

Abstract
Introduction
A deletion polymorphism in glutathione S-transferase Mu-1 (GSTM1-null) has previously been implicated to play a role in rheumatoid arthritis (RA) risk and progression, although no prior investigations have examined its associations with anticitrullinated protein antibody (ACPA) positivity. The purpose of this study was to examine the associations of GSTM1-null with ACPA positivity in RA and to assess for evidence of interaction between GSTM1 and HLA-DRB1 shared epitope (SE).
Methods
Associations of GSTM1-null with ACPA positivity were examined separately in two RA cohorts, the Veterans Affairs Rheumatoid Arthritis (VARA) registry (n = 703) and the Study of New-Onset RA (SONORA; n = 610). Interactions were examined by calculating an attributable proportion (AP) due to interaction.
Results
A majority of patients in the VARA registry (76%) and SONORA (69%) were positive for ACPA with a similar frequency of GSTM1-null (53% and 52%, respectively) and HLA-DRB1 SE positivity (76% and 71%, respectively). The parameter of patients who had ever smoked was more common in the VARA registry (80%) than in SONORA (65%). GSTM1-null was significantly associated with ACPA positivity in the VARA registry (odds ratio (OR), 1.45; 95% confidence interval (CI), 1.02 to 2.05), but not in SONORA (OR, 1.00; 95% CI, 0.71 to 1.42). There were significant additive interactions between GSTM1 and HLA-DRB1 SE in the VARA registry (AP, 0.49; 95% CI, 0.21 to 0.77; P < 0.001) in ACPA positivity, an interaction replicated in SONORA (AP, 0.38; 95% CI, 0.00 to 0.76; P = 0.050).
Conclusions
This study is the first to show that the GSTM1-null genotype, a common genetic variant, exerts significant additive interaction with HLA-DRB1 SE on the risk of ACPA positivity in RA. Since GSTM1 has known antioxidant functions, these data suggest that oxidative stress may be important in the development of RA-specific autoimmunity in genetically susceptible individuals.
Introduction
The human leukocyte antigen (HLA) region accounts for approximately one half of the genetic risk of rheumatoid arthritis (RA). This risk is attributable to alleles encoding a conserved amino acid sequence in the third hypervariable region of the DRB1 chain (commonly referred to as the shared epitope [SE]) [1]. Recent efforts have examined the importance of interactions of SE with other genetic and environmental factors in RA risk and progression. Most notably, studies have yielded evidence of significant interactions between SE and cigarette smoking in the development of anticitrullinated protein antibody (ACPA)-positive RA [2,3], although the precise mechanisms underpinning this interaction are not understood.
Genetic and environmental factors that mediate oxidative stress, including cigarette smoking, are postulated to play a central role in the pathogenesis of autoimmune disorders including RA. While oxidative stress represents a form of host defense, it can also result in tissue damage. Oxidative modification of proteins and other biologic molecules leads to the expression of neoantigens, a possible first step in the development of autoimmunity, which may herald the future onset of clinically relevant autoimmune disease [4]. Antioxidants, which mitigate tissue damage caused by reactive oxygen species, may serve important protective functions in RA. While not all studies have identified a similar protective effect [4,5], the dietary intake of small-molecule antioxidants has been reported to be inversely associated with RA risk [6-9]. Additionally, low circulating levels of antioxidants have been reported to portend the onset of RA [10].
In addition to the effects of exogenous antioxidants, oxidation is also regulated by several enzymes, including glutathione S-transferase (GST). A ubiquitous cytosolic protein, GST catalyzes the conjugation of glutathione to a variety of substrates, including reactive oxygen species and other toxins, facilitating their elimination. Four classes of GST have been identified: α, μ, π and θ. Approximately one half of all individuals of European ancestry are homozygous for a deletion at the GST Mu-1 (GSTM1) locus (GSTM1-null) [11] located on chromosome 1 (1p13.1).
The GSTM1-null genotype has been associated with an increased risk of RA and in most [12-14] but not all [15] case control studies. In addition to being implicated as a potential risk factor in RA, the GSTM1-null genotype is associated with higher levels of oxidative stress [16] and has been reported to be a risk factor for other smoking-related inflammatory diseases, including asthma, emphysema and atherosclerosis [17-21]. However, there have been no studies examining associations of GSTM1 genotypes with ACPA expression in patients with RA. This represents an important knowledge gap, since these antibodies are disease-specific, have significant prognostic and pathogenic significance and are increasingly recognized to characterize a unique subset of patients with RA [22,23]. In the present study, we have evaluated potential gene-gene interactions by exploring the GSTM1-null genotype as a risk factor for ACPA positivity in RA, providing evidence of an interaction with HLA-DRB1 shared epitope (SE)-containing alleles.
Materials and methods
Study subjects
All study subjects satisfied the American College of Rheumatology (ACR) criteria for RA [24] and were from two U.S. cohorts: the Veterans Affairs Rheumatoid Arthritis (VARA) registry [25] and the Study of New-Onset Rheumatoid Arthritis (SONORA) [26]. To limit population heterogeneity, analyses were limited to individuals self-reporting Caucasian race for whom banked samples and HLA-DRB1 data were available.
VARA is a multicenter registry with sites at nine VA medical centers in Brooklyn, NY; Dallas, TX; Denver, CO; Iowa City, IA; Jackson, MS; Omaha, NE; Portland, OR; Salt Lake City, UT; and Washington, DC. The registry has Institutional Review Board approval at each site, and patients provided informed written consent. Patients are eligible if they are U.S. Department of Veterans Affairs (VA) beneficiaries. SONORA includes patients with recent-onset RA enrolled within 12 months of diagnosis as part of a 5-year prospective follow-up study [26]. SONORA patients were recruited from 98 rheumatology practices in the U.S. and Canada, and all participants provided informed written consent. Variables abstracted from the corresponding data sets included age, gender and smoking status (never, former, or current). Smoking status in both cohorts was obtained using questionnaires reflecting exposure at the time of enrollment. Quantitative measures of smoking (pack-years and duration) were not routinely collected.
Anticitrullinated protein antibody (ACPA)
Serum ACPA (immunoglobulin G (IgG)) was measured using second-generation enzyme-linked immunosorbent assays (ELISAs) in VARA (Diastat, Axis-Shield Diagnostics Ltd., Dundee, Scotland, UK; positive ≥5 U/ml) and SONORA (Inova Diagnostics, San Diego, CA, USA; positive ≥20 U/ml) using serum samples collected at enrollment.
Determination of GSTM1 genotype
Primers "G2" and "G3" from a study by Brockmöller et al. [27] were used to amplify exons 3 through 5 of the GSTM1 gene using genomic DNA that was prepared from whole blood. These primers produce a 650-bp amplified fragment in individuals carrying at least one functional GSTM1 allele. This band is absent in GSTM1-null individuals because this mutation deletes exons 4 and 5. A 195-bp fragment of exon 7 of the CYP1a1 gene was used as an internal positive control for sample quality and polymerase chain reaction (PCR) using the primers described by Shields et al. [28]. Amplified products were resolved by electrophoresis through 1% agarose gels. Genotypes were scored independently by two investigators (KAG and KKB). On the basis of the empiric evidence for associations of this genotype across multiple conditions [17,29-31], individuals were categorized as GSTM1-null (homozygous for deletion) or GSTM1-present (one or two copies of functional allele). Individuals with an absent or faint CYP1a1 band (n = 8 from VARA and n = 25 from SONORA) were excluded from further analyses, leaving available data from 703 VARA individuals and 610 SONORA participants for analysis.
Determination of HLA-DRB1 genotypes
In VARA, HLA genotyping was performed using one of two approaches: DNA sequencing of exon 2 using the AlleleSEQR HLA-DRB1 reagent kit and protocol (Abbott Molecular, Abbott Park, IL, USA) or with a PCR-based, sequence-specific oligonucleotide probe system. In the second of these methods, a series of oligonucleotide probes corresponding to known sequence motifs in HLA-DRB1 were immobilized onto a backed nylon membrane to create a "linear array." Exon 2 of DRB1 was amplified with a set of upstream biotinylated PCR primers corresponding to known sequence motifs in the first variable region of DRB1 and a single downstream biotinylated PCR primer that amplifies all alleles. This method specifically amplified only DRB1 genes and avoided amplification of other DRB genes. The PCR product was denatured and hybridized to the 81-probe DRB1 linear array. Arrays were incubated with streptavidin-horseradish peroxidase followed by tetramethylbenzidine. Images were created by placing the arrays on a flatbed scanner, and probe intensities were measured with proprietary software. Preliminary genotypes were determined, and data were then imported into Sequence Compilation and Rearrangement Evaluation software (SCORE(tm), QIAGEN, Valencia, CA, USA) for final genotyping and data export. The following were considered to be DRB1 shared epitope (SE)-containing alleles: *0101, *0102, *0104, *0105, *0401, *0404, *0405, *0408, *0409, *1001, *1402 and *1406.
In SONORA, all participants were HLA-DRB1-typed as previously described [32] initially using the sequence-specific oligonucleotide probes (SSOP) low-resolution method [33]. Individuals with DRB1 *04 and *01 were subsequently tested using a medium-resolution panel to allow for four-digit DRB1 subtyping.
Statistical analyses
Associations of the GSTM1-null genotype with ACPA positivity were examined for each RA cohort using multivariate unconditional logistic regression. All analyses were adjusted for age (continuous variable) and gender to facilitate comparisons across the two divergent patient cohorts that differed based on these factors. Associations of HLA-DRB1 SE (positive vs. negative in addition to the number of SE alleles, 0 vs. 1 or 2) and smoking status modeled as ever versus never (and as current or former vs. never in a separate model) with ACPA positivity were also examined in separate analyses. Patients were then categorized on the basis of the presence of risk factor pairings (GSTM1-SE, GSTM1-smoking and smoking-SE), and associations of these risk factor assignments with outcomes were examined using similar regression techniques.
Gene-gene (GSTM1-SE) and gene-environment (GSTM1-smoking and SE-smoking) interactions were assessed with regard to ACPA positivity by examining for evidence of departure from additivity using the methods described by Rothman et al. [34]. Three-way interactions were not examined. Using this approach, an attributable proportion (AP) due to interaction (AP = 0 corresponds to no interaction, and AP = 1.0 corresponds to "complete" additive interaction) and 95% confidence intervals (CIs) were calculated, using the method of Hosmer and Lemeshow [35] to calculate the latter. The confidence interval serves as a statistical test of the interaction; if the null value (zero in this case) falls outside the interval, then the interaction is considered statistically significant. This method accounts for both the random variability and overlapping intervals in strata defined by the risk factors of interest [35]. Evidence of multiplicative interaction was examined by modeling the product term of interest. To optimize study power, assessments of interaction were limited to dichotomous variables (SE-positive vs. SE-negative, ever vs. never smoking) and to two-way interactions. All analyses were conducted using Stata version 10.0 software (Stata Corp., College Station, TX, USA).
Results
Patient characteristics
Patient characteristics are summarized in Table 1. Consistent with the demographic characteristics of VA beneficiaries nationally [36], VARA registry patients were predominantly men (93%) with a mean (± SD) age of 64 (± 11) years. In contrast, SONORA patients were younger, with a mean (SD) age of 53 (± 15) years, and were predominantly women (72%). A majority of patients were seropositive for ACPA (76% in VARA Registry and 69% in SONORA).
Table 1.
Characteristics of rheumatoid arthritis study patientsa
| Mean (SD) or number (%) | ||
|---|---|---|
| VARA (n = 703) |
SONORA (n = 610) |
|
| Sociodemographics | ||
| Age, yrb | 64 (11) | 53 (15) |
| Male genderb | 655 (93%) | 173 (28%) |
| ACPA-positiveb | 536 (76%) | 420 (69%) |
| RA risk factors | ||
| HLA-DRB1 SE-positive | 531 (76%) | 434 (71%) |
| One copy | 356 (51%) | 303 (50%) |
| Two copies | 175 (25%) | 131 (21%) |
| GSTM1-null | 372 (53%) | 315 (52%) |
| Smoking historyb | (n = 693) | (n = 610) |
| Never | 140 (20%) | 213 (35%) |
| Former | 371 (54%) | 257 (42%) |
| Current | 182 (26%) | 141 (23%) |
aACPA, anticitrullinated protein antibody; GSTM1, glutathione S-transferase Mu-1; SE, shared epitope; SONORA, Study of New-Onset RA; VARA, Veterans Affairs Rheumatoid Arthritis Registry. bP < 0.05 for differences between VARA and SONORA.
Risk factor prevalence
The frequency of RA-related risk factors is shown in Table 1. The prevalence of at least one HLA-DRB1 SE-containing allele was similar in the VARA Registry (76%) and SONORA (71%) (P = NS). Approximately one half of patients (53% in the VARA Registry and 52% in SONORA) were GSTM1-null (P = NS), and a majority had a history of smoking, either current or former (80% in the VARA Registry and 65% in SONORA; P < 0.05).
Age- and gender-adjusted associations
Associations of GSTM1, smoking, and HLA-DRB1 status with ACPA positivity in the VARA registry and SONORA are summarized in Table 2. In reference to patients with at least one functional GSTM1 allele, GSTM1-null was associated with a significantly higher odds ratio (OR) of ACPA positivity in the VARA Registry (OR, 1.45; 95% CI, 1.02 to 2.05), but not in SONORA (OR, 1.00; 95% CI, 0.71 to 1.42). There was a significant dose-related association of HLA-DRB1 SE with ACPA positivity in both cohorts, with more than 10-fold greater odds of ACPA positivity for those with 2 SE alleles compared with those with no SE allele (Table 2). In both cohorts, there were nonsignificant trends suggesting associations of current (vs. never) smoking with ACPA positivity, an effect that appeared to be more striking in the VARA registry (OR, 1.68; 95% CI, 0.98 to 2.88) than in SONORA (OR, 1.23; 95% CI, 0.76 to 1.99) (Table 2). Age- and gender-adjusted associations of composite risk factors with ACPA positivity are summarized in Table 3.
Table 2.
Association of GSTM1-null, HLA-DRB1 shared epitope (SE) and smoking with ACPA positivity in rheumatoid arthritisa
| VARA (n = 703) | SONORA (n = 610) | |||||
|---|---|---|---|---|---|---|
| ACPA+ (%) |
OR (95% CI) | P value | ACPA+ (%) |
OR (95% CI) | P value | |
| GSTM1-present | 73 | Ref. | - | 69 | Ref. | - |
| GSTM1-null | 79 | 1.45 (1.02 to 2.05) | 0.039 | 69 | 1.00 (0.71 to 1.42) | 0.981 |
| Never smoking | 69 | Ref. | - | 69 | Ref. | - |
| Ever smoking | 78 | 1.48 (0.97 to 2.26) | 0.067 | 68 | 0.90 (0.62 to 1.30) | 0.574 |
| Former smoking | 77 | 1.41 (0.91 to 2.19) | 0.129 | 65 | 0.77 (0.52 to 1.14) | 0.193 |
| Current smoking | 81 | 1.68 (0.98 to 2.88) | 0.059 | 74 | 1.23 (0.76 to 1.99) | 0.407 |
| SE-negative | 53 | Ref. | - | 54 | Ref. | - |
| SE-positive (one or two alleles) | 84 | 4.36 (2.98 to 6.37) | < 0.001 | 75 | 2.56 (1.77 to 3.70) | < 0.001 |
| SE-positive (one allele) | 79 | 3.23 (2.17 to 4.81) | < 0.001 | 68 | 1.77 (1.21 to 2.60) | 0.003 |
| SE-positive (two alleles) | 93 | 10.65 (5.61 to 20.20) | < 0.001 | 92 | 10.27 (5.05 to 20.89) | < 0.001 |
aACPA, anticitrullinated protein antibody; CI, confidence interval; GSTM1, glutathione S-transferase Mu-1; OR, odds ratio; SE, shared epitope; SONORA, Study of New-Onset Rheumatoid Arthritis; VARA, Veterans Affairs Rheumatoid Arthritis Registry. All analyses are age- and gender-adjusted. "Ref." = referent group in each analysis.
Table 3.
Associations of composite risk factors with ACPA positivity in patients with rheumatoid arthritisa
| VARA (n = 703) | SONORA (n = 610) | |||||
|---|---|---|---|---|---|---|
| ACPA+ (%) |
OR (95% CI) | P value | ACPA+ (%) |
OR (95% CI) | P value | |
| GSTM1/SEa,b | ||||||
| Present/Negative | 50 | Ref. | - | 59 | Ref. | - |
| Null/Negative | 56 | 1.26 (0.68 to 2.33) | 0.456 | 48 | 0.63 (0.35 to 1.15) | 0.135 |
| Present/Positive | 79 | 3.65 (2.09 to 6.40) | < 0.001 | 72 | 1.79 (1.05 to 3.03) | 0.032 |
| Null/Positive | 88 | 7.30 (4.01 to 13.29) | < 0.001 | 77 | 2.29 (1.34 to 3.90) | 0.002 |
| AP = 0.46 (0.20 to 0.73) | AP = 0.38 (0.00 to 0.76) | |||||
| Padd < 0.001 | Padd = 0.050 | |||||
| Pmult = 0.246 | Pmult = 0.063 | |||||
| GSTM1/Smokinga | ||||||
| Present/Never | 67 | Ref. | - | 72 | Ref. | - |
| Present/Ever | 75 | 1.42 (0.80 to 2.52) | 0.227 | 67 | 0.72 (0.42 to 1.22) | 0.223 |
| Null/Never | 72 | 1.35 (0.65 to 2.79) | 0.421 | 67 | 0.75 (0.42 to 1.36) | 0.349 |
| Null/Ever | 81 | 2.01 (1.13 to 3.55) | 0.017 | 70 | 0.83 (0.49 to 1.42) | 0.502 |
| AP = 0.12 (-1.41 to 1.65) | AP = 0.44 (-0.28 to 1.15) | |||||
| Padd = 0.881 | Padd = 0.231 | |||||
| Pmult = 0.917 | Pmult = 0.245 | |||||
| SE/Smokinga | ||||||
| Negative/Never | 55 | Ref. | - | 58 | Ref. | - |
| Negative/Ever | 53 | 0.90 (0.40 to 1.99) | 0.788 | 51 | 0.73 (0.39 to 1.37) | 0.329 |
| Positive/Never | 73 | 2.24 (0.97 to 5.16) | 0.058 | 74 | 2.03 (1.08 to 3.82) | 0.027 |
| Positive/Ever | 87 | 5.05 (2.31 to 11.05) | < 0.001 | 75 | 2.10 (1.17 to 3.78) | 0.013 |
| AP = 0.58 (0.31 to 0.85) | AP = 0.16 (-0.34 to 0.66) | |||||
| Padd < 0.001 | Padd = 0.530 | |||||
| Pmult = 0.054 | Pmult = 0.380 | |||||
aACPA, anticitrullinated protein antibody; AP, attributable proportion; GSTM1, glutathione S-transferase Mu-1; SE, shared epitope; SONORA, Study of New-Onset Rheumatoid Arthritis; VARA, Veterans Affairs Rheumatoid Arthritis. All analyses are age- and gender-adjusted. bCorresponding ORs and 95% for GSTM1/SE composite risk after further adjustment for ever smoking in VARA: Null/Negative OR = 1.22 (0.66 to 2.27); Present/Positive OR = 3.75 (2.13 to 6.59); Null/Positive OR = 7.34 (4.02 to 13.34); in SONORA, Null/Negative OR = 0.65 (0.36 to 1.18); Present/Positive OR = 1.80 (1.06 to 3.06); Null/Positive OR = 2.31 (1.36 to 3.95). "Ref." = referent group in each analysis.
SE-GSTM1 interactions
In the VARA registry, there was significant additive interaction between SE and GSTM1 status (AP, 0.46; 95% CI, 0.20 to 0.73; P < 0.001), an interaction that was evident, albeit of borderline significance, in SONORA (AP, 0.38; 95% CI, 0.00 to 0.76; P = 0.050). There was no evidence of a multiplicative SE-GSTM1 interaction in the VARA registry (P = 0.25), although the P value of the product term approached significance in SONORA (P = 0.06) (Table 3). These results were not changed for either cohort after further adjustments for cigarette smoking (Table 3).
In exploratory analyses stratified by SE dose (0, 1 or 2 copies) rather than SE positivity, there were marked differences in the associations of composite risk factors of GSTM1 status and SE dose with ACPA positivity. Compared to individuals lacking both risk factors, SE homozygotes carrying the GSTM1-null genotype were ~28-fold more likely to be ACPA-positive in the VARA registry (OR, 28.50; 95% CI, 8.21 to 98.87) (Figure 1) and ~21-fold more likely to be ACPA-positive in SONORA (OR, 21.04; 95% CI, 4.82 to 91.75) (Figure 2).
Figure 1.
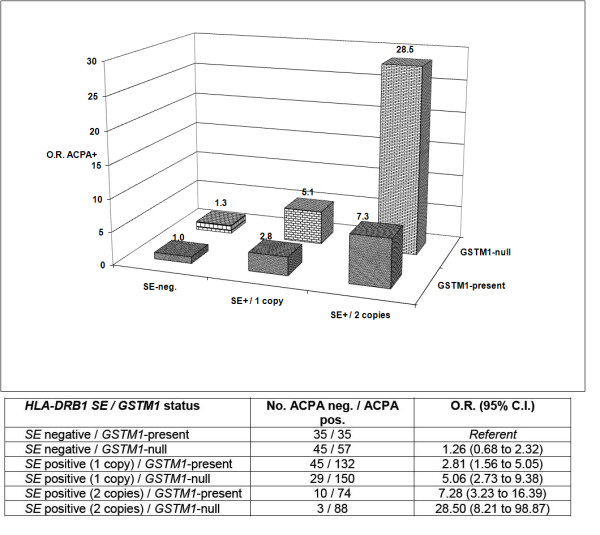
Age- and gender-adjusted associations of composite HLA-DRB1 SE dose (0, 1 or 2 alleles) and glutathione S-transferase Mu-1 (GSTM1) status with anticitrullinated protein antibody (ACPA) positivity in Caucasian patients enrolled in the Veterans Affairs Rheumatoid Arthritis (VARA) registry (n = 703).
Figure 2.
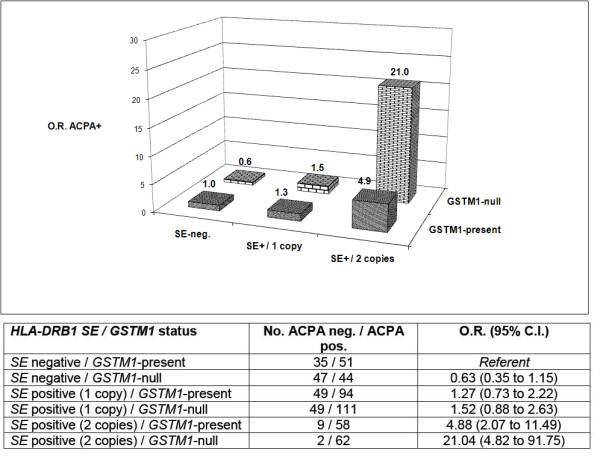
Age- and gender-adjusted associations of composite HLA-DRB1 SE dose (0, 1 or 2 alleles) and GSTM1 status with ACPA positivity in Caucasian patients enrolled in SONORA (n = 610).
GSTM1-smoking and SE-smoking interactions
There were no significant additive or multiplicative interactions between GSTM1 status and smoking for ACPA positivity in either cohort (Table 3). In contrast, there was a significant additive interaction between SE positivity and ever smoking in the VARA registry, accounting for more than 50% of the overall risk of ACPA positivity in SE-positive smokers (AP, 0.58; 95% CI, 0.31 to 0.85; P < 0.001); there was also a nonsignificant trend to suggest multiplicative interaction (P = 0.054). Consistent with prior reports in SONORA [37], we observed no evidence of additive or multiplicative interactions between SE and ever smoking referent to ACPA positivity in this cohort. To explore the effect of smoking categorization on this finding, these analyses were repeated to examine for evidence of interaction between SE and current smoking (vs. never and former smoking combined). In these analyses, there was significant additive interaction between SE and current smoking (AP, 0.47; 95% CI, 0.13 to 0.82; P = 0.008), but no evidence of multiplicative interaction (P = 0.153) (data not shown).
Discussion
Associations of glutathione S-transferase polymorphisms with RA have been the subject of several other investigations [12-15,38,39], although none of these have examined the association of GSTM1 status with ACPA-positive disease. This study is the first to show that the GSTM1-null genotype, present in approximately one half of all individuals of European ancestry, shows a significant biologic interaction with HLA-DRB1 SE-containing alleles with reference to the risk of ACPA positivity in RA. This is noteworthy, given the disease specificity (> 95%) of ACPA and the association of worse long-term outcomes in RA with ACPA seropositivity [22,23]. These results show that patients with both genetic risk factors (HLA-DRB1 SE and GSTM1-null) are two to seven times more likely to be ACPA-positive than patients lacking both risk factors. Furthermore, ~40% to 50% of the "excess" risk in this group is directly attributable to gene-gene interaction, an interaction that appears to be independent of smoking status. It is important to note that the magnitude of this interaction is similar to that previously reported to exist between HLA-DRB1 SE positivity and smoking [2,3]. The potential generalizability of these findings is further bolstered by its replication in two widely divergent RA cohorts: one composed primarily of men with long-standing disease and the other including primarily women with early-onset disease. These data are an important addition to studies showing significant additive interactions between SE and ever smoking in the risk of ACPA-positive RA [2,3]. Our results support the hypothesis that an oxidative environment promoted through the absence of functional GSTM1 enzyme potently enhances the risk of ACPA positivity in RA conferred by the presence of HLA-DRB1 SE.
Oxidative stress plays a pathogenic role in other autoimmune and inflammatory conditions, including systemic lupus erythematosus (SLE), scleroderma, diabetes and atherosclerosis [40]. Compared to those with functional GSTM1, individuals with the GSTM1-null genotype appear to be more prone to have increased levels of oxidative stress following exposure to select toxins [41]. Oxidation of nucleotides by reactive oxygen species increases the immunogenicity of DNA in SLE, generating autoantigens with significantly higher affinity for circulating autoantibodies [42]. In addition to modifying DNA and lipids, oxidative stress promotes the formation of neoantigens through posttranslational peptide modification. Bang et al. [43] have shown that oxidation of citrullinated vimentin, implicated as an autoantigen in RA, leads to substantially increased antibody reactivity to this antigen in RA.
Our results complement the prior findings of Klareskog et al. [2], who reported that patients who had ever smoked and were homozygous for SE were 21 times more likely to develop ACPA-positive RA compared to SE-negative patients who had never smoked. Results from the Swedish case control study [2] differed from an analysis of three North American cohorts including SONORA [37], which found no evidence of interaction between SE and ever smoking in SONORA and only weak evidence of interaction in one of the two other cohorts examined. In these two other cohorts, but not in SONORA, ever smoking showed a borderline association with ACPA positivity with ORs approaching 1.4 [37]. Although it was not statistically significant, we found a similar association of ever smoking with ACPA positivity in the VARA registry with an OR of 1.48, suggesting that our study was underpowered to detect this association because of the relatively small proportion of never smokers in the VARA Registry.
Differences in these reports (and differences between the VARA registry and SONORA) could relate to population heterogeneity, including differences in gender distribution and cumulative smoking exposure. Compared to women, men have been shown to have a higher penetrance of HLA-DRB1 [44], are more likely to smoke and (among smokers) are more likely to be categorized as heavy smokers [45]. Differences in smoking exposure may be salient here, given findings from a separate North American study showing that SE-smoking interactions in the risk of seropositive RA (a combined rheumatoid factor (RF)/ACPA-positive phenotype) were limited to individuals with heavy smoking (> 10 pack-years) [46]. The importance of quantifying cumulative smoking exposure has also recently been shown among African Americans with RA risk limited to those with more than 10 pack-years of exposure [47]. Cumulative smoking exposure was not available in the present study involving the VARA registry and SONORA, precluding such analyses. Underscoring the potential importance of accounting for cumulative exposure, we observed significant SE-smoking interactions in SONORA when smoking exposure was dichotomized as current vs. noncurrent rather than ever vs. never, with the "current" category likely to account for individuals with greater lifelong smoking exposure.
These results differ from a prior study showing significant multiplicative interactions between GSTM1-null status and smoking in RA disease risk [12], an effort that did not include examinations of GSTM1-SE interactions. In the present study, we found no evidence of significant interaction (multiplicative or additive) between GSTM1-null and smoking in ACPA positivity, a phenotype that was not examined in the prior nested case control analysis from the Iowa Women's Health Study [12]. It is possible that in the present study we simply lacked sufficient power to detect this interaction. Differences in study design (case only vs. case control) and study populations (smoking prevalence and predominantly male vs. female patients) may also help explain these discrepant study results.
Controversy and uncertainty remain regarding the most appropriate manner in which to model gene-gene and gene-environment interactions [48]. In contrast to prior studies that have examined smoking-SE interactions in RA risk by calculating only measures of additive interaction [2,49], we have examined measures of both additive and multiplicative interaction. Multiplicative interaction refers to the inclusion of a product term in regression analyses to generate an optimal fit of the data in the statistical model. It is important to note that the absence of multiplicative interaction does not exclude the existence of important biologic interactions. For example, the present study shows that at least one pathway to ACPA positivity in RA requires the presence of two risk factors (that is, GSTM1-null and HLA-DRB1 SE).
Although they involved two large independent cohorts, our analyses were limited to two-way interactions. We lacked the sample sizes even after combining cohorts that would be necessary to examine more complex interactions, including analyses of GSTM1-SE stratified by smoking status. Future analyses of this sort with larger patient populations will be essential not only in replicating our findings but also in providing critical insight into mechanisms underpinning these observed interactions. Although this study included a case-only approach, ACPA positivity is increasingly recognized as a distinct disease phenotype in RA. Indeed, the well-defined associations of cigarette smoking and HLA-DRB1 SE with RA in European populations apply only to ACPA-positive disease and do not apply to seronegative disease [2]. Because of the limited sample sizes in subgroups of interest, our study did not include analyses of interactions of distinct HLA-DRB1 subtypes with GSTM1. Recent findings have shown that different *01 and *04 subtypes appear to contribute equally to SE-smoking interactions in ACPA-positive RA [50], suggesting that analyses of specific SE subtypes may yield limited incremental information.
Conclusions
The GSTM1-null genotype, observed in approximately 50% of individuals of European ancestry, shows significant interactions with HLA-DRB1 SE alleles in ACPA positivity among patients with RA. Future studies will be needed to explore precisely how GSTM1 and other antioxidant enzymes influence disease expression in RA. Along with other recent reports, this work emphasizes the need for the simultaneous investigation of multiple genetic and environmental factors to better understand the pathogenic contributions of these elements to the development and progression of RA with potential application to other autoimmune diseases.
Abbreviations
ACPA: anticitrullinated protein antibody; ACR: American College of Rheumatology; AP: attributable proportion; CI: confidence interval; CYP1a1: cytochrome p450 1a1; GSTM1: glutathione S-transferase Mu-1; HLA: human leukocyte antigen; OR: odds ratio; PCR: polymerase chain reaction; RA: rheumatoid arthritis; SE: shared epitope; SLE: systemic lupus erythematosus; SONORA: Study of New-Onset Rheumatoid Arthritis; SSOP: specific oligonucleotide probes; VA: Veterans Affairs; VARA: Veterans Affairs Rheumatoid Arthritis.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
TRM was involved in all aspects of study conception, design, analysis, interpretation and report generation and provided final approval of the version of the submitted manuscript. TRM had full access to all of the study data and had final responsibility for the decision to submit the manuscript. SLB, LH, PKG, JAN, FY, KAG, TDLV, GMT, KDM, JRO'D, AMR, RH, LC, DSJ, GK, JSR, GWC and KKB were involved in data acquisition, analysis and report drafting and provided final approval of the submitted manuscript. LAC was involved in data interpretation and report generation and also provided final approval of the submitted manuscript draft.
Contributor Information
Ted R Mikuls, Email: tmikuls@unmc.edu.
Karen A Gould, Email: kagould@unmc.edu.
Kimberly K Bynoté, Email: kbynote@unmc.edu.
Fang Yu, Email: fangyu@unmc.edu.
Tricia D LeVan, Email: tlevan@unmc.edu.
Geoffrey M Thiele, Email: gthiele@unmc.edu.
Kaleb D Michaud, Email: kmichaud@unmc.edu.
James R O'Dell, Email: jrodell@unmc.edu.
Andreas M Reimold, Email: Andreas.Reimold@UTSouthwestern.edu.
Roderick Hooker, Email: roderick.hooker@va.gov.
Liron Caplan, Email: liron.caplan@UCDenver.edu.
Dannette S Johnson, Email: djohnson3@umc.edu.
Gail Kerr, Email: gail.kerr@va.gov.
J Steuart Richards, Email: john.richards1@va.gov.
Grant W Cannon, Email: grant.cannon@va.gov.
Lindsey A Criswell, Email: Lindsey.Criswell@ucsf.edu.
Janelle A Noble, Email: jnoble@chori.org.
S Louis Bridges, Jr, Email: LBridges@uab.edu.
Laura Hughes, Email: laura.hughes@ccc.uab.edu.
Peter K Gregersen, Email: peterg@nshs.edu.
Acknowledgements
This work was funded by a grant from the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant R03 AR054539). The VARA Registry has received research support from the Health Services Research & Development (HSR&D) Program of the Veterans Health Administration (VHA) in addition to unrestricted research funds from Abbott Laboratories and Bristol-Myers Squibb. Dr Mikuls receives research support from the VHA (VA Merit) and the American College of Rheumatology Research and Education Foundation. The authors thank Debra Bergman and Bart Hamilton for their assistance in this work and the many U.S. veterans who have generously participated in this research.
References
- Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis: an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- Klareskog L, Stolt P, Lundberg K, Kallberg H, Bengtsson C, Grunewald J, Ronnelid J, Harris HE, Ulfgren AK, Rantapaa-Dahlqvist S, Eklund A, Padyukov L, Alfredsson L. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006;54:38–46. doi: 10.1002/art.21575. [DOI] [PubMed] [Google Scholar]
- Pedersen M, Jacobsen S, Garred P, Madsen HO, Klarlund M, Svejgaard A, Pedersen BV, Wohlfahrt J, Frisch M. Strong combined gene-environment effects in anti-cyclic citrullinated peptide-positive rheumatoid arthritis: a nationwide case-control study in Denmark. Arthritis Rheum. 2007;56:1446–1453. doi: 10.1002/art.22597. [DOI] [PubMed] [Google Scholar]
- Pattison DJ, Winyard PG. Dietary antioxidants in inflammatory arthritis: do they have any role in etiology or therapy? Nat Clin Pract Rheumatol. 2008;4:590–596. doi: 10.1038/ncprheum0920. [DOI] [PubMed] [Google Scholar]
- Bae SC, Jung WJ, Lee EJ, Yu R, Sung MK. Effects of antioxidant supplements intervention on the level of plasma inflammatory molecules and disease severity of rheumatoid arthritis patients. J Am Coll Nutr. 2009;28:56–62. doi: 10.1080/07315724.2009.10719762. [DOI] [PubMed] [Google Scholar]
- Cerhan JR, Saag KG, Merlino LA, Mikuls TR, Criswell LA. Antioxidant micronutrients and risk of rheumatoid arthritis in a cohort of older women. Am J Epidemiol. 2003;157:345–354. doi: 10.1093/aje/kwf205. [DOI] [PubMed] [Google Scholar]
- Pattison DJ, Silman AJ, Goodson NJ, Lunt M, Bunn D, Luben R, Welch A, Bingham S, Khaw KT, Day N, Symmons DP. Vitamin C and the risk of developing inflammatory polyarthritis: prospective nested case-control study. Ann Rheum Dis. 2004;63:843–847. doi: 10.1136/ard.2003.016097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison DJ, Symmons DP, Lunt M, Welch A, Bingham SA, Day NE, Silman AJ. Dietary β-cryptoxanthin and inflammatory polyarthritis: results from a population-based prospective study. Am J Clin Nutr. 2005;82:451–455. doi: 10.1093/ajcn.82.2.451. [DOI] [PubMed] [Google Scholar]
- Shapiro JA, Koepsell TD, Voigt LF, Dugowson CE, Kestin M, Nelson JL. Diet and rheumatoid arthritis in women: a possible protective effect of fish consumption. Epidemiology. 1996;7:256–263. doi: 10.1097/00001648-199605000-00007. [DOI] [PubMed] [Google Scholar]
- Heliovaara M, Knekt P, Aho K, Aaran RK, Alfthan G, Aromaa A. Serum antioxidants and risk of rheumatoid arthritis. Ann Rheum Dis. 1994;53:51–53. doi: 10.1136/ard.53.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HJ, Han CY, Bernstein DA, Hsiao W, Lin BK, Hardy S. Ethnic distribution of the glutathione transferase Mu 1-1 (GSTM1) null genotype in 1473 individuals and application to bladder cancer susceptibility. Carcinogenesis. 1994;15:1077–1081. doi: 10.1093/carcin/15.5.1077. [DOI] [PubMed] [Google Scholar]
- Criswell LA, Saag KG, Mikuls TR, Cerhan JR, Merlino LA, Lum RF, Pfeiffer KA, Woehl B, Seldin MF. Smoking interacts with genetic risk factors in the development of rheumatoid arthritis among older Caucasian women. Ann Rheum Dis. 2006;65:1163–1167. doi: 10.1136/ard.2005.049676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinobu S, Morinobu A, Kanagawa S, Hayashi N, Nishimura K, Kumagai S. Glutathione S-transferase gene polymorphisms in Japanese patients with rheumatoid arthritis. Clin Exp Rheumatol. 2006;24:268–273. [PubMed] [Google Scholar]
- Yun BR, El-Sohemy A, Cornelis MC, Bae SC. Glutathione S-transferase M1, T, and P1 genotypes and rheumatoid arthritis. J Rheumatol. 2005;32:992–997. [PubMed] [Google Scholar]
- Ghelani AM, Samanta A, Jones AC, Mastana SS. Association analysis of TNFR2, VDR, A2M, GSTT1, GSTM1, and ACE genes with rheumatoid arthritis in South Asians and Caucasians of East Midlands in the United Kingdom. Rheumatol Int. 2010. in press . [DOI] [PubMed]
- Datta SK, Kumar V, Pathak R, Tripathi AK, Ahmed RS, Kalra OP, Banerjee BD. Association of glutathione S-transferase M1 and T1 gene polymorphism with oxidative stress in diabetic and nondiabetic chronic kidney disease. Ren Fail. 2010;32:1189–1195. doi: 10.3109/0886022X.2010.517348. [DOI] [PubMed] [Google Scholar]
- De Waart FG, Kok FJ, Smilde TJ, Hijmans A, Wollersheim H, Stalenhoef AF. Effect of glutathione S-transferase M1 genotype on progression of atherosclerosis in lifelong male smokers. Atherosclerosis. 2001;158:227–231. doi: 10.1016/S0021-9150(01)00420-8. [DOI] [PubMed] [Google Scholar]
- Lakhdar R, Denden S, Knani J, Leban N, Daimi H, Hassine M, Lefranc G, Ben Chibani J, Haj Khelil A. Association of GSTM1 and GSTT1 polymorphisms with chronic obstructive pulmonary disease in a Tunisian population. Biochem Genet. 2010;48:647–657. doi: 10.1007/s10528-010-9346-z. [DOI] [PubMed] [Google Scholar]
- Minelli C, Granell R, Newson R, Rose-Zerilli MJ, Torrent M, Ring SM, Holloway JW, Shaheen SO, Henderson JA. Glutathione-S-transferase genes and asthma phenotypes: a Human Genome Epidemiology (HuGE) systematic review and meta-analysis including unpublished data. Int J Epidemiol. 2010;39:539–562. doi: 10.1093/ije/dyp337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers AJ, Brasch-Andersen C, Ionita-Laza I, Murphy A, Sharma S, Klanderman BJ, Raby BA. The interaction of glutathione S-transferase M1-null variants with tobacco smoke exposure and the development of childhood asthma. Clin Exp Allergy. 2009;39:1721–1729. doi: 10.1111/j.1365-2222.2009.03372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamer L, Ercan B, Camsari A, Yildirim H, Cicek D, Sucu N, Ates NA, Atik U. Glutathione S-transferase gene polymorphism as a susceptibility factor in smoking-related coronary artery disease. Basic Res Cardiol. 2004;99:223–229. doi: 10.1007/s00395-004-0465-8. [DOI] [PubMed] [Google Scholar]
- Kastbom A, Strandberg G, Lindroos A, Skogh T. Anti-CCP antibody test predicts the diseases course during 3 years in early rheumatoid arthritis (the Swedish TIRA project) Ann Rheum Dis. 2004;63:1085–1089. doi: 10.1136/ard.2003.016808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellekens GA, Visser H, de Jong BA, van den Hoogen FH, Hazes JM, Breedveld FC, van Venrooij WJ. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000;43:155–163. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Arnett F, Edworthy S, Bloch D, McShane D, Fries J, Cooper N, Healy L, Kaplan S, Liang M, Luthra H, Medsger T, Mitchell D, Neustadt D, Pinals R, Schaller J, Sharp J, Wilder R, Hunder G. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- Mikuls TR, Kazi S, Cipher D, Hooker R, Kerr GS, Richards JS, Cannon GW. The association of race and ethnicity with disease expression in male US veterans with rheumatoid arthritis. J Rheumatol. 2007;34:1480–1484. [PubMed] [Google Scholar]
- Sokka T, Willoughby J, Yazici Y, Pincus T. Databases of patients with early rheumatoid arthritis in the USA. Clin Exp Rheumatol. 2003;21(5 Suppl 31):S146–S153. [PubMed] [Google Scholar]
- Brockmoller J, Kerb R, Drakoulis N, Nitz M, Roots I. Genotype and phenotype of glutathione S-transferase class mu isoenzymes mu and psi in lung cancer patients and controls. Cancer Res. 1993;53:1004–1011. [PubMed] [Google Scholar]
- Shields PG, Bowman ED, Harrington AM, Doan VT, Weston A. Polycyclic aromatic hydrocarbon-DNA adducts in human lung and cancer susceptibility genes. Cancer Res. 1993;53:3486–3492. [PubMed] [Google Scholar]
- Benhamou S, Lee WJ, Alexandrie AK, Boffetta P, Bouchardy C, Butkiewicz D, Brockmoller J, Clapper ML, Daly A, Dolzan V, Ford J, Gaspari L, Haugen A, Hirvonen A, Husgafvel-Pursiainen K, Ingelman-Sundberg M, Kalina I, Kihara M, Kremers P, Le Marchand L, London D, Seidegard J, Shields P, Strange RC, Stucker I, To-Figueras J, Brennan P, Taioli E. Meta- and pooled analyses of the effects of glutathione S- transferase M1 polymorphisms and smoking on lung cancer risk. Carcinogenesis. 2002;23:1343–1350. doi: 10.1093/carcin/23.8.1343. [DOI] [PubMed] [Google Scholar]
- Mann CL, Davies MB, Boggild MD, Alldersea J, Fryer AA, Jones PW, Ko Ko C, Young C, Strange RC, Hawkins CP. Glutathione S-transferase polymorphisms in MS: their relationship to disability. Neurology. 2000;54:552–557. doi: 10.1212/wnl.54.3.552. [DOI] [PubMed] [Google Scholar]
- Morinobu A, Kanagawa S, Koshiba M, Sugai S, Kumagai S. Association of the glutathione S-transferase M1 homozygous null genotype with susceptibility to Sjogren's syndrome in Japanese individuals. Arthritis Rheum. 1999;42:2612–2615. doi: 10.1002/1529-0131(199912)42:12<2612::AID-ANR15>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Lee HS, Lee AT, Criswell L, Seldin MF, Amos CI, Carulli JP, Navarrete C, Remmers EF, Kastner DL, Plenge RM, Li W, Gregersen PK. Several regions in the major histocompatibility complex confer risk for anti-CCP-antibody positive rheumatoid arthritis, independent of the DRB1 locus. Mol Med. 2008;14:293–300. doi: 10.2119/2007-00123.Lee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich H, Bugawan T, Begovich AB, Scharf S, Griffith R, Saiki R, Higuchi R, Walsh PS. HLA-DR, DQ and DP typing using PCR amplification and immobilized probes. Eur J Immunogenet. 1991;18:33–55. doi: 10.1111/j.1744-313X.1991.tb00005.x. [DOI] [PubMed] [Google Scholar]
- Rothman K, Greenland S, Walker A. Concepts of interaction. Am J Epidemiol. 1980;112:467–470. doi: 10.1093/oxfordjournals.aje.a113015. [DOI] [PubMed] [Google Scholar]
- Hosmer D, Lemeshow S. Confidence interval estimation of interaction. Epidemiology. 1992;3:452–456. doi: 10.1097/00001648-199209000-00012. [DOI] [PubMed] [Google Scholar]
- 2001 National Survey of Veterans. http://www1.va.gov/VETDATA/docs/SurveysAndStudies/NSV_Final_Report.pdf
- Lee HS, Irigoyen P, Kern M, Lee A, Batliwalla F, Khalili H, Wolfe F, Lum RF, Massarotti E, Weisman M, Bombardier C, Karlson EW, Criswell LA, Vlietinck R, Gregersen PK. Interaction between smoking, the shared epitope, and anti-cyclic citrullinated peptide: a mixed picture in three large North American rheumatoid arthritis cohorts. Arthritis Rheum. 2007;56:1745–1753. doi: 10.1002/art.22703. [DOI] [PubMed] [Google Scholar]
- Mattey DL, Hassell AB, Plant M, Dawes PT, Ollier WR, Jones PW, Fryer AA, Alldersea J, Strange RC. Association of polymorphism in glutathione S-transferase loci with susceptibility and outcome in rheumatoid arthritis: comparison with the shared epitope. Ann Rheum Dis. 1999;58:164–168. doi: 10.1136/ard.58.3.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattey DL, Hutchinson D, Dawes PT, Nixon NB, Clarke S, Fisher J, Brownfield A, Alldersea J, Fryer AA, Strange RC. Smoking and disease severity in rheumatoid arthritis: association with polymorphism at the glutathione S-transferase M1 locus. Arthritis Rheum. 2002;46:640–646. doi: 10.1002/art.10174. [DOI] [PubMed] [Google Scholar]
- Griffiths HR. Is the generation of neo-antigenic determinants by free radicals central to the development of autoimmune rheumatoid disease? Autoimmun Rev. 2008;7:544–549. doi: 10.1016/j.autrev.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Breton CV, Kile ML, Catalano PJ, Hoffman E, Quamruzzaman Q, Rahman M, Mahiuddin G, Christiani DC. GSTM1 and APE1 genotypes affect arsenic-induced oxidative stress: a repeated measures study. Environ Health. 2007;6:39. doi: 10.1186/1476-069X-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount S, Griffiths H, Emery P, Lunec J. Reactive oxygen species modify human DNA, eliciting a more discriminating antigen for the diagnosis of systemic lupus erythematosus. Clin Exp Immunol. 1990;81:384–389. doi: 10.1111/j.1365-2249.1990.tb05343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang H, Egerer K, Gauliard A, Luthke K, Rudolph PE, Fredenhagen G, Berg W, Feist E, Burmester GR. Mutation and citrullination modifies vimentin to a novel autoantigen for rheumatoid arthritis. Arthritis Rheum. 2007;56:2503–2511. doi: 10.1002/art.22817. [DOI] [PubMed] [Google Scholar]
- Jawaheer D, Lum RF, Gregersen PK, Criswell LA. Influence of male sex on disease phenotype in familial rheumatoid arthritis. Arthritis Rheum. 2006;54:3087–3094. doi: 10.1002/art.22120. [DOI] [PubMed] [Google Scholar]
- Russell MA, Wilson C, Taylor C, Baker CD. Smoking habits of men and women. Br Med J. 1980;281:17–20. doi: 10.1136/bmj.281.6232.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlson EW, Chang SC, Cui J, Chibnik LB, Fraser PA, De Vivo I, Costenbader KH. Gene-environment interaction between HLA-DRB1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Ann Rheum Dis. 2010;69:54–60. doi: 10.1136/ard.2008.102962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikuls TR, Sayles H, Yu F, Levan T, Gould KA, Thiele GM, Conn D, Jonas BL, Callahan LF, Smith E, Brasington R, Moreland LW, Reynolds R, Bridges SL Jr. Associations of cigarette smoking with rheumatoid arthritis in African Americans. Arthritis Rheum. 2010;62:3560–3568. doi: 10.1002/art.27716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlbom A, Alfredsson L. Interaction: a word with two meanings creates confusion. Eur J Epidemiol. 2005;20:563–564. doi: 10.1007/s10654-005-4410-4. [DOI] [PubMed] [Google Scholar]
- Padyukov L, Silva C, Stolt P, Alfredsson L, Klareskog L. A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 2004;50:3085–3092. doi: 10.1002/art.20553. [DOI] [PubMed] [Google Scholar]
- Lundstrom E, Kallberg H, Alfredsson L, Klareskog L, Padyukov L. Gene-environment interaction between the DRB1 shared epitope and smoking in the risk of anti-citrullinated protein antibody-positive rheumatoid arthritis: all alleles are important. Arthritis Rheum. 2009;60:1597–1603. doi: 10.1002/art.24572. [DOI] [PMC free article] [PubMed] [Google Scholar]