel enhancement of p70S6K phosphorylation in α7BX2-mdx/utr−/− mice. p70S6K phosphorylation is associated with increased initiation of protein translation necessary for increased muscle growth. Bcl-2/Bcl-XL-associated death promoter (BAD) phosphorylation and a concomitant decrease in apoptosis were also observed in dystrophic mice expressing higher levels of the α7 integrin. In addition, specific association of ILK with a critical tyrosine residue on the α7 integrin subunit is demonstrated. These results reveal that increasing the amount of α7β1 integrin results in the activation of the ILK–AKT signaling pathway which promotes cell survival and hypertrophy and plays an important role in the integrin-mediated rescue of α7BX2-mdx/utr−/− transgenic mice.
2. Materials and methods
2.1. Transgenic mice
The generation of transgenic mouse lines expressing the rat α7BX2 protein has been previously described [11]. Transgenic mdx/utr+/− male mice were bred with mdx/utr+/− females to produce transgenic α7BX2-mdx/utr−/− animals. Transgenic mice used in this study were 5 weeks of age and have an approximately 4-fold increase in α7BX2 protein in their hindlimb muscle compared to non-transgenic wild type mice and a 2-fold increase over that detected in mdx/utr−/− mice [11]. Procedures for animal use and care were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Illinois at Urbana-Champaign.
2.2. Antibodies
ILK polyclonal antibody was purchased from Santa Cruz Biotechnology, Santa Cruz, CA, for determination of total ILK expression. AKT, phospho-AKT (Ser 473), phosphor-mTOR (Ser2448), mTOR, phospho-p70S6K, p70S6K, phospho-BAD (Ser 112), BAD, ERK (Thr202/Tyr204), ERK, phospho-p38 (Thr180/Tyr182), and p38 antibodies were purchased from Cell Signaling Technology, Beverly, MA. HRP-labeled donkey anti-mouse and anti-rabbit antibodies were purchased from Jackson ImmunoResearch Laboratories Inc., West Grove, PA.
2.3. ILK activity assay
Hindlimb muscles were powdered in liquid nitrogen using a mortar and pestle. Proteins were extracted in ice-cold lysis buffer containing 2% Triton X-100, 20 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA (pH 8.0), 1 mM EGTA (pH 7.5), 2.5 mM Na pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium vanadate, 1:200 protease inhibitor cocktail III (Calbiochem), and 1 mM PMSF. Homogenates were rotated 30 min at 4 °C and centrifuged for 5 min at 10,000 g. Supernatants were collected and protein concentrations were determined using Bradford assays. Muscle extracts (1 mg) were precleared with 20 μL prewashed protein G beads prior to immunoprecipitation with 10 μL of ILK antibody (Millipore [Upstate Biotechnology, 06-592], Billerica, MA) in a total volume of 1 mL of fresh lysis buffer overnight at 4 °C, followed by the addition of 20 μL protein G beads for 2 h. Supernatants were discarded and beads were washed twice with lysis buffer and once with 2× lysis buffer (62.5 mM MOPS (pH 7.5), 37.5 mM MgCl2, 2.5 mM EGTA, 0.1 M β-glycerophosphate, 0.1 M sodium fluoride, 0.1 M sodium vanadate, and 330 mM DTT) diluted 1:1. In vitro kinase buffer containing 2× kinase buffer, 1 mg/mL basic myelin protein, and 10 μCi of adenosine 5′ triphosphate (γ-32P) (PerkinElmer, Boston, MA) was added to each sample for 20 min at 30 °C and the reaction was terminated with 10 μL 4× Laemmli buffer containing 330 mM DTT, followed by boiling for 5 min. Samples were loaded onto a 12% gel for approximately 1 h at 200 V. Gels were rinsed, stained with 1:1 fast green stain (Sigma), destained, and then dried. Gels were exposed in a PhosphoImager cassette for 1–2 h and then bands were quantified using a PhosphoImager and Image Reader software. Muscle samples were also run as negative controls without ILK antibody or MBP to verify the validity of this assay.
2.4. ILK immunoprecipitation
Approximately 3 × 105 C2C12 mouse myoblasts, stably transfected to express the α7BX2 integrin chain or a cytoplasmic domain mutant α7BX2-YTF (tyrosine to phenylalanine mutation), were cultured on fibronectin-coated dishes in Dulbecco’s medium (low glucose) containing 20% fetal calf serum, 0.5% chicken embryo extract, 2 mM glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin and 10 μg/mL kanamycin [19]. G418 was added to ensure selection of stably transfected cells. At 80% confluence, Dulbecco’s medium containing 2% horse serum (no embryo extract) was added to induce differentiation. Association of ILK with the α7BX2 integrin was induced by engaging the integrin with 15 μg/mL of purified anti-α7 (O26) antibody [18], a concentration that also induces acetylcholine receptor clustering [19]. Following antibody stimulation, cells were washed once in ice-cold phosphate buffered saline containing 2 mM PMSF, collected and extracted in ice-cold lysis buffer as described above for the ILK activity assay, except that 2% NP-40 was used in place of Triton-X. The Bradford assay was used to determine protein concentration. Protein extracts (0.5 g) were incubated overnight with 10 μL ILK antibody (Millipore [Upstate Biotechnology, 06-592]) and incubated for 2 h with 20 μL prewashed protein G beads. The beads were washed 3 times with NP-40 lysis buffer, boiled with 40 μL 2× Laemmli buffer and samples were loaded onto 8% gels. Following electrophoresis, proteins were transferred to nitrocellulose, blocked with 5% BSA, and examined for α7BX2 using CDB antibody, reactive with the cytoplasmic α7B cytoplasmic domain [20]. Blocking peptide experiments verified the α7 integrin immunoreactive bands following detection with secondary antibody and enhanced chemiluminescence (ECL) (Amersham Pharmacia Biotech, Piscataway, NJ).
2.5. PI3K activity assay
Extracts (2 mg) prepared from hindlimb muscles (as described for the ILK activity assay above) were pre-cleared with 20 μL pre-cleared protein G beads prior to immunoprecipitation with 5 μL of p85 antibody (Millipore [Upstate Biotechnology, 06-195], Billerica, MA) in a total volume of 1 mL of fresh lysis buffer overnight at 4 °C, followed by the addition of 60 μL protein A beads for 2 h. Supernatants were discarded and beads were washed twice with each of the following buffers: Buffer 1 [10× PBS, 1% NP-40, 100 μM sodium vanadate], Buffer 2 [100 mM Tris–HCl (pH 7.5), 500 mM LiCl, 100 mM sodium vanadate], and Buffer 3 [10 mM Tris (pH 7.5), 100 mM NaCl, 100 mM sodium vanadate, and 1 mM EDTA]. Phosphatidylinositol (PI) substrate (10 μL; Aventi Polar Lipids, Alabaster, AL) and 10 μL of 100 mM MgCl2 were added to each tube prior to initiation of reaction with adenosine 5′ triphosphate (γ-32P) (7 μCi/sample), 100 mM MgCl2, and ATP (10 mM; Sigma, A9187) at 30 °C for 30 min, vortexing every 2 min. The reactions were terminated with 20 μL 8 N HCl. Lipids were extracted in a 1:1 choloroform:methanol and spotted onto thin layer chromatography plates (Silica gel 60A plates, 250 μm layer thickness, Fisher Scientific). Migration of lipids occurred by capillary action in a TLC tank and TLC plates were air dried for 30 min and exposed to a PhosphoImager cassette. Lipids were quantified using a PhosphoImager and Image Reader software. Omission of antibody and substrate were used as controls.
2.6. Western analysis
Hindlimb muscles were powdered in liquid nitrogen using a mortar and pestle. Extraction of proteins was completed at 4 °C in ice-cold lysis buffer containing either 200 mM octyl-β-d-glucopyranoside or 2% Triton X-100 in 100 mM Tris–HCl (pH 7.4), 150 mM NaCl, 2 mM PMSF, 1:200 dilution of Protease Cocktail Set III (Calbiochem, La Jolla, CA), 1 mM CaCl2, and 1 mM MgCl2 at 4 °C for 1 h. Supernatants were collected and protein concentrations were determined using Bradford assays. Equal amounts of extracted muscle proteins were separated on 8–12% SDS polyacrylamide gels at 40 mA for 50 min. Proteins were transferred to nitrocellulose membranes for 1 h at 100 V and membranes were stained with Ponceau S (Sigma) to ensure equal protein loading. Membranes were blocked in Tris-buffered saline (pH 7.8) containing 5% BSA and membranes were incubated overnight with the following antibodies at a concentration of 1:1000 unless otherwise indicated: ILK, phospho-AKT (1:2000), AKT, phospho-mTOR, mTOR, phospho-p70S6K (1:500), p70S6K (1:500), phospho-BAD, BAD, phospho-ERK, ERK, phospho-p38, and p38. Anti-rabbit or anti-goat antibodies coupled with horseradish peroxidase were used to detect primary antibodies (1:2000). Immuno-reactive bands were detected using an enhanced chemiluminescence kit (ECL) (Amersham Pharmacia Biotech, Piscataway, NJ). ImageQuant software (Amersham Pharmacia Biotech) was used to measure the intensities of immunoreactive bands on scanned films and intensity of the 43 kDa actin band on digital photos of Ponceau S-stained blots.
2.7. Apoptosis
Apoptotic nuclei were detected by using the DEAD END™ Fluorometric TUNEL System (Promega, Madison, WI). Briefly, hindlimb was frozen in liquid nitrogen cooled isopentane and stored at −80 °C until needed. Using a CM1900 series cryostat (Leica Microsystems, Germany), 10 μm-thick sections were cut and put on microscope slides. Multiple sections at least 50 μm apart were obtained from each animal. Sections were treated as instructed according to the DEAD END™ Fluorometric TUNEL System. Apoptotic nuclei were scored by counting the total number of TUNEL+ nuclei per 50 40× fields.
2.8. Statistics
All averaged data are presented as the means ± SE. Comparisons of signaling and TUNEL staining between groups were performed by one-way ANOVA, followed by Tukey’s post hoc analysis (SigmaStat). Differences were considered significant at P<0.05.
3. Results
3.1. Integrin linked kinase (ILK) associates with the α7 integrin and is increased in the skeletal muscle of mice with elevated α7 integrin
Integrins have a dual functional capacity in cell adhesion and cell signaling [21]. Therefore, increasing α7β1 integrin in skeletal muscle may result in changes in cell signaling that could ameliorate the development of dystrophic muscle. To investigate if altered cell signaling played a role in the rescue of mdx/utr−/− mice we examined the expression and activity of candidate downstream integrin signaling proteins in α7BX2-mdx/utr−/− mice.
Integrin linked kinase (ILK) is a serine/threonine protein kinase that interacts with β1 integrin cytoplasmic domains and mediates protein–protein interactions, cell signaling, and adhesion [22,23]. ILK protein and activity were analyzed in 5 week-old mice with enhanced levels of the α7β1 integrin. ILK activity was measured by in vitro assay using MBP as a substrate. Western blot analysis revealed significant increases in ILK protein in hindlimb of mdx (2-fold), mdx/utr−/− (2.5-fold) and α7BX2-mdx/utr−/− (2.9-fold) mice compared to wild type animals (Fig. 1A). ILK activity was increased in mdx/utr−/− (2.0-fold) and α7BX2-mdx/utr−/− (2.5-fold) mice compared to wild type animals (Fig. 1A). ILK activity and expression were elevated but not statistically higher in α7BX2-mdx/utr−/− compared to mdx-utr−/− mice. Interestingly, the amount of ILK protein was increased 2.0-fold in mdx mice yet ILK activity remained unchanged. The coincident elevations in ILK and ILK activity in mice overexpressing the α7 integrin prompted us to examine whether ILK specifically associates with the α7BX2 isoform. We evaluated this interaction using C2C12 myoblasts stably transfected with α7BX2 or a mutant form of α7BX2 (tyrosine to phenylalanine mutation). In the absence of activation of the integrin with anti-α7 antibody (O26), ILK did not associate with the integrin. However, stimulation with anti-α7 antibody resulted in the rapid (5 s) phosphorylation of the α7BX2 integrin chain (data not shown) and its association with ILK, an interaction that remained stable for at least 1 min (Fig. 1B). Interestingly, both phosphorylation and the association with ILK were suppressed in the absence of this critical tyrosine residue.
Fig. 1.
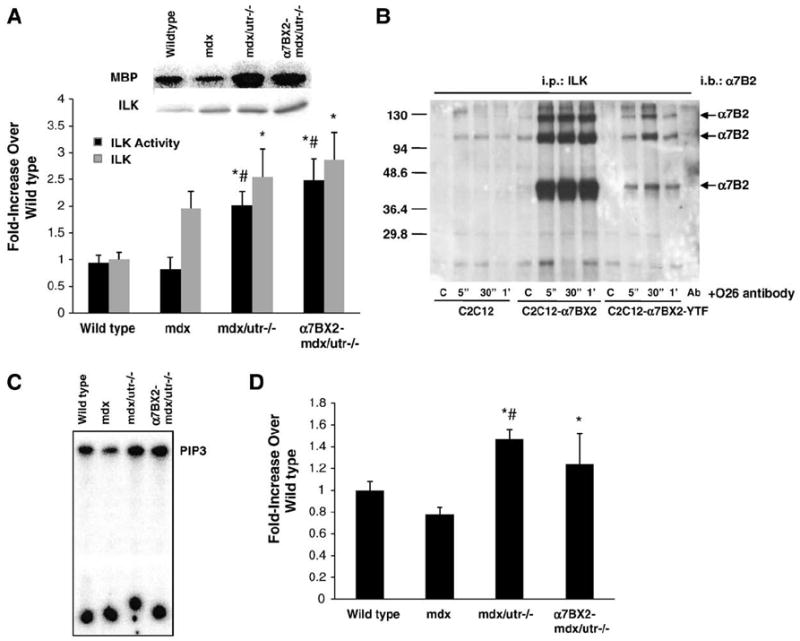
Integrin-linked kinase (ILK) and PI3 kinase (PI3K) activities are increased in transgenic mice overexpressing the α7BX2 integrin. (A) ILK protein and activity were assessed using MBP as a substrate in muscle from 5 week-old wild type, mdx, mdx/utr−/−, and α7BX2-mdx/utr−/− mice (n=3–6/grp; mdx group n=3). ILK activity was increased in both mdx/utr−/− and α7BX2-mdx/utr−/− mice with corresponding increases in ILK protein. (B) ILK association with α7BX2 was examined in vitro in C2C12 cells transfected with α7BX2 or α7BX2 containing a cytoplasmic Tyr to Phe substitution (C2C12-α7BX2-YTF). ILK was not bound to α7BX2 in the absence of stimulation with anti-α7 antibody (O26) in control cells (C), but quickly associates (within 5 s) in its presence. Ab=antibody only lane. (C) Total PI3K activity was evaluated in 5 week-old wild type, mdx, mdx/utr−/−, and α7BX2-mdx/utr−/− mice. (D) PI3K activity was increased in both mdx/utr−/− and α7BX2-mdx/utr−/− mice (n=5–7/grp). *=vs. wild type and #=vs. mdx (P<0.05).
Phosphoinositide-3-OH kinase (PI3K) appears to be important for activation of downstream signaling by ILK [24]. PI3K was not activated in mdx, yet increased in both mdx/utr−/− (47%) and α7BX2-mdx/utr−/− (24%) compared to wild type, consistent with our results for ILK activity in these mice (Fig. 1C and D).
3.2. The AKT signaling pathway is enhanced by increased α7β1 integrin
The active form of AKT is phosphorylated and acts downstream from ILK in a pathway that inhibits apoptosis and promotes cell survival [24,25] via the anti-apoptotic protein BAD [26]. We have previously reported resistance to apoptosis in C2C12 mouse myoblasts overexpressing the α7 integrin [12]. To further determine if integrin-mediated anti-apoptotic signaling is enhanced in the transgenic animals, the amount of phosphorylated and total AKT levels were measured in extracts of hindlimb muscle from wild type, mdx, mdx/utr−/− and α7BX2-mdx/utr−/− mice. Total AKT increased 2.4-fold, 3.9-fold, and 3.4-fold in mdx, mdx/utr−/−, and α7BX2-mdx/utr−/− mice, respectively, compared to wild type animals (Fig. 2A). A 6.3-fold increase in phospho-AKT (Ser 473) was detected in mdx/utr−/− mice compared to wild type animals and a 10-fold increase in phospho-AKT (Ser 473) was found in α7BX2-mdx/utr−/− mice (Fig. 2A). No differences were observed in the proportions of AKT that were active in wild type, mdx, or mdx/utr−/− mice. In contrast, α7BX2-mdx/utr−/− mice have a significant increase in the proportion of active AKT compared to all the groups (Fig. 2B).
Fig. 2.
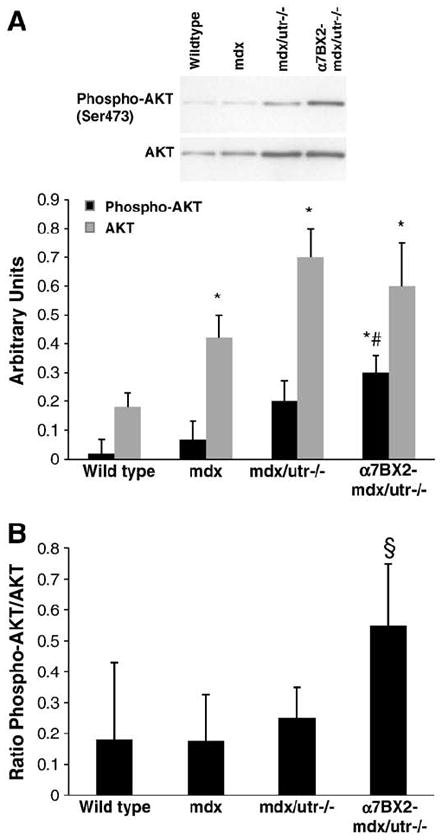
Total AKT and phosphorylated AKT are increased in α7BX2-mdx/utr−/− mice. (A) AKT phosphorylation (Ser473) and expression were examined in 5 week-old wild type, mdx, mdx/utr−/−, and α7BX2-mdx/utr−/− mice (n=4–6/grp) and reported as a percentage of an internal control sample. AKT expression is increased in all groups compared to wild type, however, AKT phosphorylation was only significantly increased in α7BX2-mdx/utr−/− mice. (B) The ratio of phosphorylated to total expression of AKT was measured. The α7BX2-mdx/utr−/− mice have higher proportions of activated AKT compared to all other groups. *=vs. wild type, #=vs. mdx, and §=vs. all groups (P<0.05).
3.3. Increased α7β1 integrin results in reduced apoptosis in α7BX2-mdx/utr−/− mice
Active AKT is pivotal in regulating proteins involved in cell survival, growth, and muscle integrity, including the Bcl-2/Bcl-XL-associated death promoter (BAD). Phosphorylation of pro-apoptotic BAD on Ser136 or 112 results in loss of the ability of BAD to heterodimerize with the survival proteins Bcl-XL or Bcl-2. Phosphorylated BAD then associates with the eukaryotic regulatory binding protein 14-3-3 and remains sequestered in the cytoplasm. This results in the loss of ability of BAD to initiate apoptosis and thereby promotes cell survival. Phosphorylation of BAD on Ser136, a preferred substrate for AKT, was not detected in our samples using the methods described (data not shown), despite confirmation of BAD expression which was unaltered. On the other hand, phosphorylation of BAD on Ser112 was increased 2.8-fold, 3.0-fold, and 5-fold in mdx, mdx/utr−/−, and α7BX2-mdx/utr−/− mice, respectively, compared to wild type animals (Fig. 3A). Similar to results for ILK activity and expression, BAD Ser112 phosphorylation was elevated but not significantly higher in α7BX2-mdx/utr−/− compared to mdx/utr−/− mice. ERK is an upstream regulator of BAD phosphorylation on Ser112. Phosphorylation of ERK was elevated in all dystrophic mice and correlated with levels of BAD Ser112 phosphorylation (Fig. 3B). No changes in ERK protein were observed between groups.
Fig. 3.
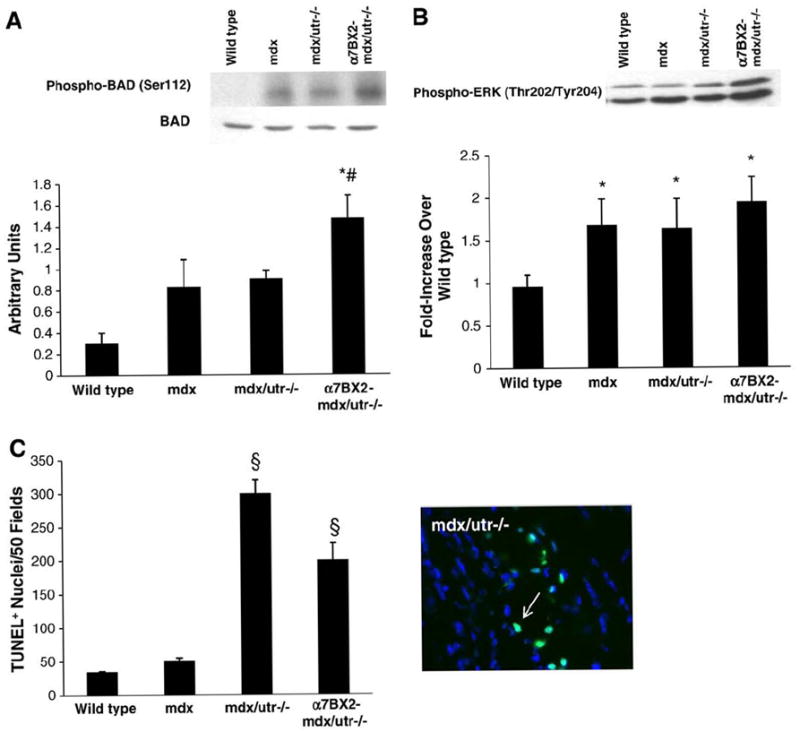
Phosphorylation of BAD and ERK is increased and apoptosis is inhibited in α7BX2-mdx/utr−/− mice. (A) BAD phosphorylation (Ser112) was examined in 5 week-old wild type, mdx, mdx/utr−/−, and α7BX2-mdx/utr−/− mice (n=4–8/grp) and reported as a percentage of an internal control sample. BAD phosphorylation was significantly increased only in α7BX2-mdx/utr−/− compared to wild type mice. Total BAD levels were unchanged (not shown). (B) ERK is an upstream regulator of BAD Ser112 phosphorylation. ERK phosphorylation was increased in all groups (n=4–8/grp). (C) TUNEL+ nuclei were counted as an indication of apoptosis in skeletal muscle of 5 week-old wild type, mdx, mdx/utr−/−, and α7BX2-mdx/utr−/− mice (n=4–7/grp). A representative image of TUNEL staining is shown. Apoptosis was significantly increased in mdx/utr−/− mice compared to wild type and mdx, and apoptosis was suppressed in dystrophic mice expressing the α7 integrin transgene. *=vs. wild type, #=vs. mdx, and §=vs. all groups (P<0.05).
The increase in BAD phosphorylation prompted us to examine apoptosis in these mice. The number of TUNEL+ apoptotic nuclei was not increased in mdx mice but was markedly increased in mdx/utr−/− mice compared to wild type mice (Fig. 3C). Although still elevated compared to wild type, the number of TUNEL+ nuclei was attenuated in α7BX2-mdx/utr−/− mice compared to mdx/utr−/−. Thus, the increased amount of α7β1 integrin may maintain muscle integrity in α7BX2-mdx/utr−/− mice in part by preventing apoptosis.
3.4. p70S6K is enhanced by increased α7β1 integrin
The serine kinase activity of AKT also promotes fiber growth by activation of mTOR and p70S6K [27]. We previously reported a 4-fold increase in hypertrophic fibers in α7BX2-mdx/utr−/− compared to mdx/utr−/− mice in both soleus and the tibialis anterior muscles [28]. While total expression of mTOR and p70S6K were not altered in any group, mTOR phosphorylation was increased (57%) in mdx/utr−/− mice compared to wild type (Fig. 4A). mTOR phosphorylation was not altered in α7BX2-mdx/utr−/− mice compared to any other group. Strikingly, p70S6K phosphorylation was increased 6.7-fold, 11.7-fold, and 18-fold in mdx, mdx/utr−/−, and α7BX2-mdx/utr−/− mice, respectively, compared to wild type animals. p70S6K phosphorylation was significantly higher in α7BX-mdx/utr−/− compared to mdx/utr−/− mice (Fig. 4B). Together, these data suggest that the AKT pathway is enhanced in severely dystrophic mdx/utr−/− mice and that it is further activated by increased integrin in α7BX2-mdx/utr−/− mice. This likely contributes to the formation of hypertrophic fibers previously observed in these animals.
Fig. 4.
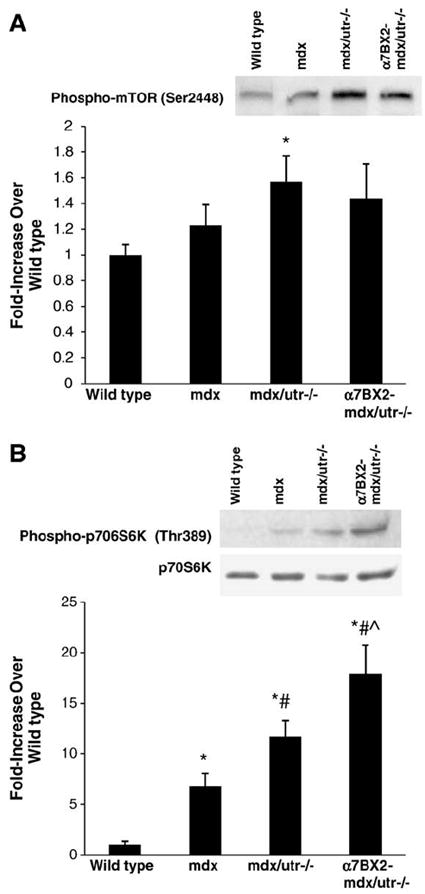
Signaling associated with muscle hypertrophy is activated in α7BX2-mdx/utr−/− mice. mTOR and p70S6K phosphorylation were measured in muscle from 5 week-old wild type, mdx, mdx/utr−/−, and α7BX2-mdx/utr−/− mice (4–6/grp). (A) mTOR phosphorylation was significantly increased only in α7BX2-mdx/utr−/− muscle compared to wild type mice. (B) p70S6K phosphorylation was significantly increased in all groups compared to wild type muscle, and was higher in α7BX2-mdx/utr−/− mice compared to mdx/utr−/− mice that did not express the transgene. *=vs. wild type, #=vs. mdx, and ˆ=vs. mdx/utr−/− (P<0.05).
3.5. Dystrophy decreases phosphorylation of p38
Reductions in p38 phosphorylation have been reported in diaphragm muscle of mdx mice [29]. In this study, p38 levels were not altered (data not shown), but p38 phosphorylation was significantly decreased in hindlimb muscle to the same extent in all dystrophic mice examined (Fig. 5). The significance of p38 suppression in the dystrophic mice is not clear, but the level of α7 integrin in muscle does not seem to influence p38 activity.
Fig. 5.
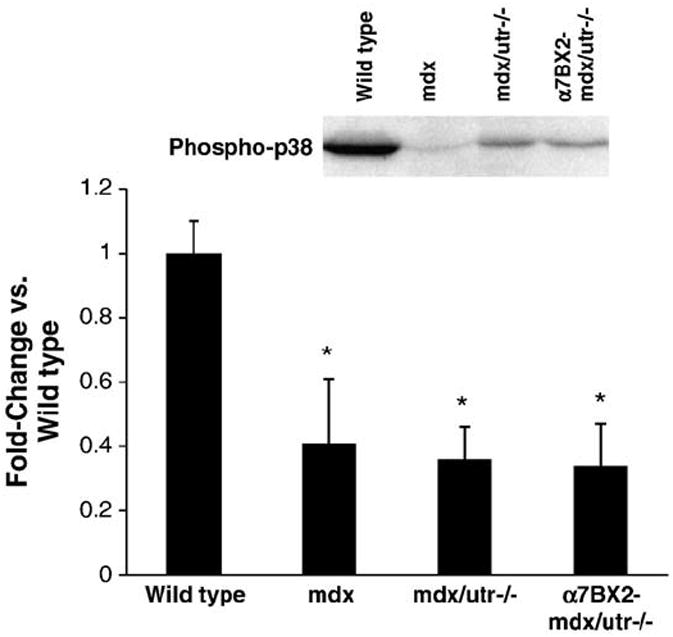
p38 phosphorylation is reduced in dystrophic mice and increasing α7 integrin did not restore activation. p38 phosphorylation was measured in muscle from 5 week-old wild type, mdx, mdx/utr−/−, and α7BX2-mdx/utr−/− mice (n=4–6). p38 phosphorylation was decreased to the same extent in all mice evaluated. *=vs. wild type (P<0.05).
4. Discussion
Duchenne muscular dystrophy (DMD) is an X-linked human disease affecting 1/3500 newborn boys. Mutations in the dystrophin gene result in a lack of the dystrophin protein in both patients and mdx mice. The loss of dystrophin leads to a breakdown of a transmembrane system linking laminin in the extracellular matrix to the actin cytoskeleton resulting in muscle disease. Attempts to treat muscular dystrophy have involved both direct gene replacement and gene repair [30-32]. Recently, several additional approaches have emerged based on the study of proteins with complementary functions to the dystrophin glycoprotein complex [11,33]. The α7β1 integrin is a laminin receptor on the surface of muscle cells that interacts with the actin cytoskeleton and has complementary functions to the dystrophin glycoprotein complex. Enhanced expression of the α7β1 integrin in mice lacking dystrophin and utrophin results in a three-fold increase in life span and partial rescue of muscle pathology in mice that would normally develop severe muscle disease and die [11]. The mechanisms by which enhanced levels of the α7β1 integrin rescued these mice are unclear and have been the focus of this investigation.
The α7 integrin and ILK are localized to similar structures in muscle, including costameres and myotendinous junctions [15,34]. Previous groups reported binding of ILK with the β1 integrin subunit and this association results in the activation of AKT in vitro [24,35,36]. Loss of either the α7 integrin or ILK independently results in mild muscular dystrophies characterized by progressive loss of integrity at myotendinous and neuromuscular junctions that worsens with age or mechanical stress [15,37]. In this paper, we show that the amount of ILK protein and activity are increased in both mdx/utr−/− and α7BX2-mdx/utr−/− compared to wild type and mdx mice. Although ILK activity is not significantly higher in α7BX2-mdx/utr−/− compared to mdx/utr−/−, an elevation in activation is observed. Interestingly, association of ILK with the α7BX2 integrin subunit upon stimulation with an anti-α7 integrin antibody was observed in vitro. Thus, engagement of the α7 integrin in vivo may facilitate ILK binding and allow ILK to stably interact with proteins at focal adhesions within the sarcolemma. Although we did not evaluate binding between α7 and ILK in vivo, it is tempting to speculate that fibrosis associated with dystrophy prevents the α7 integrin–laminin interaction necessary for α7 integrin activation, ILK binding to the α7 integrin, and concomitant initiation of anti-apoptotic and regenerative signaling. We have previously demonstrated the ability of the α7 integrin transgene to decrease inflammation and improve the microenvironment in α7BX2-mdx/utr−/− mice [11,28]. These beneficial changes may facilitate greater adhesion of the α7 integrin with the laminin matrix and subsequently activate the ILK survival pathway (Fig. 6). In addition, elimination of α7 integrin in mdx mice (α7/mdx−/−) results in severe muscle disease that may in part result from loss of integrin-mediated ILK activation [38].
Fig. 6.
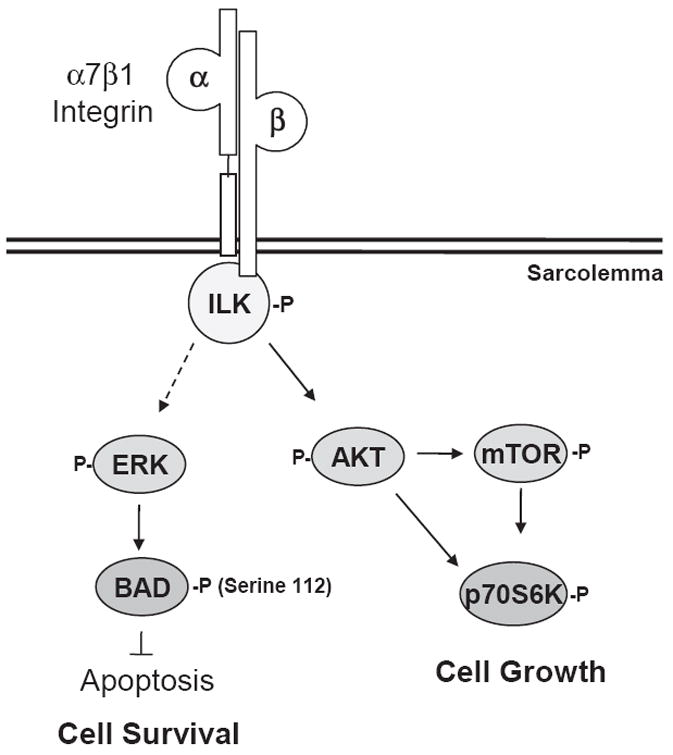
Signaling pathways by which the α7 integrin decreases pathology in dystrophic mice. The α7 integrin–ILK complex is activated in response to extracellular matrix attachment, resulting in phosphorylation of AKT and p70S6K in an mTOR-dependent or independent manner, resulting in increased muscle growth. The α7–ILK complex may also stimulate phosphorylation of BAD on Ser112 (via ERK), suppressing the pro-apoptotic actions of BAD and enhancing cell survival.
An increase in the level of apoptosis has been reported in mdx mice, suggesting programmed cell death plays a role in the pathology underlying muscular dystrophy [14]. Since the α7β1 integrin has been implicated in muscle cell survival [10-13,39], increased transgenic expression of α7β1 integrin may inhibit apoptosis in mice that would normally develop severe muscular dystrophy. We have previously reported that overexpression of the α7 integrin transgene can prevent staurosporine-induced apoptosis in C2C12 myoblasts in culture [12]. In this study, expression of the α7 integrin transgene did suppress apoptosis in α7BX2-mdx/utr−/− mice compared to mdx/utr−/− mice. Inhibition of the pro-apoptotic BAD by AKT-mediated phosphorylation on Ser136 is important for limiting cell death, presumably by allowing association of BAD with 14-3-3 which sequesters it in the cytoplasm. We did not detect BAD phosphorylation at Ser136, but phosphorylation at Ser112 was enhanced in α7BX2-mdx/utr−/− mice, perhaps due to ERK which strongly influences phosphorylation at this site [40,41]. Since Ser112 phosphorylation is also important for inhibiting the pro-apoptotic function of BAD [26], it is likely this signaling event played a significant role in the suppression of apoptosis and protection from developing pathology in mice overexpressing the α7 integrin transgene. Further studies of α7 integrin-mediated regulation of the apoptotic signaling pathway in muscle are warranted.
In addition to promoting cell survival, AKT is a key regulator of the compensatory hypertrophy that is a hallmark of muscular dystrophy. AKT phosphorylation and expression are consistently activated in both diaphragm [42] and skeletal muscle [43,44] in mouse models of disease and in patients with muscular dystrophy. Moreover, conditional overexpression of muscle-specific AKT can enhance muscle mass and fiber cross-sectional area in mdx mice [45]. p70S6K, an initiator of protein translation, lies downstream within the AKT signaling pathway and is upregulated in mdx hindlimb [43] and diaphragm muscles [29]. Consistent with these findings, hypertrophy is increased in hindlimbs of α7BX2-mdx/utr−/− mice compared to both wild type and mdx/utr−/− mice [28]. In this study, we report a marked increase in p70S6K phosphorylation in severely dystrophic α7BX2-mdx/utr−/− mice that overexpress the α7 integrin transgene compared to mice that do not. Although mTOR phosphorylation was not elevated to the same extent in these mice, it is possible that AKT activates p70S6K independently of mTOR or perhaps p70S6K is directly activated by the integrin via a mechanoregulatory mechanism as a result of enhanced adhesion to the surrounding basal lamina. Amino acid uptake and protein synthesis can occur in cultured myotubes in the complete absence of systemic factors as a result of stretch, yet the mechanism responsible for this hypertrophic effect is still not known [46]. Our data suggest that p70S6K may be activated as a result of increased α7 integrin and this event contributes to hypertrophic responses necessary to combat loss of myofibrillar proteins and disease progression.
p38 is a member of the mitogen-activated protein kinase family that is ubiquitously expressed in a variety of tissues and activated by stressors, such as proinflammatory cytokines, osmotic shock, shear stress, and stretch [47,48]. Although the precise role of p38 in skeletal muscle is not clear, p38 has been suggested to positively influence myogenic cell differentiation [48] and mitochondrial biosynthesis [49,50], and it plays a protective role against apoptosis [51] and Foxo3a-induced muscle atrophy [52]. In this study, we observe a consistent decline in p38 phosphorylation in all models of muscular dystrophy, including mice overexpressing the α7 integrin. These results are similar to several studies that report significant decreases in p38 phosphorylation in dystrophic diaphragm and hindlimb muscle [29,53,54]. We were surprised that increasing the amount of α7 integrin did not restore p38 phosphorylation levels given our previous data showed elevated activation of p38 3 h following exercise in mice overexpressing the α7 integrin in wild type mice [55].
This study reveals for the first time that the α7β1 integrin plays an important role in modulating the activation of several signaling molecules important for promoting muscle cell survival and hypertrophy in mice with severe muscular dystrophy. Elevated expression and activation of the α7β1 integrin or molecules in its pathway may provide novel avenues for treatment of neuromuscular diseases.
Acknowledgments
We thank Eric Chaney and James Mulligan for their technical assistance. This work was supported by grants from the NIH (RO1-AG014632) and Muscular Dystrophy Association (to SJK), Ellison Medical Foundation (to MDB), and NIH (R01AR053697) (to DJB).
References
- 1.Monaco AP, Neve RL, Colletti-Feener C, Bertelson CJ, Kurnit DM, Kunkel LM. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323:646–650. doi: 10.1038/323646a0. [DOI] [PubMed] [Google Scholar]
- 2.Sicinski P, Geng Y, Tyder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 3.Matsumura K, Campbell KP. Dystrophin–glycoprotein complex: its role in the molecular pathogenesis of muscular dystrophies. Muscle Nerve. 1994;17:2–15. doi: 10.1002/mus.880170103. [DOI] [PubMed] [Google Scholar]
- 4.Campbell KP. Three muscular dystrophies: loss of cytoskeleton–extracellular matrix linkage. Cell. 1995;80:675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- 5.Rybakova IN, Amann KJ, Ervasti JM. A new model for the interaction of dystorphin with F-actin. J Cell Biol. 1996;135:661–672. doi: 10.1083/jcb.135.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD, Campbell KP. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- 8.Deconinck AE, Potter AC, Tinsley JM, Wood SJ, Vater R, Young C, Metzinger L, Vincent A, Slater CR, Davies KE. Utrophin-dystrophin deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- 9.Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RW, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 10.Hodges BL, Hayashi YK, Nonaka I, Wang W, Arahata K, Kaufman SJ. Altered expression of the α7β1 integrin in human and murine muscular dystrophies. J Cell Sci. 1997;110:2873–2881. doi: 10.1242/jcs.110.22.2873. [DOI] [PubMed] [Google Scholar]
- 11.Burkin DJ, Wallace GQ, Nichol KJ, Kaufman DJ, Kaufman SJ. Enhanced expression of the α7β1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J Cell Biol. 2001;152:1207–1218. doi: 10.1083/jcb.152.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Burkin DJ, Kaufman SJ. Increasing α7β1-integrin promotes muscle cell proliferation, adhesion, and resistance to apoptosis without changing gene expression. Am J Physiol Cell Physiol. 2008;294:C627–C640. doi: 10.1152/ajpcell.00329.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gurpur P, Liu J, Burkin DJ, Kaufman SJ. Valproic acid activates the PI3K/Akt/mTOR pathway in muscle and ameliorates pathology in a mouse model of Duchenne muscular dystrophy. Am J Pathol. 2009;174:999–1008. doi: 10.2353/ajpath.2009.080537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tidball JG, Albrecht DE, Lokensgard BE, Spencer MJ. Apoptosis precedes necrosis of dystrophin-deficient mice. J Cell Sci. 1995;108:2197–2204. doi: 10.1242/jcs.108.6.2197. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Chang L, Brixius K, Wickstrom SA, Montanez E, Thievessen I, Schwander M, Muller U, Bloch W, Mayer U, Fassler R. Integrin-linked kinase stabilizes myotendinous junctions and protects muscle from stress-induced damage. J Cell Biol. 2008;180:1037–1049. doi: 10.1083/jcb.200707175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bendig G, Grimmler M, Huttner IG, Wessels G, Dahme T, Just S, Trano N, Katus HA, Fishman MC, Rottbauer W. Integrin-linked kinase, a novel component of the cardiac mechanical stretch sensor, controls contractility in the zebrafish heart. Genes Dev. 2006;20:2361–2372. doi: 10.1101/gad.1448306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White DE, Coutu P, Shi YF, Tardif JC, Nattel S, St Arnaud R, Dedhar S, Muller WJ. Targeted ablation of ILK from the murine heart results in dilated cardiomyopathy and spontaneous heart failure. Genes Dev. 2006;20:2355–2360. doi: 10.1101/gad.1458906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song WK, Wang W, Foster RF, Bielser DA, Kaufman SJ. H36-α7 is a novel integrin alpha chain that is developmentally regulated during skeletal myogenesis. J Cell Biol. 1992;117:643–667. doi: 10.1083/jcb.117.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burkin DJ, Gu M, Hodges BL, Campanelli JT, Kaufman SJ. A functional role for specific spliced variants of the alpha7 beta1 integrin in acetylcholine receptor clustering. J Cell Biol. 1998;143:1067–1075. doi: 10.1083/jcb.143.4.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song WK, Wang W, Sato H, Bielser DA, Kaufman SJ. Expression of α7 integrin cytoplasmic domains during skeletal muscle development: alternate forms, conformational change, and homologies with serine/threonine kinases and tyrosine phosphatases. J Cell Sci. 1993;106:1139–1152. doi: 10.1242/jcs.106.4.1139. [DOI] [PubMed] [Google Scholar]
- 21.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 22.Dedhar S. Cell–substrate interactions and signaling through ILK. Curr Opin Cell Biol. 2000;12:250–256. doi: 10.1016/s0955-0674(99)00083-6. [DOI] [PubMed] [Google Scholar]
- 23.Wu C, Dedhar S. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton ad signaling complexes. J Cell Biol. 2001;155:505–510. doi: 10.1083/jcb.200108077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B-AKT by the integrin-linked kinase. Proc Natl Acad Sci USA. 1998;95:11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langenbach KJ, Rando TA. Inhibition of dystroglycan binding to laminin disrupts the PI3K/AKT pathway and survival signaling in muscle cells. Muscle Nerve. 2002;26:644–653. doi: 10.1002/mus.10258. [DOI] [PubMed] [Google Scholar]
- 26.Chiang CW, Kanies C, Kim KW, Fang WB, Parkhurst C, Xie M, Henry T, Yang E. Protein phosphatase 2A dephosphorylation of phosphoserine 112 plays the gatekeeper role for BAD-mediated apoptosis. Mol Cell Biol. 2003;23:6350–6362. doi: 10.1128/MCB.23.18.6350-6362.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3:1014–1019. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- 28.Burkin DJ, Wallace GQ, Milner DJ, Chaney EJ, Mulligan JA, Kaufman SJ. Transgenic expression of α7β1 integrin maintains muscle integrity, increases regenerative capacity, promotes hypertrophy, and reduces cardiomyopathy in dystrophic mice. Am J Pathol. 2005;166:253–263. doi: 10.1016/s0002-9440(10)62249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lang JM, Esser KA, Dupont-Versteegden EE. Altered activity of signaling pathways in diaphragm and tibialis anterior muscle of dystrophic mice. Exp Biol Med (Maywood) 2004;229:503–511. doi: 10.1177/153537020422900608. [DOI] [PubMed] [Google Scholar]
- 30.Incitti T, De Angelis FG, Cazzella V, Sthandier O, Pinnaro C, Legnini I, Bozzoni I. Exon skipping and Duchenne muscular dystrophy therapy: selection of the most active U1 snRNA antisense able to induce dystrophin exon 51 skipping. Mol Ther. 2010;18:1675–1682. doi: 10.1038/mt.2010.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koppanati BM, Li J, Wang B, Daood M, Zheng H, Xiao X, Watchko JF, Clemens PR. Improvement of the mdx mouse dystrophic phenotype by systemic in utero AAV8 delivery of a minidystrophin gene. Gene Ther. 2010;17:1355–1362. doi: 10.1038/gt.2010.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Odom GL, Banks GB, Shultz BR, Gregorevic P, Chamberlain JS. Preclinical studies for gene therapy of Duchenne muscular dystrophy. J Child Neurol. 2010;25:1149–1157. doi: 10.1177/0883073810371006. [DOI] [PubMed] [Google Scholar]
- 33.Odom GL, Gregorevic P, Allen JM, Finn E, Chamberlain JS. Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin deficient mice. Mol Ther. 2008;16:1539–1545. doi: 10.1038/mt.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paul AC, Sheard PW, Kaufman SJ, Duxson MJ. Localization of alpha 7 integrins and dystrophin suggest potential for both lateral and longitudinal transmission of tension in large mammalian muscles. Cell Tissue Res. 2002;308:255–265. doi: 10.1007/s00441-002-0526-y. [DOI] [PubMed] [Google Scholar]
- 35.Persad S, Attwell S, Gray V, Mawji N, Deng JT, Leung D, Yan J, Sanghera J, Walsh MP, Dedhar S. Regulation of protein kinase B/Akt-Serine 473 phosphorylation by integrin-linked kinase. J Biol Chem. 2001;276:27462–27469. doi: 10.1074/jbc.M102940200. [DOI] [PubMed] [Google Scholar]
- 36.Troussard AA, Mawji NM, Ong C, Mui A, St Arnaud R, Dedhar S. Conditional knock-out of integrin-linked kinase (ILK) demonstrates an essential role in PKB/Akt activation. J Biol Chem. 2003;278:22374–22378. doi: 10.1074/jbc.M303083200. [DOI] [PubMed] [Google Scholar]
- 37.Mayer U, Saher G, Fassler R, Bornemann A, Echtermeyer F, von der Mark H, Miosge N, Poschl E, von der Mark K. Absence of integrin alpha 7 causes a novel form of muscular dystrophy. Nat Genet. 1997;17:318–323. doi: 10.1038/ng1197-318. [DOI] [PubMed] [Google Scholar]
- 38.Rooney JE, Welser JV, Dechert MA, Flintoff-Dye NL, Kaufman SJ, Burkin DJ. Severe muscular dystrophy in mice that lack dystrophin and alpha7 integrin. J Cell Sci. 2006;119:2185–2195. doi: 10.1242/jcs.02952. [DOI] [PubMed] [Google Scholar]
- 39.Vachon PH, Xu H, Liu L, Loechel F, Hayashi Y, Arahata K, Reed JC, Wewer UM, Engvall E. Integrins (alpha7beta 1) in muscle function and survival. Disrupted expression in merosin-deficient congenital muscular dystrophy. J Clin Invest. 1997;100:1870–1881. doi: 10.1172/JCI119716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alejandro EU, Johnson JD. Inhibitin of Raf-1 alters multiple downstream pathways to induce pancreatic beta-cell apoptosis. J Biol Chem. 2008;283:2407–2417. doi: 10.1074/jbc.M703612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mabuchi S, Ohmichi M, Kimura A, Hisamoto K, Hayakawa J, Nishio Y, Adachi K, Takahashi K, Arimoto-Ishida E, Nakatsuji Y, Tasaka K, Murata Y. Inhibition of phosphorylation of BAD and Raf-1 by Akt sensitizes human ovarian cancer cells to paclitaxel. J Biol Chem. 2002;227:33490–33500. doi: 10.1074/jbc.M204042200. [DOI] [PubMed] [Google Scholar]
- 42.Dogra C, Changotra H, Wergedal JE, Kumar A. Regulation of phosphatidylinositol 3-kinase (PI3K)/Akt and nuclear factor-kappa B signaling pathways in dystrophin-deficient skeletal muscle in response to mechanical stretch. J Cell Physiol. 2006;208:575–585. doi: 10.1002/jcp.20696. [DOI] [PubMed] [Google Scholar]
- 43.Peter AK, Crosbie RH. Hypertrophic response of Duchenne and limb-girdle muscular dystrophies is associated with activation of Akt pathway. Exp Cell Res. 2006;312:2580–2591. doi: 10.1016/j.yexcr.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 44.Xiong Y, Zhou Y, Jarrett HW. Dystrophin glycoprotein complex-associated Gbetagamma subunits activate phosphatidylinositol-3-kinase/Akt signaling in skeletal muscle in a laminin-dependent manner. J Cell Physiol. 2009;219:402–414. doi: 10.1002/jcp.21684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peter AK, Ko CY, Kim MH, Hsu N, Ouchi N, Rhie S, Izumiya Y, Zeng L, Walsh K, Crosbie RH. Myogenic Akt signaling upregulates the utrophin–glycoprotein complex and promotes sarcolemma stability in muscular dystrophy. Hum Mol Genet. 2009;18:318–327. doi: 10.1093/hmg/ddn358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vandenburgh H, Kaufman S. In vitro model for stretch-induced hypertrophy of skeletal muscle. Science. 1979;203:265–268. doi: 10.1126/science.569901. [DOI] [PubMed] [Google Scholar]
- 47.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 48.Boppart MD, Hirshman MF, Sakamoto K, Fielding RA, Goodyear LJ. Static stretch increases c-Jun NH2-terminal kinase activity and p38 phosphorylation in rat skeletal muscle. Am J Physiol Cell Physiol. 2001;280:C352–C358. doi: 10.1152/ajpcell.2001.280.2.C352. [DOI] [PubMed] [Google Scholar]
- 49.Zetser A, Gredinger E, Bengal E. p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. J Biol Chem. 1999;274:5193–5200. doi: 10.1074/jbc.274.8.5193. [DOI] [PubMed] [Google Scholar]
- 50.Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB, Williams RS, Yan Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem. 2005;280:19587–19593. doi: 10.1074/jbc.M408862200. [DOI] [PubMed] [Google Scholar]
- 51.Pogozelski AR, Geng T, Li P, Yin X, Lira VA, Zhang M, Chi JT, Yan Z. p38gamma mitogen-activated protein kinase in as key regulator in skeletal muscle metabolic adaptation in mice. PLoS ONE. 2009;4:e7934. doi: 10.1371/journal.pone.0007934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim MJ, Choi SY, Park IC, Hwang SG, Kim C, Choi YH, Kim H, Lee KH, Lee SJ. Opposing roles of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase in the cellular response to ionizing radiation in human cervical cancer cells. Mol Cancer Res. 2008;6:1718–1731. doi: 10.1158/1541-7786.MCR-08-0032. [DOI] [PubMed] [Google Scholar]
- 53.Hnia K, Hugon G, Rivier F, Masmoudi A, Mercier J, Mornet D. Modulation of p38 mitogen-activated protein kinase cascade and metalloproteinase activity in diaphragm muscle in response to free radical scavenger administration in dystrophin-deficient mdx mice. Am J Pathol. 2007;170:633–643. doi: 10.2353/ajpath.2007.060344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feron M, Guevel L, Rouger K, Dubreil L, Arnaud M-C, Ledevin M, Megeney LA, Cherel Y, Sakanyan V. PTEN contributes to profound PI3K/Akt signaling pathway deregulation in dystrophin-deficient dog muscle. Am J Pathol. 2009;174:1459–1470. doi: 10.2353/ajpath.2009.080460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boppart MD, Burkin DJ, Kaufman SJ. α7β1-Integrin regulates mechanotransduction and prevents skeletal muscle injury. Am J Physiol Cell Physiol. 2006;290:1660–1665. doi: 10.1152/ajpcell.00317.2005. [DOI] [PubMed] [Google Scholar]