

Abstract
Gout is the most prevalent form of inflammatory arthropathy. Several studies suggest that its prevalence and incidence have risen in recent decades. Numerous risk factors for the development of gout have been established, including hyperuricaemia, genetic factors, dietary factors, alcohol consumption, metabolic syndrome, hypertension, obesity, diuretic use and chronic renal disease. Osteoarthritis predisposes to local crystal deposition. Gout appears to be an independent risk factor for all-cause mortality and cardiovascular mortality and morbidity, additional to the risk conferred by its association with traditional cardiovascular risk factors.
Introduction
Gout is the most prevalent form of inflammatory arthritis and is associated with impaired quality of life [1-3]. Elevation of serum uric acid (SUA) levels, or hyperuricaemia, is an essential prerequisite for the development of gout. As SUA levels rise and the physiological saturation threshold for uric acid is exceeded in body fluids, the formation and deposition of monosodium urate (MSU) crystals occurs in and around joints. Clinical manifestations of MSU crystal deposition include acute attacks of severe pain and inflammation affecting peripheral joints, most commonly the first metatarsophalangeal (MTP) joint, chronic joint damage, and tophaceous deposits of MSU crystals in the joints and skin. Recent epidemiological studies have described trends in the prevalence and incidence of gout, and have increased our understanding of risk factors for its development and the implications of co-morbid disease on mortality and cardiovascular morbidity.
Prevalence of gout
Epidemiological evidence from New Zealand, the USA, the UK and China suggests that gout is becoming more prevalent (Table 1) [4-20]. The findings of three similarly-conducted successive surveys from New Zealand show an increase in the prevalence of gout, diagnosed by interview and physical examination, in both European US and Maori subjects [4-6].
Table 1.
Increasing prevalence of gout: epidemiological data from New Zealand, the USA, the UK and China
| Study | Survey date | Study population | Case definition | Prevalence |
|---|---|---|---|---|
| New Zealand | ||||
| Lennane and colleagues [4] | 1958 | Random community sample | Personal interview and examination | European 3/1,000, Maori 27/1,000 |
| Prior and Rose [5] | 1966 | Random community sample | Personal interview and examination | European 9/1,000, Maori 60/1,000 |
| Klemp and colleagues [6] | 1992 | Random community sample | Personal interview and examination; 1977 ARA criteria | European 29/1,000, Maori 64/1,000 |
| USA | ||||
| Wallace and colleagues [7] | 1990 | Medical claims database | Claims with gout diagnosis or urate-lowering drugs | 2.9/1,000 |
| 1999 | 5.2/1,000 | |||
| Lawrence and colleagues [8,9] | 1969 | National Health Interview Surveys | Self-reported gout | 4.8/1,000 |
| 1976 | 7.8/1,000 | |||
| 1980 | 8.3/1,000 | |||
| 1983 to 1985 | 9.9/1,000 | |||
| 1988 | 8.5/1,000 | |||
| 1992 | 8.4/1,000 | |||
| UK | ||||
| Currie [10] | 1975 | GP records | GP diagnosis | 2.6/1,000 |
| Steven [11] | 1987 | GP records (Scotland) | Specialist review of clinical records | 3.4/1,000 |
| Harris and colleagues [12] | 1993 | GP records (England) | GP diagnosis | 9.5/1,000 |
| Mikuls and colleagues [13] | 1999 | UK-GPRD | GP diagnosis | 14/1,000 |
| Annemans and colleagues [14] | 2000 to 2005 | IMS Disease Analyzer | GP diagnosis | 14/1,000 |
| Elliot and colleagues [15] | 2001 | RCGP Weekly Returns Service | GP diagnosis | 4.3/1,000 |
| 2003 | 4.9/1,000 | |||
| 2005 | 4.8/1,000 | |||
| 2007 | 4.7/1,000 | |||
| China | ||||
| Nan and colleagues [16] | 2002 | Random community sample | Self-report, confirmed in medical records | 3.6/1,000 |
| Miao and colleagues [17] | 2004 | Random community sample | Questionnaire, physical examination | 5.3/1,000 |
| 1977 ARA criteria | ||||
| Taiwan | ||||
| Chou and colleagues [18] | Not stated | Random community sample | Questionnaire, physical examination. Hyperuricaemia/synovial fluid analysis | Rural 1.6/1,000. suburban 6.7/1,000, urban 6.7/1,000 |
| Chang and colleagues [19] | 1993 to 1995 | Random community sample | Personal interview, 1977 ARA criteria | Aborigine 91/1,000, non-Aborigine 3/1,000 |
| Lin and colleagues [20] | 1993 to 1996 | Nutrition and Health Survey | Personal interview, physical examination | 34/1,000 |
ARA, American Rheumatism Association; GP, general practitioner; RCGP, Royal College of General Practitioners; UK-GPRD, UK General Practice Research Database.
In the USA, the prevalence of gout in a managed-care administrative claims database increased from 2.9/1,000 in 1990 to 5.2/1,000 in 1999 [7], most notably in men aged over 75 years. Successive National Health Interview Surveys in the USA show an increasing prevalence of self-reported gout, starting from a low of 4.8/1,000 in 1969, increasing steadily to peak at 9.9/1,000 in 1983 to 1985, and then decreasing slightly to 8.4/1,000 in 1992 [8,9].
Epidemiological surveys from the UK also suggest that gout is becoming more prevalent. Surveys undertaken in general practice diagnostic indices reported gout prevalence per 1,000 of 2.6 in 1975 [10], 3.4 in 1987 [11], and 9.5 in 1993 [12]. Subsequent studies conducted in the UK-General Practice Research Database (UK-GPRD) in 1999 [13] and the IMS Disease Analyzer from 2000 to 2005 [14] both found the prevalence of gout to be 1.4%. A further UK study undertaken at the Royal College of General Practitioners Weekly Returns Service between 2001 and 2007 reports a lower annual prevalence of primary care gout consultation ranging from 4.2/1,000 in 2002 to 4.9/1,000 in both 2003 and 2006, although there was a suggestion of a slight increase in prevalence over the study period [15].
Finally, successive random population surveys undertaken in the city of Qingdao, China report an increase in gout prevalence from 3.6/1,000 in 2002 [16] to 5.3/1,000 in 2004 [17].
Several studies have demonstrated suboptimal management of gout in primary care [21-23], which may contribute to the rising prevalence of clinically significant symptomatic gout. It should also be noted that the diagnosis of gout in these studies was based upon clinical assessment, patient self-report, general practice diagnosis, medical record/database review, or fulfilment of the 1977 American Rheumatism Association preliminary criteria for the acute arthritis of primary gout [24] (Table 1) - rather than on microscopic identification of MSU crystals, the gold standard for gout diagnosis [25]. Clinical diagnosis has been shown to have poor sensitivity and specificity compared with MSU crystal identification [26,27], and hence such methods risk over-ascertainment of gout cases.
Incidence of gout
Several studies have examined the incidence of gout. The John Hopkins Precursors Study followed 1,216 male medical students for a median of 29 years, identifying 60 cases of incident gout (incidence 1.73 per 1,000 patient-years) [28]. In the Framingham Heart Study, which followed 5,209 people for a median of 28 years, 104 incident cases of gout in women and 200 cases in men were documented [29] - giving an incidence of gout per 1,000 person-years of 1.4 in women and 4.0 in men.
Data from two general practice consultation databases show the incidence of gout to be stable in the UK. The earlier study, undertaken in the UK-GPRD between 1990 and 1999, found gout incidence per 10,000 patient-years to range from a low of 11.9 cases in 1991 to a peak of 18.0 cases in 1994, before stabilising at 13.1 cases in 1999 [13]. In a later study, undertaken at the Royal College of General Practitioners Weekly Returns Service between 1994 and 2007, the mean annual incidence was 18.6 per 10,000 population [15]. In both studies, gout incidence was higher in men than in women and increased with age. In contrast, successive surveys from the USA undertaken as part of the Rochester Epidemiology Project computerised medical records system found the age-adjusted and sex-adjusted annual incidence of acute gout to increase from 45.0/100,000 in 1977/78 to 62.3/100,000 in 1995/96 [30]. The largest increase in incidence occurred in elderly men and in primary gout (that is, no history of diuretic use).
Risk factors for the development of gout
Hyperuricaemia
Hyperuricaemia is considered the most important risk factor for the development of gout. In a community-based, cross-sectional Taiwanese study of 3,185 adults aged over 30 years, the odds ratio (OR) for prevalent gout was 3.65 (95% confidence interval (CI) = 2.72, 5.09) between men with and without hyperuricaemia (SUA ≥ 7.0 mg/dl) [31]. The 5-year cumulative incidence of gout was 18.8% in 223 men who had asymptomatic hyperuricaemia at baseline [32]. A dose-dependent effect on the 5-year cumulative incidence of gout was seen with increasing SUA level (SUA 7.0 to 7.9 mg/dl, 10.8%; SUA 8.0 to 8.9 mg/dl, 27.7%; SUA ≥ 9.0 mg/dl, 61.1%) [32]. The Normative Aging Study followed 2,046 male veterans aged 21 to 81 years over a period of 15 years, identifying 84 new cases of acute gouty arthritis [33]. The 5-year cumulative incidence of gout increased as SUA increased (Figure 1). In the Framingham Heart Study, the risk of developing gout increased with increasing SUA level in both men and women (Figure 2) [29]. Gout incidence rates increased exponentially with increasing SUA levels in both studies (Table 2) [29,33].
Figure 1.
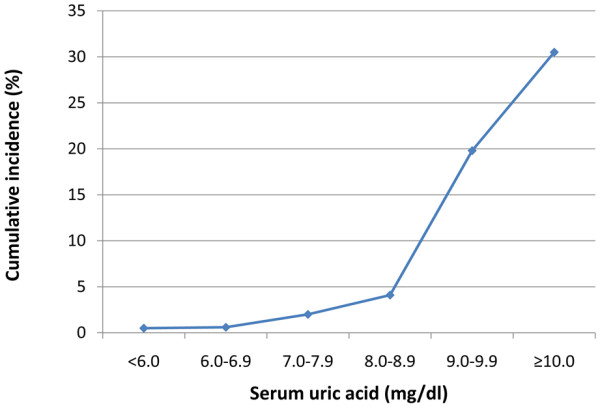
Incidence of gout according to serum uric acid level. Five-year cumulative incidence of gout according to serum uric acid level in men in the Normative Aging Study [33].
Figure 2.
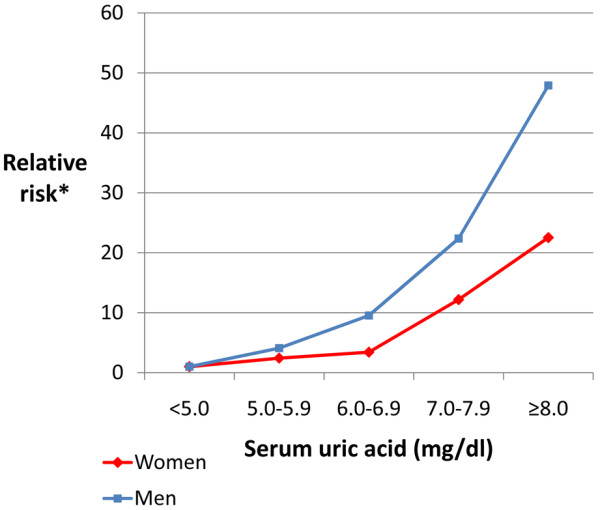
Risk of developing gout according to serum uric acid level. Relative risk of developing gout according to serum uric acid level in men and women in the Framingham Heart Study [29]. Referent group: serum uric acid <5.0 mg/dl. *Adjusted for age, education, body mass index, alcohol consumption, hypertension, diuretic use, blood glucose level, blood cholesterol level, and menopausal status.
Table 2.
Incidence rate of gout in relation to serum uric acid levels
| Incidence rate of gout per 1,000 person-years | |||
|---|---|---|---|
| Serum uric acid level (mg/dl) | Normative Aging Study (men) [33] | Framingham Heart Study (men) [29] | Framingham Heart Study (women) [29] |
| < 5.0 | 0.8 | 0.8 | |
| 5.0 to 5.9 | 0.8a | 3.4 | 2.5 |
| 6.0 to 6.9 | 0.9 | 8.0 | 4.2 |
| 7.0 to 7.9 | 4.1 | 17.8 | 13.1 |
| 8.0 to 8.9 | 8.4 | 32.9b | 27.3b |
| 9.0 to 9.9 | 43.2 | ||
| ≥ 10.0 | 70.2 | ||
aIncidence rate for serum uric acid < 6.0 mg/dl. bIncidence rate for serum uric acid ≥ 8.0 mg/dl.
Genetic factors
Familial clustering is often evident in common primary gout, and twin studies show high heritability for both uric acid renal clearance (60%) and uric acid:creatinine ratio (87%) [34,35]. The usual mechanism of hyperuricaemia in primary gout relates predominantly to relative inefficiency in excretion rather than over-production. It is estimated that approximately 30% of the body's uric acid is excreted into the intestine by ill-defined mechanisms, and is broken down by colonic bacteria (which possess uricase) to allantoin. The kidney excretes the majority (70%) of uric acid excretion, however, and renal mechanisms appear key to the understanding of hyperuricaemia. Recent interest has therefore particularly focused on genes regulating renal urate transport [36].
The SLC22A12 gene codes for human urate transporter 1 (URAT1), a member of the organic anion transporter family that, together with other recently identified transporters, is important in controlling reabsorption of uric acid from the proximal renal tubules. URAT1 is the site of action for many drugs and ions that influence SUA. For example, lactate, nicotinate and pyrazinoate act as a substrate for URAT1 and increase reabsorption of uric acid (causing an increase in SUA), whereas benzbromarone, probenecid and losartan inhibit URAT1 to cause increased uricosuria and reduction in SUA [37]. A polymorphism of this gene has been associated with relative under-excretion of uric acid and hyperuricaemia in German Caucasians [38], and inactivating mutations of URAT1 have been shown in Japanese patients to cause marked hypouricaemia [39].
The glucose and fructose transporter SLC2A9 (GLUT9) is another urate transporter in proximal renal tubules [40], and polymorphisms of this gene have been associated with increased SUA [41,42] and with self-reported gout [43]. The association between polymorphisms in SLC2A9 and both high SUA levels and risk of gout was confirmed in a genome-wide association study of three large cohorts [44]. This study also identified two further gene associations in ABCG2 (a urate efflux transporter in proximal collecting duct cells) and SLC17A3 (encoding NPT4 - a proximal tubule sodium/phosphate co-transporter), allowing development of a genetic score to predict risk of gout. Polymorphisms in the SLC17A1 gene, which codes for NPT1, a sodium-dependent phosphate co-transporter, have also been shown to associate with gout [45]. Two recent meta-analyses of genome-wide association studies have confirmed these associations, although except for SCL2A9, which may account for up to 5% of variance of uric acid levels, the other genetic associations each appear to account for less than 1% of variance [46,47].
Two other reported genetic associations with hyperuricaemia are of special interest. Firstly, the 64Arg variant of the β3-adrenergic receptor (ADRB3) gene - which may also induce insulin resistance through reduced lipolysis and increase in adipocytes, thus possibly providing an explanation for the link between these facets of the metabolic syndrome [48]. Secondly, the 677T allele of the methylene tetrahydrofolate reductase (MTHFR) gene - which may facilitate availability of methylene tetrahydrofolate for de novo purine synthesis [49]. Single gene mutations that cause hyperuricaemia and gout are very rare, but examples of these include, uromodulin, renin, the aldolase B (ALDOB) gene and hypoxanthine guanine phosphoribosylpyrophosphate - the cause of Lesch-Nyhan syndrome [49].
Given its high heritability, further genetic studies are warranted. However, future studies require careful phenotypic characterisation. There should be clear distinction between studies examining the genetics of hyperuricaemia, which probably are best judged at a young age before development of co-morbidities, drug intake and age-related renal impairment, and those linking genetic associations with crystal deposition and gout, since different associations may emerge.
Dietary factors
An association between gout and dietary factors has been recognised for centuries. Only recently, however, has this been confirmed in large, well-designed epidemiological studies (Table 3). The Health Professionals Follow-up Study (HPFS) was a large, prospective cohort study that followed 51,529 male health professionals, documenting 757 incident cases of gout over a 12-year period. Gout cases were required to meet the 1977 American Rheumatism Association preliminary criteria [24]. Dietary intake was assessed using a semiquantitative food frequency questionnaire administered at baseline and at 4-year and 8-year follow-up [50]. Dietary consumption of meat and seafood was associated with an increased risk of gout, whereas consumption of dairy products appeared to be protective [51]. After adjustment for age, body mass index (BMI), diuretic use, hypertension, renal failure, alcohol intake, and other dietary factors, the multivariate relative risk (RR) of developing gout was 1.41 among men in the highest quintile of total meat intake compared with those in the lowest quintile (95% CI = 1.07, 1.86). The multivariate RR of developing gout in those among the highest quintile of seafood consumption versus the lowest quintile was 1.51 (95% CI = 1.17, 1.95). Consumption of purine-rich vegetables was not associated with the development of gout. The risk of developing gout decreased with increasing consumption of dairy products (highest versus lowest quintile; multivariate RR = 0.56, 95% CI = 0.42, 0.74). This association was seen for low-fat dairy products (multivariate RR = 0.58, 95% CI = 0.45, 0.77) but not for high-fat dairy products (highest versus lowest quintile; multivariate RR = 1.00, 95% CI = 0.77, 1.29).
Table 3.
Risk of incident gout in men with diet and alcohol intake: Health Professionals Follow-up Study
| Dietary factor | Comparison | Multivariate RR (95% CI) |
|---|---|---|
| Total meat intake [51] | Highest versus lowest quintile | 1.41 (1.07, 1.86) |
| Seafood [51] | Highest versus lowest quintile | 1.51 (1.17,1.95) |
| Vegetable purines [51] | Highest versus lowest quintile | 0.96 (0.74, 1.24) |
| Dairy products [51] | Highest versus lowest quintile | 0.56 (0.42, 0.74) |
| Low-fat dairy products [51] | Highest versus lowest quintile | 0.58 (0.45, 0.76) |
| High-fat dairy products [51] | Highest versus lowest quintile | 1.00 (0.77, 1.29) |
| Coffee [52] | ≥ 6 cups per day versus none | 0.41 (0.19, 0.88) |
| Decaffeinated coffee [52] | ≥ 4 cups per day versus none | 0.73 (0.46, 1.17) |
| Tea [52] | ≥ 4 cups per day versus none | 0.82 (0.38, 1.75) |
| Total caffeine [52] | Highest versus lowest quintile | 0.83 (0.64, 1.08) |
| Sugar-sweetened soft drinks [53] | ≥ 2 drinks per day versus none | 1.85 (1.08, 3.16) |
| Diet soft drinks [53] | ≥ 2 drinks per day versus none | 1.12 (0.82, 1.52) |
| Total fructose [53] | Highest versus lowest quintile | 1.81 (1.31, 2.50) |
| Total vitamin C [55] | ≥ 1,500 mg versus <250 mg/day | 0.55 (0.38, 0.80) |
| Total alcohol [58] | ≥ 50 g per day versus none | 2.53 (1.73, 3.70) |
| Beer [58] | ≥ 2 drinks per day versus none | 2.51 (1.77, 3.55) |
| Spirits [58] | ≥ 2 drinks per day versus none | 1.60 (1.19, 2.16) |
| Wine [58] | ≥ 2 drinks per day versus none | 1.05 (0.64, 1.72) |
CI, confidence interval; RR, relative risk.
In a subsequent HPFS study, the same authors examined the relationship between coffee consumption and the risk of developing gout [52]. Consumption of six or more cups of coffee per day appeared to be protective against the development of gout (multivariate RR = 0.41, 95% CI = 0.19, 0.88) compared with no coffee consumption. Although the risk of developing gout was not significantly decreased in those drinking four or more cups of decaffeinated coffee per day compared with no consumption (multivariate RR = 0.73, 95% CI = 0.46, 1.17), a significant protective effect was seen in those drinking smaller amounts of decaeinated coffee and a statistically significant trend was seen across the groups. Tea consumption and total caffeine intake were not associated with development of gout. Subsequently, the authors explored the relationship between intake of sugar-sweetened soft drinks and fructose and the risk of incident gout [53]. Fructose increases degradation of purine nucleotides, which act as a substrate for uric acid production [54]. Consumption of two or more sugar-sweetened soft drinks per day was a risk factor for the development of gout (multivariate RR = 1.85, 95% CI = 1.08, 3.16) compared with less than one per month [53]. Diet soft drinks did not appear to confer risk of developing gout. Increasing total fructose intake, however, increased the risk of incident gout (highest versus lowest quintile; multivariate RR = 1.81, 95% CI = 1.31, 2.50).
Most recently, the authors have examined the risk of developing gout with vitamin C consumption using 20-year follow-up data from the HPFS (including 1,317 incident gout cases) [55]. Higher total vitamin C consumption appeared to be protective against the development of gout. The multivariate RR of developing gout was 0.55 (95% CI = 0.38, 0.80) in those consuming >1,500 mg dietary vitamin C per day compared with those consuming < 250 mg/day.
Local variation in the prevalence of gout has been suggested to be influenced by dietary habits. A cross-sectional study of 5,003 adults found the prevalence of gout across five coastal cities in the Shandong province of China to range from 0.50% to 2.55% [17]. Consumption of meat and seafood was significantly higher in the city of Yantai where gout prevalence was the highest, compared with the city of Dongying that had the lowest prevalence of gout - raising the possibility that variations in the prevalence of gout might be directly attributable to lifestyle factors [56].
Alcohol consumption
Similar to dietary factors, an association between gout and excess alcohol consumption has long been recognised. Although it is now thought that the epidemic of gout in nineteenth-century England was secondary to lead poisoning resulting from the high lead content of wines and port at this time [57], recent epidemiological data support the important relationship between alcohol consumption and risk of developing gout today.
In the HPFS, a graded association was observed between alcohol intake and the risk of developing gout on multivariate analysis (no alcohol intake, RR = 1.00; 0.1 to 4.9 g/day, RR = 1.09; 5.0 to 9.9 g/day, RR = 1.25; 10.0 to 14.9 g/day, RR = 1.32; 15.0 to 29.9 g/day, RR = 1.49; 30.0 to 49.9 g/day, RR = 1.96; ≥ 50.0 g/day, RR = 2.53) [58]. The multivariate RR was 1.17 (95% CI = 1.11, 1.22) per 10 g increase in alcohol intake per day. Comparing those who drank two or more drinks per day with those who did not drink, the risk of developing gout was greatest for beer consumption (RR = 2.51, 95% CI = 1.77, 3.55) followed by spirits (RR = 1.60, 95% CI = 1.19, 2.16), whereas wine consumption conferred no risk (RR = 1.05, 95% CI = 0.64, 1.72) (Table 3). The multivariate RR per serving per day was 1.49 (95% CI = 1.32, 1.70) for beer, 1.15 (95% CI = 1.04, 1.28) for spirits, and 1.04 (95% CI = 0.88, 1.22) for wine.
Excess alcohol consumption has also been shown to be an important risk factor for the development of gout in women. In the Framingham Heart Study, alcohol consumption was categorised as heavy, moderate and abstinent/light [29]. Compared with people whose alcohol intake was abstinent/light (0 to 1 ounce per week), the multivariate RR of developing gout in the heavy alcohol consumption category (≥ 7 ounces per week) was 3.10 (95% CI = 1.69, 5.68) in women and 2.21 (95% CI = 1.56, 3.14) in men.
Alcohol consumption has also been shown to trigger attacks of acute gout. In an Internet cross-over study of 321 acute attacks in 197 subjects, a dose-dependent relationship was found between the number of alcoholic drinks consumed in the previous 48 hours and an acute attack of gout (seven alcoholic drinks in 48 hours: OR = 2.5, 95% CI = 1.1, 5.9) [59].
Several mechanisms by which alcohol predisposes to hyperuricaemia have been proposed, including reduced renal urate excretion via lactic acidosis or lead poisoning, increased urate production via ethanol-induced accelerated degradation of purine nucleotides or the high purine content of beer enhancing substrate provision, and poor compliance with urate-lowering therapy [60].
Metabolic syndrome
Gout has an important association with the metabolic syndrome. A study undertaken using data from the Third National Health and Nutrition Examination Survey, conducted between 1988 and 1994, compared the prevalence of the metabolic syndrome - defined using the revised National Cholesterol Education Program Adult Treatment Panel III criteria [61] - between individuals with gout and control subjects without gout [61,62]. Amongst individuals with gout the prevalence of metabolic syndrome was 62.8%, compared with 25.4% among those without gout (age-adjusted and sex-adjusted OR = 3.05, 95% CI = 2.01, 4.61).
Other studies have reported the relationship between gout and various individual components of the metabolic syndrome. In the HPFS, on multivariate analysis, there was a clear graded association between increasing BMI and the risk of incident gout in men (BMI <21 kg/m2, RR = 0.85; BMI 21 to 22.9 kg/m2, RR = 1.00; BMI 23 to 24.9 kg/m2, RR = 1.31; BMI 25 to 29.9 kg/m2, RR = 1.95; BMI 30 to 34.9 kg/m2, RR = 2.33; BMI ≥ 35 kg/m2, RR = 2.97) [63]. A similar relationship was seen between incident gout and waist:hip ratio. Compared with men who had maintained their weight, graded associations were also seen between weight gain since study entry and increased risk of gout (weight gain ≥ 30 lbs, multivariate RR = 1.72, 95% CI = 1.02, 2.91). Weight loss since study entry decreased the risk of gout (weight loss ≥ 10 lbs, multivariate RR = 0.61, 95% CI = 0.40, 0.92). The Framingham Heart Study also identified obesity as a risk factor for developing gout [29]. The multivariate RR of developing gout was 2.74 (95% CI = 1.65, 4.58) in obese women (BMI ≥ 30 kg/m2) and 2.90 (95% CI = 1.89, 4.44) in obese men, compared with those with BMI <25 kg/m2.
Several studies have examined the relationship between gout and hypertension. In a case-control study undertaken in the UK-GPRD that compared 56,483 gout cases with 150,867 controls subjects with osteoarthritis (OA), the age-adjusted and sex-adjusted OR for gout in patients with hypertension was 1.52 (95% CI = 1.48, 1.56) [13]. The HPFS and Framingham Heart Study found multivariate RRs of incident gout with hypertension of 2.31 (95% CI = 1.96, 2.72) and 1.59 (95% CI = 1.12, 2.24), respectively, in men and 1.82 (95% CI = 1.06, 3.14) in women [29,63].
The UK-GPRD study found a small but statistically significant association between gout and diabetes mellitus (age-adjusted and sex-adjusted OR = 1.11, 95% CI = 1.06, 1.16) [13]. Interestingly, whereas most epidemiological studies concerning gout and components of the metabolic syndrome have focused on the risk of incident gout in people with individual co-morbid conditions, gout itself has been shown to be a risk factor for incident type 2 diabetes mellitus in men [64]. A prospective cohort study of 11,351 men was nested within the Multiple Risk Factor Intervention Trial, a randomised clinical trial of a coronary risk reduction programme in men at high risk of cardiovascular disease (CVD). After exclusion of men with diabetes at baseline, the multivariate RR of incident type 2 diabetes mellitus was 1.34 (95% CI = 1.09, 1.64) in men who self-reported gout at baseline compared with those without a history of gout [64].
Diuretic use
Diuretic use is a significant risk factor for the development of gout but this relationship is potentially confounded by the indication for diuretic therapy, such as hypertension, renal disease and cardiac failure, which may also predispose to gout. Diuretic-induced hyperuricaemia occurs via inhibition of renal urate excretion at the low-affinity asymmetric urate transporter, organic anion transporter 4 (OAT4) [65]. Diuretics have been shown to be a risk factor for incident gout in men in the HPFS (RR = 1.77, 95% CI = 1.42, 2.20) [63] and in both sexes in the Framingham Heart Study (men RR = 3.41, 95% CI = 2.38, 4.89; women RR = 2.39, 95% CI = 1.53, 3.74) [29] after adjustment for multiple confounders, including hypertension. An association between diuretic use and gout was also seen in the UK-GPRD (OR = 1.72, 95% CI = 1.67, 1.76, adjusted for age and sex but not hypertension) [13]. In contrast, a Dutch case-control study did not find an association between incident gout and prior diuretic use (incidence rate ratio = 0.6, 95% CI = 0.2, 2.0), although the study was small (70 gout cases) and several important confounding variables were not adjusted for [66]. Diuretic use also predisposes to recurrent acute gout. In the Internet cross-over study described above, diuretic use in the preceding 48 hours was associated with acute attacks of gout (OR = 3.6, 95% CI = 1.4, 9.7) [67].
Renal disease
Chronic renal disease is an important risk factor for gout. It was associated with gout in both the HPFS (RR = 3.61, 95% CI = 1.60, 8.14, adjusted for multiple confounders including diuretic use) [63] and the UK-GPRD (age-adjusted and sex-adjusted OR = 4.95, 95% CI = 4.28, 5.72) [13]. Gout can be particularly challenging in patients with end-stage renal disease or following renal transplantation. A retrospective study of 259,209 patients registered in the US Renal Data System found the incidence of gout to be 5.4% in the first year of dialysis and 15.4% in the first 5 years [68]. In the UK-GPRD, gout was associated with both renal transplantation (OR = 25.13, 95% CI = 12.97, 48.68) and cyclosporin use (OR = 7.93, 95% CI = 5.97, 10.54), although risk estimates were only adjusted for age and sex [13].
Osteoarthritis
Gout shows a striking predilection to affect certain joints, most strikingly the first MTP joint. Although it is not known why gout demonstrates such a characteristic pattern, the first MTP joint is also a target joint for OA and it has been postulated that MSU crystals may deposit more readily in osteoarthritic cartilage [69]. Radiographic and clinical studies support the tendency of gout to occur at joints already affected by OA. A Polish hospital-based study of 262 subjects with gout found the presence of gout and radiographic OA to be significantly correlated at the first MTP joints, tarsal joints and knees [70]. More recently, a community-based study of 164 subjects with gout found a strong association between sites of acute attacks of gout and the presence of clinical OA (multivariate OR = 7.94, 95% CI = 6.27, 10.05), especially at the first MTP joints, tarsal joints, knees and finger distal interphalangeal joints [71]. Such cross-sectional associations cannot differentiate whether OA predisposes to or arises from MSU crystal deposition. The association between gout and OA at individual joint sites was not affected by gout duration [71], however, which does not support the latter hypothesis. Nodal OA was no more frequent in gout subjects than control subjects without gout from the same community [72]. These findings suggest that OA predisposes to local MSU crystal deposition in individuals with gout, but is not a risk factor for gout per se.
Mortality and cardiovascular disease associated with gout
As discussed above, there is a strong association between gout and co-morbid disease, including traditional cardiovascular risk factors. Gout has a well-recognised association with CVD, but there has been much debate as to whether this risk is independent of traditional cardiovascular risk factors (Table 4).
Table 4.
Risk of mortality and cardiovascular disease associated with gout
| Study estimate (95% CI) | Design | Population | Comparison | Multivariate risk |
|---|---|---|---|---|
| All-cause mortality | ||||
| HPFS [73] | Cohort: stratified by baseline CHD status | 51,927 male health professionals, 40 to 75 years old | CHD: gout vs. no gout | RR = 1.25 (1.09, 1.45) |
| No CHD: gout vs. no gout | RR = 1.28 (1.15, 1.41) | |||
| MRFIT [74] | MRFIT RCT participants | 9,105 men, 41 to 63 years old | Gout vs. no gout | HR = 1.09 (1.00, 1.19) |
| CGMH [75] | Cohort: health screening programme participants | 61,527 men and women, 30 to 74 years old | Gout vs. normouricaemia | HR = 1.46 (1.12, 1.91) |
| All cardiovascular death | ||||
| HPFS [73] | Cohort: stratified by baseline CHD status | 51,927 male health professionals, 40 to 75 years old | CHD: gout vs. no gout | RR = 1.26 (1.07, 1.50) |
| No CHD: gout vs. no gout | RR = 1.38 (1.15, 1.66) | |||
| MRFIT [74] | MRFIT RCT participants | 9,105 men, 41 to 63 years old | Gout vs. no gout | HR = 1.21 (0.99, 1.49) |
| CGMH [75] | Cohort: health screening programme participants | 61,527 men and women, 30 to 74 years old | Gout vs. normouricaemia | HR = 1.97 (1.08, 3.59) |
| Fatal CHD | ||||
| HPFS [73] | Cohort: stratified by baseline CHD status | 51,927 male health professionals, 40 to 75 years old | CHD: gout vs. no gout | RR = 1.24 (1.04, 1.49) |
| No CHD: gout vs. no gout | RR = 1.55 (1.24, 1.93) | |||
| MRFIT [74] | MRFIT RCT participants | 9,105 men, 41 to 63 years old | Gout vs. no gout | HR = 1.35 (1.06, 1.72) |
| Fatal myocardial infarction | ||||
| MRFIT [74] | MRFIT RCT participants | 9,105 men, 41 to 63 years old | Gout vs. no gout | HR = 1.35 (0.94, 1.93) |
| Cardiovascular disease | ||||
| Dutch primary care [76] | Follow-up of case-control study participants | 261 men and women | Gout vs. no gout | RR = 0.98 (0.65, 1.47) |
| Coronary heart disease | ||||
| Framingham Study [78] | Cohort | 2,316 men | Gout vs. no gout | RR = 1.6 (1.1, 2.5) |
| Meharry-Hopkins Study [77] | Cohort | 1,552 male physicians | Gout vs. no gout | RR = 0.59 (0.24, 1.46) |
| GPRD [13] | Case-control | 207,350 men and women | Gout vs. osteoarthritis | OR = 1.75 (1.70, 1.79) |
| Myocardial infarction | ||||
| Framingham Study [78] | Cohort | 2,316 men | Gout vs. no gout | RR = 1.5 (0.9, 2.6) |
| MRFIT [79] | MRFIT RCT participants | 12,866 men | Gout vs. no gout | OR = 1.26 (1.14, 1.40) |
| Angina pectoris | ||||
| Framingham Study [78] | Cohort | 2,316 men | Gout vs. no gout | RR = 1.8 (1.1, 3.2) |
CGMH, Chang Gung Memorial Hospital; CHD, coronary heart disease; CI, confidence interval; GPRD, General Practice Research Database; HPFS, Health Professionals Follow-up Study; HR, hazard ratio; MRFIT, Multiple Risk Factor Intervention Trial; OR, odds ratio; RCT, randomised controlled trial; RR, relative risk.
The association between gout and all-cause and cardiovascular mortality was examined in men in the HPFS [73]. The multivariate RR of death from any cause was 1.28 (95% CI = 1.15, 1.41) in men with self-reported physician-diagnosed gout but no history of CVD at baseline, compared with those with neither gout nor CVD. Death from both CVD (RR = 1.38, 95% CI = 1.15, 1.66) and coronary heart disease (CHD) (RR = 1.55, 95% CI = 1.24, 1.93) were also more common in those with gout. Similar risk estimates were seen when, amongst men with pre-existing CHD at baseline, mortality was compared between those who had self-reported physician-diagnosed gout and those who did not: all-cause mortality, RR = 1.25 (95% CI = 1.09, 1.45); CVD death, RR = 1.26 (95% CI = 1.07, 1.50); and fatal CHD, RR = 1.24 (95% CI = 1.04, 1.49). Gout was associated with death from any cause (multivariate RR = 1.49, 95% CI = 1.43, 1.55) and with CVD death (multivariate RR = 1.47, 95% CI = 1.25, 1.59) in the study of gout in renal disease undertaken in the US Renal Data System described above [68]. In the Multiple Risk Factor Intervention Trial cohort, there was an increased risk of CHD death (hazard ratio = 1.35, 95% CI = 1.06, 1.72) in men with self-reported gout at baseline compared with those without no history of gout [74]. However, gout was not associated with death from acute myocardial infarction or CVD.
These studies inform about all-cause and cardiovascular mortality in men. A further study examined all-cause and cardiovascular mortality in 61,527 people of either sex identified from health screening participants attending a Taiwanese hospital between 2000 and 2006 [75]. Multivariate hazard ratios of all-cause and cardiovascular mortality between 1,311 subjects with gout compared with 48,021 subjects with normouricaemia were 1.46 (95% CI = 1.12, 1.91) and 1.97 (95% CI = 1.08, 3.59), respectively. Hyperuricaemia per se was not associated with mortality.
Several studies have examined the relationship between gout and cardiovascular morbidity. A Dutch general practice study compared incident CVD (a composite outcome of angina, myocardial infarction, heart failure, cerebrovascular accident, transient ischaemic attack, and peripheral vascular disease) in 170 gout cases and 340 control subjects without prevalent CVD at baseline [76]. Gout was not an independent risk factor for CVD (risk ratio = 0.98, 95% CI = 0.65, 1.47), although only a small number of potential confounders were accounted for. Similarly in the Meharry-Hopkins Study of 1,552 male This article is part of a review series on Gout, edited by Alex So. Other articles in the series can be found online at physicians, self-reported gout was not an independent risk factor for CHD over a mean follow-up period of 30 years (multivariate RR = 0.59, 95% CI = 0.24, 1.46) [77]. In contrast, in the Framingham Study, the 2-year age-adjusted incidence of CHD was 5.8/1,000 in men with gout compared with 3.8/1,000 in those without gout (multivariate RR = 1.6, 95% CI = 1.1, 1.25) [78]. Interestingly, increased risk of CHD was primarily attributable to a twofold risk of angina. In the UK-GPRD study, an association was observed between gout and coronary artery disease (age-adjusted and sex-adjusted OR = 1.75, 95% CI = 1.70, 1.79) [13]. Within the Multiple Risk Factor Intervention Trial cohort, gout was found to be a significant independent predictor of subsequent nonfatal acute myocardial infarction (OR = 1.31, 95% CI = 1.24, 1.38) after adjustment for multiple confounding variables including co-morbidity and hyperuricaemia [79]. An association between gout and coronary heart disease was also seen in the UK-GPRD (age- adjusted and sex-adjusted OR = 1.75, 95% CI = 1.70, 1.79) [13].
Conclusions
Gout is the most common form of inflammatory arthritis, and the findings of several studies suggest that the prevalence and incidence of gout has risen in recent decades. Numerous risk factors for the development of gout in men and women have now been established in prospective epidemiological studies, including hyperuricaemia, genetic factors, dietary factors, alcohol consumption, metabolic syndrome (including hypertension and obesity), diuretic use and renal disease. OA appears to predispose to local MSU crystal deposition but is not a risk factor for the development of gout per se. Gout appears to be an independent risk factor for all-cause mortality and CVD mortality and morbidity beyond that which would be expected from its strong association with traditional co-morbid cardiovascular risk factors.
Abbreviations
BMI: body mass index; CHD: coronary heart disease; CI: confidence interval; CVD: cardiovascular disease; HPFS: Health Professionals Follow-up Study; MSU: monosodium urate; MTP: metatarsophalangeal; OA: osteoarthritis; OR: odds ratio; RR: relative risk; SUA: serum uric acid; UK-GPRD: UK General Practice Research Database; URAT1: urate transporter 1.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Edward Roddy, Email: e.roddy@cphc.keele.ac.uk.
Michael Doherty, Email: michael.doherty@nottingham.ac.uk.
Acknowledgements
ER is supported by an Arthritis Research UK Primary Care Centre Grant (18139).
References
- Roddy E, Zhang W, Doherty M. Is gout associated with reduced quality of life? A case-control study. Rheumatology. 2007;46:1441–1444. doi: 10.1093/rheumatology/kem150. [DOI] [PubMed] [Google Scholar]
- Singh JA, Strand V. Gout is associated with more comorbidities, poorer health-related quality of life and higher healthcare utilisation in US veterans. Ann Rheum Dis. 2008;67:1310–1316. doi: 10.1136/ard.2007.081604. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Hirsch JD, Terkeltaub R, Khanna D, Singh JA, Sarkin A, Kavanaugh A. Perceptions of disease and health-related quality of life among patients with gout. Rheumatology. 2009;48:582–586. doi: 10.1093/rheumatology/kep047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennane GAQ, Rose BS, Isdale IC. Gout in the Maori. Ann Rheum Dis. 1960;19:120–125. doi: 10.1136/ard.19.2.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Rose BS. Uric acid, gout and public health in the South Pacific. N Z Med J. 1966;65:295–300. [PubMed] [Google Scholar]
- Klemp P, Stansfield SA, Castle B, Robertson MC. Gout is on the increase in New Zealand. Ann Rheum Dis. 1997;56:22–26. doi: 10.1136/ard.56.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace KL, Riedel AA, Joseph-Ridge N, Wortmann R. Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004;31:1582–1587. [PubMed] [Google Scholar]
- Lawrence RC, Hochberg MC, Kelsey JL, McDuffie FC, Medsger TA Jr, Felts WR, Shulman LE. Estimates of the prevalence of selected arthritic and musculoskeletal diseases in the United States. J Rheumatol. 1989;16:427–441. [PubMed] [Google Scholar]
- Lawrence RC, Helmick CG, Arnett FC, Deyo RA, Felson DT, Giannini EH, Heyse SP, Hirsch R, Hochberg MC, Hunder GG, Liang MH, Pillemer SR, Steen VD, Wolfe F. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778–799. doi: 10.1002/1529-0131(199805)41:5<778::AID-ART4>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Currie WJ. Prevalence and incidence of the diagnosis of gout in Great Britain. Ann Rheum Dis. 1979;38:101–106. doi: 10.1136/ard.38.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven MM. Prevalence of chronic arthritis in four geographical areas of the Scottish Highlands. Ann Rheum Dis. 1992;51:186–194. doi: 10.1136/ard.51.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CM, Lloyd DC, Lewis J. The prevalence and prophylaxis of gout in England. J Clin Epidemiol. 1995;48:1153–1158. doi: 10.1016/0895-4356(94)00244-K. [DOI] [PubMed] [Google Scholar]
- Mikuls TR, Farrar JT, Bilker WB, Fernandes S, Schumacher HR Jr, Saag KG. Gout epidemiology: results from the UK General Practice Research Database, 1990-1999. Ann Rheum Dis. 2005;64:267–272. doi: 10.1136/ard.2004.024091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annemans L, Spaepen E, Gaskin M, Bonnemaire M, Malier V, Gilbert T, Nuki G. Gout in the UK and Germany: prevalence, comorbidities and management in general practice 2000-2005. Ann Rheum Dis. 2008;67:960–966. doi: 10.1136/ard.2007.076232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliot AJ, Cross KW, Fleming DM. Seasonality and trends in the incidence and prevalence of gout in England and Wales 1994-2007. Ann Rheum Dis. 2009;68:1728–1733. doi: 10.1136/ard.2008.096693. [DOI] [PubMed] [Google Scholar]
- Nan H, Qiao Q, Dong Y, Gao W, Tang B, Qian R, Tuomilehto J. The prevalence of hyperuricemia in a population of the coastal city of Qingdao, China. J Rheumatol. 2006;33:1346–1350. [PubMed] [Google Scholar]
- Miao Z, Li C, Chen Y, Zhao S, Wang Y, Wang Z, Chen X, Xu F, Wang F, Sun R, Hu J, Song W, Yan S, Wang C. Dietary and lifestyle changes associated with high prevalence of hyperuricaemia and gout in the Shandong coastal cities of Eastern China. J Rheumatol. 2008;35:1859–1864. [PubMed] [Google Scholar]
- Chou CT, Pei L, Chang DM, Lee CF, Schumacher HR, Liang MH. Prevalence of rheumatic diseases in Taiwan: a population study of urban, suburban, rural differences. J Rheumatol. 1994;21:302–306. [PubMed] [Google Scholar]
- Chang SJ, Ko YC, Wang TN, Chang FT, Cinkotai FF, Chen CJ. High prevalence of gout and related risk factors in Taiwan's Aborigines. J Rheumatol. 1997;24:1364–1369. [PubMed] [Google Scholar]
- Lin CJ, Liu JT, Chang CH, Nowalk MP. Association of obesity and chronic diseases in Taiwan. Asia Pac J Public Health. 2006;18:8–14. doi: 10.1177/10105395060180030301. [DOI] [PubMed] [Google Scholar]
- Roddy E, Zhang W, Doherty M. Concordance of the management of chronic gout in a UK primary-care population with the EULAR gout recommendations. Ann Rheum Dis. 2007;66:1311–1315. doi: 10.1136/ard.2007.070755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal B, Foxall M, Dysart T, Carey F, Whittaker M. How is gout managed in primary care? A review of current practice and proposed guidelines. Clin Rheumatol. 2000;19:21–25. doi: 10.1007/s100670050005. [DOI] [PubMed] [Google Scholar]
- Singh JA, Hodges JS, Toscano JP, Asch SM. Quality of care for gout in the US needs improvement. Arthritis Rheum. 2007;57:822–829. doi: 10.1002/art.22767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace SL, Robinson H, Masi AT, Decker JL, Mccarty DJ. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20:895–900. doi: 10.1002/art.1780200320. [DOI] [PubMed] [Google Scholar]
- Zhang W, Doherty M, Pascual E, Bardin T, Barskova V, Conaghan P, Gerster J, Jacobs J, Leeb B, Liote F, McCarthy G, Netter P, Nuki G, Perez-Ruiz F, Pignone A, Pimentao J, Punzi L, Roddy E, Uhlig T, Zimmermann-Gorska I. EULAR evidence based recommendations for gout. Part I: diagnosis. Report of a task force of the Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT) Ann Rheum Dis. 2006;65:1301–1311. doi: 10.1136/ard.2006.055251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens HJ, Janssen M, van de Lisdonk EH, Fransen J, van Riel PL, van Weel C. Limited validity of the American College of Rheumatology criteria for classifying patients with gout in primary care. Ann Rheum Dis. 2010;69:1255–1256. doi: 10.1136/ard.2009.123687. [DOI] [PubMed] [Google Scholar]
- Malik A, Schumacher HR, Dinnella JE, Clayburne GM. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol. 2009;15:22–24. doi: 10.1097/RHU.0b013e3181945b79. [DOI] [PubMed] [Google Scholar]
- Roubenoff R, Klag MJ, Mead LA, Liang KY, Seidler AJ, Hochberg MC. Incidence and risk factors for gout in white men. JAMA. 1991;266:3004–3007. doi: 10.1001/jama.266.21.3004. [DOI] [PubMed] [Google Scholar]
- Bhole V, de Vera M, Rahman MM, Krishnan E, Choi H. Epidemiology of gout in women: fifty-two-year followup of a prospective cohort. Arthritis Rheum. 2010;62:1069–1076. doi: 10.1002/art.27338. [DOI] [PubMed] [Google Scholar]
- Arromdee E, Michet CJ, Crowson CS, O'Fallon WM, Gabriel SE. Epidemiology of gout: is the incidence rising? J Rheumatol. 2002;29:2403–2406. [PubMed] [Google Scholar]
- Lin KC, Lin HY, Chou P. Community based epidemiological study on hyperuricemia and gout in Kin-Hu, Kinmen. J Rheumatol. 2000;27:1045–1050. [PubMed] [Google Scholar]
- Lin KC, Lin HY, Chou P. The interaction between uric acid level and other risk factors on the development of gout among asymptomatic hyperuricemic men in a prospective study. J Rheumatol. 2000;27:1501–1505. [PubMed] [Google Scholar]
- Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med. 1987;82:421–426. doi: 10.1016/0002-9343(87)90441-4. [DOI] [PubMed] [Google Scholar]
- Scott JT, Pollard AC. Uric acid excretion in the relatives of patients with gout. Ann Rheum Dis. 1970;29:397–400. doi: 10.1136/ard.29.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerson BT, Nagel SL, Duff y DL, Martin NG. Genetic control of the renal clearance of urate: a study of twins. Ann Rheum Dis. 1992;51:375–377. doi: 10.1136/ard.51.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So A, Thorens B. Uric acid transport and disease. J Clin Invest. 2010;120:1791–1799. doi: 10.1172/JCI42344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T, Matsuo H, Kikuchi Y, Oda T, Ichida K, Hosoya T, Shimokata K, Niwa T, Kanai Y, Endou H. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417:447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- Graessler J, Graessler A, Unger S, Kopprasch S, Tausche AK, Kuhlisch E, Schroeder HE. Association of the human urate transporter 1 with reduced renal uric acid excretion and hyperuricemia in a German Caucasian population. Arthritis Rheum. 2006;54:292–300. doi: 10.1002/art.21499. [DOI] [PubMed] [Google Scholar]
- Taniguchi A, Urano W, Yamanaka M, Yamanaka H, Hosoyamada M, Endou H, Kamatani N. A common mutation in an organic anion transporter gene, SLC22A12, is a suppressing factor for the development of gout. Arthritis Rheum. 2005;52:2576–2577. doi: 10.1002/art.21242. [DOI] [PubMed] [Google Scholar]
- Le MPT, Shafiu M, Mu W, Johnson RJ. SLC2A9 - a fructose transporter identified as a novel uric acid transporter. Nephrol Dial Transplant. 2008;23:2746–2749. doi: 10.1093/ndt/gfn349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield MJ, Munroe PB, O'Neill D, Witkowska K, Charchar FJ, Doblado M, Evans S, Eyheramendy S, Onipinla A, Howard P. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008;5:e197. doi: 10.1371/journal.pmed.0050197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Sanna S, Maschio A, Busonero F, Usala G, Mulas A, Lai S, Dei M, Orru M, Albai G, Bandinelli S, Schlessinger D, Lakatta E, Scuteri A, Najjar SS, Guralnik J, Naitza S, Crisponi L, Cao A, Abecasis G, Ferrucci L, Uda M, Chen WM, Nagaraja R. The GLUT9 gene is associated with serum uric acid levels in Sardinia and Chianti cohorts. PLoS Genet. 2007;3:e194. doi: 10.1371/journal.pgen.0030194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doring A, Gieger C, Mehta D, Gohlke H, Prokisch H, Coassin S, Fischer G, Henke K, Klopp N, Kronenberg F, Paulweber B, Pfeufer A, Rosskopf D, Volzke H, Illig T, Meitinger T, Wichmann HE, Meisinger C. SLC2A9 influences uric acid concentrations with pronounced sex-specific effects. Nat Genet. 2008;40:430–436. doi: 10.1038/ng.107. [DOI] [PubMed] [Google Scholar]
- Dehghan A, Kottgen A, Yang Q, Hwang SJ, Kao WL, Rivadeneira F, Boerwinkle E, Levy D, Hofman A, Astor BC, Benjamin EJ, van Duijn CM, Witteman JC, Coresh J, Fox CS. Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet. 2008;372:1953–1961. doi: 10.1016/S0140-6736(08)61343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano W, Taniguchi A, Anzai N, Inoue E, Kanai Y, Yamanaka M, Endou H, Kamatani N, Yamanaka H. Sodium-dependent phosphate cotransporter type 1 sequence polymorphisms in male patients with gout. Ann Rheum Dis. 2010;69:1232–1234. doi: 10.1136/ard.2008.106856. [DOI] [PubMed] [Google Scholar]
- Kolz M, Johnson T, Sanna S, Teumer A, Vitart V, Perola M, Mangino M, Albrecht E, Wallace C, Farrall M, Johansson A, Nyholt DR, Aulchenko Y, Beckmann JS, Bergmann S, Bochud M, Brown M, Campbell H. EUROSPAN Consortium; Connell J, Dominiczak A, Homuth G, Lamina C, McCarthy MI. ENGAGE Consortium. Meitinger T, Mooser V, Munroe P, Nauck M, Peden J. et al. Metaanalysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009;5:e1000504. doi: 10.1371/journal.pgen.1000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Harst P, Bakker SJ, de Boer RA, Wolffenbuttel BH, Johnson T, Caulfield MJ, Navis G. Replication of the five novel loci for uric acid concentrations and potential mediating mechanisms. Hum Mol Genet. 2010;19:387–395. doi: 10.1093/hmg/ddp489. [DOI] [PubMed] [Google Scholar]
- Morcillo S, Rojo-Martinez G, Martin-Nunez G, Gómez-Zumaquero JM, García-Fuentes E, de Adana MR. Trp64Arg polymorphism of the ADRB3 gene predicts hyperuricemia risk in a population from southern Spain. J Rheumatol. 2010;37:417–421. doi: 10.3899/jrheum.090637. [DOI] [PubMed] [Google Scholar]
- Merriman TR, Dalbeth N. The genetic basis of hyperuricaemia and gout. Joint Bone Spine. 2010. in press . [DOI] [PubMed]
- Willett WC, Sampson L, Stampfer MJ, Rosner B, Bain C, Witschi J, Hennekens CH, Speizer FE. Reproducibility and validity of a semiquantitative food frequency questionnaire. Am J Epidemiol. 1985;122:51–65. doi: 10.1093/oxfordjournals.aje.a114086. [DOI] [PubMed] [Google Scholar]
- Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med. 2004;350:1093–1103. doi: 10.1056/NEJMoa035700. [DOI] [PubMed] [Google Scholar]
- Choi HK, Willett W, Curhan G. Coffee consumption and risk of incident gout in men: a prospective study. Arthritis Rheum. 2007;56:2049–2055. doi: 10.1002/art.22712. [DOI] [PubMed] [Google Scholar]
- Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ. 2008;336:309–312. doi: 10.1136/bmj.39449.819271.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivio KO, Becker A, Meyer LJ, Greene ML, Nuki G, Seegmiller JE. Stimulation of human purine synthesis de novo by fructose infusion. Metabolism. 1975;24:861–869. doi: 10.1016/0026-0495(75)90133-X. [DOI] [PubMed] [Google Scholar]
- Choi HK, Gao X, Curhan G. Vitamin C intake and the risk of gout in men: a prospective study. Arch Intern Med. 2009;169:502–507. doi: 10.1001/archinternmed.2008.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roddy E. Hyperuricemia, gout, and lifestyle factors. J Rheumatol. 2008;35:1689–1691. [PubMed] [Google Scholar]
- Ball GV. Two epidemics of gout. Bull Hist Med. 1971;45:401–408. [PubMed] [Google Scholar]
- Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Alcohol intake and risk of incident gout in men: a prospective study. Lancet. 2004;363:1277–1281. doi: 10.1016/S0140-6736(04)16000-5. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Woods R, Chaisson CE, Neogi T, Niu J, McAlindon TE, Hunter D. Alcohol consumption as a trigger of recurrent gout attacks. Am J Med. 2006;119:800–808. doi: 10.1016/j.amjmed.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Fam AG. Gout, diet, and the insulin resistance syndrome. J Rheumatol. 2002;29:1350–1355. [PubMed] [Google Scholar]
- Grundy SM, Brewer HB Jr, Cleeman JI, Smith SC Jr, Lenfant C. American Heart Association; National Heart, Lung, and Blood Institute. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109:433–438. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- Choi HK, Ford ES, Li C, Curhan G. Prevalence of the metabolic syndrome in patients with gout: the Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2007;57:109–115. doi: 10.1002/art.22466. [DOI] [PubMed] [Google Scholar]
- Choi HK, Atkinson K, Karlson EW, Curhan G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: the health professionals follow-up study. Arch Intern Med. 2005;165:742–748. doi: 10.1001/archinte.165.7.742. [DOI] [PubMed] [Google Scholar]
- Choi HK, De Vera MA, Krishnan E. Gout and the risk of type 2 diabetes among men with a high cardiovascular risk profile. Rheumatology. 2008;47:1567–1570. doi: 10.1093/rheumatology/ken305. [DOI] [PubMed] [Google Scholar]
- Hagos Y, Stein D, Ugele B, Burckhardt G, Bahn A. Human renal organic anion transporter 4 operates as an asymmetric urate transporter. J Am Soc Nephrol. 2007;18:430–439. doi: 10.1681/ASN.2006040415. [DOI] [PubMed] [Google Scholar]
- Janssens HJ, van de Lisdonk EH, Janssen M, van den Hoogen HJ, Verbeek AL. Gout, not induced by diuretics? A case-control study from primary care. Ann Rheum Dis. 2006;65:1080–1083. doi: 10.1136/ard.2005.040360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DJ, York M, Chaisson CE, Woods R, Niu J, Zhang Y. Recent diuretic use and the risk of recurrent gout attacks: the online case-crossover gout study. J Rheumatol. 2006;33:1341–1345. [PubMed] [Google Scholar]
- Cohen SD, Kimmel PL, Neff R, Agodoa L, Abbott KC. Association of incident gout and mortality in dialysis patients. J Am Soc Nephrol. 2008;19:2204–2210. doi: 10.1681/ASN.2007111256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simkin PA. The pathogenesis of podagra. Ann Intern Med. 1977;86:230–233. doi: 10.7326/0003-4819-86-2-230. [DOI] [PubMed] [Google Scholar]
- Kawenoki-Minc E, Eyman E, Leo W, Werynska-Przybylska J. Osteoarthrosis and spondylosis in gouty patients. Analysis of 262 cases of gout. Reumatologia. 1974;12:267–277. [PubMed] [Google Scholar]
- Roddy E, Zhang W, Doherty M. Are joints affected by gout also affected by osteoarthritis? Ann Rheum Dis. 2007;66:1374–1377. doi: 10.1136/ard.2006.063768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roddy E, Zhang W, Doherty M. Gout and nodal osteoarthritis: a case-control study. Rheumatology. 2008;47:732–733. doi: 10.1093/rheumatology/ken087. [DOI] [PubMed] [Google Scholar]
- Choi HK, Curhan G. Independent impact of gout on mortality and risk for coronary heart disease. Circulation. 2007;116:894–900. doi: 10.1161/CIRCULATIONAHA.107.703389. [DOI] [PubMed] [Google Scholar]
- Krishnan E, Svendsen K, Neaton JD, Grandits G, Kuller LH. Long-term cardiovascular mortality among middle-aged men with gout. Arch Intern Med. 2008;168:1104–1110. doi: 10.1001/archinte.168.10.1104. [DOI] [PubMed] [Google Scholar]
- Kuo CF, See LC, Luo SF, Ko YS, Lin YS, Hwang JS, Lin CM, Chen HW, Yu KH. Gout: an independent risk factor for all-cause and cardiovascular mortality. Rheumatology. 2010;49:141–146. doi: 10.1093/rheumatology/kep364. [DOI] [PubMed] [Google Scholar]
- Janssens HJ, van de Lisdonk EH, Bor H, van den Hoogen HJ, Janssen M. Gout, just a nasty event or a cardiovascular signal? A study from primary care. Fam Pract. 2003;20:413–416. doi: 10.1093/fampra/cmg413. [DOI] [PubMed] [Google Scholar]
- Gelber AC, Klag MJ, Mead LA, Thomas J, Thomas DJ, Pearson TA, Hochberg MC. Gout and risk for subsequent coronary heart disease: the Meharry-Hopkins study. Arch Intern Med. 1997;157:1436–1440. doi: 10.1001/archinte.157.13.1436. [DOI] [PubMed] [Google Scholar]
- Abbott RD, Brand FN, Kannel WB, Castelli WP. Gout and coronary heart disease: the Framingham Study. J Clin Epidemiol. 1988;41:237–242. doi: 10.1016/0895-4356(88)90127-8. [DOI] [PubMed] [Google Scholar]
- Krishnan E, Baker JF, Furst DE, Schumacher HR. Gout and the risk of acute myocardial infarction. Arthritis Rheum. 2006;54:2688–2696. doi: 10.1002/art.22014. [DOI] [PubMed] [Google Scholar]