

Abstract
Introduction
Rheumatoid arthritis is an autoimmune disease in which joint inflammation leads to progressive cartilage and bone erosion. Matrix metalloproteinases (MMPs) implicated in homeostasis of the extracellular matrix play a central role in cartilage degradation. However, the role of specific MMPs in arthritis pathogenesis is largely unknown. The aim of the present study was to investigate the role of Mmp-8 (collagenase-2) in an arthritis model.
Methods
Arthritis was induced in Mmp8-deficient and wildtype mice by K/BxN serum transfer. Arthritis severity was measured by a clinical index and ankle sections were scored for synovial inflammation, cartilage damage and bone erosion. cDNA microarray analysis, real-time PCR and western blot were performed to identify differential changes in gene expression between mice lacking Mmp8 and controls.
Results
Mmp8 deficiency increased the severity of arthritis, although the incidence of disease was similar in control and deficient mice. Increased clinical score was associated with exacerbated synovial inflammation and bone erosion. We also found that the absence of Mmp8 led to increased expression of IL-1β, pentraxin-3 (PTX3) and prokineticin receptor 2 (PROKR2) in arthritic mice joints.
Conclusions
Lack of Mmp-8 is accompanied by exacerbated synovial inflammation and bone erosion in the K/BxN serum-transfer arthritis model, indicating that this Mmp has a protective role in arthritis.
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by joint inflammation and progressive destruction of cartilage and bone. Current knowledge of joint destruction indicates that matrix metalloproteinases (MMPs) have a pivotal role in cartilage damage. Articular cartilage is composed of the extracellular matrix and a small number of chondrocytes. Aggrecan and fibrillar type II collagen are the main components of the cartilage extracellular matrix. In RA, depletion of proteoglycans and the subsequent degradation of collagen lead to destruction of articular cartilage. The metalloproteinases induced by IL-1β, TNF, IL-17 and IL-18 are pivotal in this process [1-4].
Multiple pieces of evidence support the relevance of MMPs in the pathogenesis of RA. Several MMPs are highly expressed in the synovial lining and sublining of RA patients and high levels of these proteins have been detected in their sera and synovial fluid [5-7]. Specifically, the high serum levels of MMP-1 and MMP-3 have been proposed as predictors of joint destruction [8]. The role of a few of the MMPs has been analyzed in experimental arthritis models using deficient mice, and the results were variable depending on the MMP analyzed. The effect of Mmp-2 was analyzed in an antibody-induced arthritis model [9]. The Mmp2-deficient mice showed significantly exacerbated arthritis compared with wildtype mice, suggesting a suppressive role of Mmp-2 in this model. In contrast, the absence of Mmp-9 was associated with reduced severity of arthritis, indicating the need of Mmp-9 for the development of arthritis [9]. The role of Mmp-3 was analyzed in antigen-induced arthritis and collagen-induced arthritis models [10,11], and a similar incidence and severity of arthritis was displayed by Mmp3-deficient and control mice in both arthritis models. This range of results indicates the need to investigate the specific role of individual MMPs in the pathogenesis of RA to identify specific targets.
MMP-8 (collagenase-2) is mainly produced by neutrophils, although it is also expressed by a wide range of cells including chondrocytes [12] and synovial fibroblasts [13]. MMP-8 is a potent collagenolytic enzyme that is involved in the pathogenesis of several inflammatory conditions. Van Lint and colleagues showed that Mmp8-deficient mice were protected against TNF-induced lethal hepatitis [14]. Livers of knockout mice did not show the massive influx of neutrophils seen in wildtype mice, probably due to the functional link between Mmp-8 and lipopolysaccharide-induced CXC chemokine, a PMN chemokine. Their work suggests that Mmp-8 is involved in lipopolysaccharide-induced CXC chemokine release and, in turn, in neutrophil recruitment during inflammation. Likewise, the pivotal role of MMP-8 in lipopolysaccharide-induced CXC chemokine, CXCL5 and CXCL8 activation was recently reported [15]. An increased neutrophil accumulation was found, however, in induced skin carcinomas and during wound healing in mice lacking Mmp8 [16]. Also, Mmp8-deficient mice developed more severe inflammation than wildtype mice in an allergen-induced airway inflammation model and showed more neutrophils in the bronchoalveolar lavage fluid [17]. Overall, these studies indicate that the role of MMP-8 in the inflammatory process is complex and difficult to predict in advance, probably due to specific features of the tissue and stimulus involved in each situation.
Several findings suggest that MMP-8 has a role in RA pathogenesis. It is expressed in serum and synovial fluid from patients with RA. Fibroblast-like synoviocyte cultures from RA patients produce MMP-8 after TNFα stimulation [6,13]. In addition, MMP-8 regulates the activity of several chemokines implicated in RA [18,19]. In the present study we have therefore investigated the impact of Mmp8 deficiency in the induced arthritis using the K/BxN serum transfer model. We have also performed a cDNA microarray analysis to investigate differences in the transcriptional profiles from Mmp8-deficient and wildtype mice. According to our data, we conclude that Mmp-8 has a protective role in arthritis derived from the ability of this metalloprotease to induce changes in a series of inflammatory mediators.
Materials and methods
Mice
Mice lacking Mmp8 have been previously described [19] and the KRN T-cell-receptor transgenic mice were a kind gift from C Benoist and D Mathis (Harvard Medical School, Boston, MA, USA; and IGBMC, Strasbourg, France). NOD and C57BL/6 mice were purchased from Charles River (Barcelona, Spain).
Mmp8+/- (mixed C57BL/6 × 129Sv background) mice were backcrossed into the C57BL/6 background for 12 breedings. Mmp8+/- mice were then intercrossed to generate the Mmp8-/-, Mmp8+/- and Mmp8+/+ mice used for arthritis induction.
Genotypes were assessed by PCR of tail DNA. PCR reactions were made using DNA 100 ng, dNTPS 200 μM (Roche, Mannheim, Germany), specific primers 0.5 μM, MgCl2 1.5 mM, PCR buffer reaction (Roche) and Taq DNA polymerase 0.5 U (Roche). Amplification was performed using the following conditions: 94°C for 2 minutes, 30 cycles at 94°C for 1 minute, 60°C for 1 minute and 72°C for 1 minute, and a final cycle of elongation at 72°C for 4 minutes. The size of the amplified DNA products was determined in a 1.5% agarose gel in Tris-acetate-EDTA. Amplification of the wildtype allele was carried out with the primer pairs 5'-GTGGATGAATCCCCAGACTC-3' (forward) and 5'-CAAGCAATCAATTCCGGTCT-3' (reverse) [EMBL: AK089234]; and for amplification of the knockout allele, the primers 5'-GTGGATGAATCCCCAGACTC-3' [EMBL: AK089234] (forward) and 5'-TCGCCTTCTTGACGAGTTCT-3' (reverse) [GenBank: DQ890917.2] were used.
K/BxN mice that spontaneously develop arthritis were generated by crossing KRN T-cell-transgenic mice with NOD mice, as previously described [20].
Mice were maintained in the conventional mouse facility of the Medical School of the University of Santiago de Compostela. Animal care was in compliance with Spanish regulations on the protection of animals used for experimental and other scientific purposes (Real Decreto 223/1998). The experimental protocols were approved by the Animal Care and Use Committee of the University of Santiago de Compostela.
Generation of serum-transferred arthritis and clinical scoring
K/BxN serum was collected from 4-week-old to 8-week-old arthritic K/BxN mice. The serum samples were pooled and stored at -80°C until use. Arthritis was induced by transfer of this pool of sera in 6-week-old to 8-week-old mice in three different experimental groups.
In Group 1, arthritis was induced in 10 Mmp8-/- mice, 10 Mmp8+/+ mice, and 10 Mmp8+/- mice by intraperitoneal injection of 200 μl K/BxN serum on days 0 and 2. These mice were killed on day 14 after serum transfer.
In Group 2, arthritis was induced in 17 Mmp8-/- mice and 17 control mice (Mmp8+/+ and Mmp8+/-) by intraperitoneal injection on days 0 and 2 of 100 μl K/BxN serum. These mice were killed for histological assessment on day 9 after serum transfer.
In Group 3, arthritis was induced in nine Mmp8-/- male mice and nine Mmp8+/+ male mice by injection on days 0 and 2 of 150 μl K/BxN mice serum. These mice were killed for RNA and protein isolation on day 7 after serum transfer.
Arthritis was assessed in each of the four limbs every other day by two blinded observers, using a semiquantitative clinical score (0 = no swelling; 1 = slight swelling and erythema of the ankle, wrist or digits; 2 = moderate swelling and erythema; 3 = severe swelling and erythema; and 4 = maximal inflammation with joint rigidity). The maximum possible score was 16 per mouse.
Histological analysis
Hind limbs were prepared for histology by dissecting the skin and muscle, and then sectioning ankle joints. Specimens were fixed for 24 hours and demineralized in PBS-0.5 M ethylenediamine tetraacetic acid for 10 days. Ankle joints were embedded in paraffin and sections were cut and stained with hematoxylin and eosin for evaluation of inflammation and bone erosion, as previously described [21]. For analysis of the damage in cartilage, ankle sections were stained with Toluidine blue and Safranin-O following the standard methodology. To determinate osteoclast activity, staining for tartrate-resistant acid phosphatase (TRAP) was performed using the Acid Phosphatase, Leukocyte (TRAP) kit (Sigma, St Louis, MO, USA) following the manufacturer's instructions. Synovial inflammation was scored as previously described [21]: 0 = no inflammation; 1 = slight thickening of synovial cell layer and/or some inflammatory cells in the sublining; 2 = thickening of synovial lining and moderate infiltration of the sublining; 3 = thickening of synovial lining and marked infiltration; and 4 = thickening of synovial lining and severe infiltration.
Cartilage damage was evaluated following a 0 to 4 scale, as previously described [21]: 0 = normal cartilage; 1 = cartilage surface irregularities and loss of metachromasia adjacent to superficial chondrocytes; 2 = fibrillation of cartilage with minor loss of surface cartilage; 3 = moderate cartilage abnormalities including loss of superficial cartilage and moderate multifocal chondrocyte loss; and 4 = marked cartilage destruction with extension of fissures close to subchondral bone.
Bone erosion was scored on a 0 to 4 scale, as previously described [22]: 0 = normal bone; 1 = small areas of resorption; 2 = more numerous areas of resorption; 3 = obvious resorption; and 4 = full-thickness resorption areas in the bone.
Osteoclast activity was evaluated following a scale from 0 to 4 regarding TRAP staining, as previously described [23]: 0 = no staining; 1 = rare positive cells; 2 = some foci of positive cells; 3 = multiple foci; and 4 = diffuse staining. All scores were performed blind with respect to the mouse group.
Microarray analysis
Total RNA was obtained from ankle joints of three male mice from each of the following groups: Mmp8+/+ arthritic mice, Mmp8+/+ control mice, Mmp8-/- arthritic mice, and Mmp8-/- control mice. Male mice were used because they showed a trend to higher arthritis severity compared with female mice. The joints were taken 7 days after serum transfer and immediately frozen in liquid nitrogen. Subsequent processing was done at Progenika BioPharma SA (Bilbao, Spain).
Total RNA was isolated using the RNeasy Mini Kit and the QIAshredder (Qiagen GmbH, Hilden, Germany) according to the manufacturer's instructions. Integrity of RNA was assessed with the Agilent 2100 Bioanalyzer (Agilent Technologies, Duesseldorf, Germany). Total RNA (300 ng) was subjected to cDNA synthesis and labeling using the Whole Transcrit cDNA synthesis and amplification kit (Affymetrix, Santa Clara, CA, USA). This procedure involves synthesis of cDNA using T7-promoter-containing random primers, which is transcribed subsequently to cRNA. cRNA was quantified and used to generate dUTP-containing cDNA. The enzymes uracyl DNA glycosylase (UDG) and apurinic/apyramidinic endonuclease-1 (APE1) were used to fragment the dUTP-containing cDNA. Complete fragmentation was checked in the Bioanalyzer. Fragmented cDNA was labeled with the terminal transferase-based Whole Transcript Terminal Labeling kit from Affymetrix. Gene expression was evaluated using the Mouse Gene 1.0 ST array (Affymetrix) that contains about 27 probes for hybridization with each of the 28,853 mouse genome transcripts. Quality control procedures recommended by Affymetrix were followed. Intensity raw data were processed following the Robust Multichip Average method. Expression values below background were discarded, leaving information for 18,495 transcripts - of which 11,524 showed variable expression in at least one sample in relation with the others.
Real-time PCR analysis
Total RNA was obtained from knee joints of six Mmp8+/+ and six Mmp8-/- male mice 7 days after serum transfer, and from joints of three Mmp8+/+ and three Mmp8-/- control mice without arthritis, with the RNeasy Kit and RNase-Free DNase Set (Qiagen GmbH) according to the manufacturer's instructions. Total RNA (500 ng) was subjected to cDNA synthesis using the RT2 First Strand Kit (SABiosciences, Frederick, MD, USA). Quantitative real-time PCR was performed in duplicate in a Rotor Gene 6000 thermal cycler (Corbett Research, Cambridge, UK), using the RT2 SYBR Green/Rox qPCR Master Mix (SABiosciences), according to the manufacturer's protocol. The specific primers used in these reactions are listed in Table 1.
Table 1.
Primer sets used for quantitative PCR study
| Gene | Band size (bp) | SABiosciences catalog number | EMBL accession number | Reference position |
|---|---|---|---|---|
| IL-1β | 156 | PPM03109E | AK168047 | 1,059 to 1,080 |
| PROKR2 | 136 | PPM39370A | AF487279 | 851 to 870 |
| PTX3 | 99 | PMM03342E | BC022176 | 1,593 to 1,612 |
| C1QTNF3 | 132 | PPM37236A | AF246265 | 811 to 830 |
| CALPAIN6 | 98 | PMM27781A | AK145116 | 2,029 to 2,049 |
| MMP-3 | 94 | PMM03673A | AK148467 | 1,154 to 1,175 |
| TenascinN | 116 | PMM30367A | AF455756 | 4,477 to 4,499 |
| β-Actin | 154 | PMM02945A | AB028847 | 163 to 182 |
Relative levels of gene expression were normalized to the β-actin gene using the comparative Ct method, where Ct is the cycle at which the amplification is initially detected. The relative amount of mRNA from the different genes was calculated using the formula 2-ΔΔCt, where:
For wildtype and Mmp8-deficient samples without arthritis, ΔΔCt = 0 and 20 = 1. For wildtype and knockout samples with arthritis, the value of 2-ΔΔCt indicates the fold change in gene expression relative to the wildtype and knockout controls, respectively. Melting curves and agarose gel electrophoresis were used to assess the amplified band.
Western blot analysis
Total proteins were obtained from ankle joints of six Mmp8+/+ mice and six Mmp8-/- mice after 7 days of serum transfer. Whole protein lysates (40 to 50 μg protein) were fractionated by Tris-glycine buffered 10% SDS-PAGE, transferred to Polyvinylidene difluoride membrane (Roche) and probed with antibodies to prokineticin receptor 2 (PROKR2) (Santa Cruz, Santa Cruz, CA, USA) and β-actin (Sigma). Bound antibodies were revealed with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz) and the blot developed using a SuperSignal West Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL, USA).
Statistical analysis
Differences between experimental groups were assessed by repeated-measures analysis of covariance (ANCOVA) and two-sided Mann-Whitney U tests. P < 0.05 was considered significant. Correlation of histological parameters with clinical scores was determined with the Spearman RS.
Statistical analysis of the microarray expression results was performed with the Partek Genomics Suite v7.3.1 (Partek, Saint Louis, MO, USA) after normalization with the Robust Multichip Average method and filtering of values below background. Comparisons of expression levels between sample groups were carried out with lineal regression. Significance thresholds were considered applying a False Discovery Rate (FDR) approach or the more conservative Bonferroni correction by the number of independent tests. Functional classification of genes that showed differential expression was done with the DAVID functional annotation clustering utility [24,25]. The default set of 13 gene annotation databases, including three of each of the following functional categories, gene ontology, protein domains and pathways, was used for this clustering. An enrichment score of 3.0 was taken as the threshold for reporting clusters of genes, given that this level corresponds to significant enrichment of the included categories according to a FDR of 0.05. The fold change in expression levels of one group in relation to the other was also obtained after normalization of hybridization signals by the geometric mean of expression levels in all of the arrays.
Results
Increased severity of arthritis in mice lacking MMP-8
To ascertain the role of MMP-8 in experimental arthritis, we induced passive K/BxN arthritis in 12-generation B6-backrossed Mmp8-deficient (Mmp8-/-) mice, and their matched wildtype (Mmp8+/+) and heterozygous (Mmp8+/-) littermate controls.
In a first experimental group, male and female Mmp8+/+ (n = 10), Mmp8+/- (n = 10) and Mmp8-/- (n = 10) mice were injected intraperitoneally at day 0 and 2 with 200 μl K/BxN mice serum and monitored for signs of arthritis. Evolution of arthritis was evaluated by two blinded observers on a 0 to 4 scale, as described in Materials and methods.
An incidence of 100% of arthritis was observed in Mmp8-/-, Mmp8+/+ or Mmp8+/- mice (Figure 1a). The time course of arthritis was also similar in the three groups of mice. The disease developed rapidly and the maximum of severity was observed between 9 and 12 days. Surprisingly, Mmp8-deficient mice displayed significantly higher severity of arthritis than Mmp8+/+ and Mmp8+/- mice (P = 0.025 by repeated-measures one-way ANCOVA test) all through the follow-up. As the severity of arthritis was similar in Mmp8+/+ and Mmp8+/- mice, these mice were considered a unique control group (Mmp8+).
Figure 1.
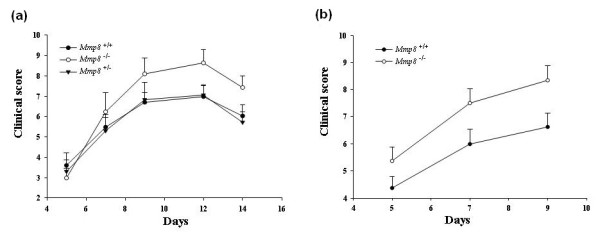
Increased arthritis severity in Mmp8-deficient mice. (a) Clinical score measured in 10 Mmp8-/- mice, 10 Mmp8+/+ mice and 10 Mmp8+/- mice after intraperitoneal injection of 200 μl K/BxN mice serum at days 0 and 2. Values expressed as mean ± standard error of the mean (SEM); P = 0.025, by analysis of covariance (ANCOVA) test. (b) Clinical score measured in 17 Mmp8-/- mice and 17 Mmp8+ mice after intraperitoneal injection of 100 μl K/BxN mice serum at days 0 and 2. Values expressed as mean ± SEM; P = 0.04, by ANCOVA test.
To exclude that the system was overloaded by using 200 μl K/BxN serum and to further evaluate the observed differences between Mmp8 control and deficient mice, a second experimental group composed of male and female Mmp8-/- mice (n = 17) and control mice (n = 17) was injected intraperitoneally at day 0 and 2 with 100 μl K/BxN serum (Figure 1b). Arthritis was monitored until day 9 and the results confirmed those previously obtained - arthritis severity was significantly higher in Mmp8-deficient mice compared with control mice (P = 0.04 by repeated-measures one-way ANCOVA test; Figure 1b).
Increased joint inflammation and bone erosion in Mmp8-deficient mice
To quantify joint involvement, we assessed synovial inflammation and bone erosion in hematoxylin and eosin stained sections of ankle joints, and cartilage damage was evaluated in Toluidine blue and Safranin-O stained sections. Right ankles were taken on day 9 after serum transfer from seven Mmp8-/- and seven Mmp8+ male and female mice of the group injected intraperitoneally with 100 μl K/BxN serum, and a blinded observer scored the histological sections. The clinical score of the Mmp8-/- mice was higher than in the Mmp8+ mice (P = 0.027 by Mann-Whitney U test).
Synovial inflammation was scored on a 0 to 4 scale, corresponding to the degree of thickening of the synovial lining and sublining infiltration. A significant increase in synovial inflammation score in Mmp8-/- mice compared with Mmp8+ mice was observed (P = 0.04 by Mann-Whitney U test; Figures 2 and 3a,d). Changes in cellular infiltrate composition, however, were not observed in mice lacking Mmp8 compared with wildtype mice. Specifically, a similar rate of neutrophils and mononuclear cells were seen in both groups of mice.
Figure 2.
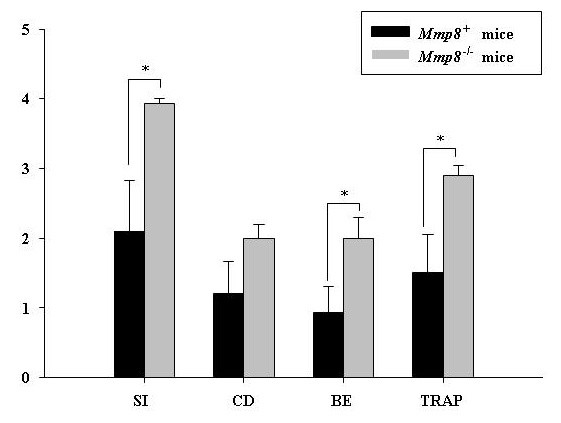
Increased joint inflammation and bone erosion in mice lacking Mmp8. Histologic scores of synovial inflammation (SI), cartilage damage (CD), bone erosion (BE) and tartrate-resistant acid phosphatase (TRAP) staining of ankle sections of Mmp8+ mice (n = 8) and Mmp8-/- mice (n = 7) at day 9 after intraperitoneal arthritis induction. Values expressed as mean ± standard error of the mean; *P < 0.05 by two-sided Mann-Whitney U test.
Figure 3.
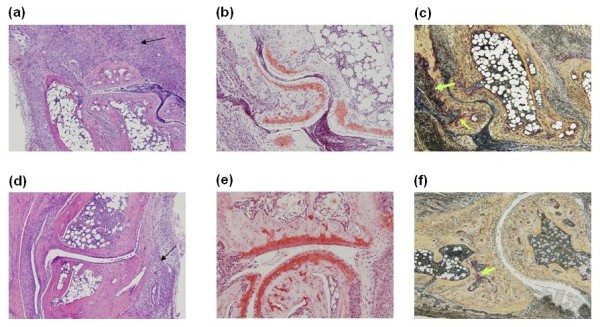
Higher synovial inflammation and bone erosion in arthritic joints of Mmp8-deficient mice than control mice. Representative sections of ankle joints of Mmp8-/- mice (a) to (c) and Mmp8+ mice (d) to (f) 9 days after intraperitoneal injection of K/BxN serum. More severe inflammation and pannus formation (arrows) were observed on hematoxylin and eosin stained sections from Mmp8-deficient mice (a) compared with control mice (d). More severe loss of proteoglycans indicated by destained cartilage was observed on Safranin-O stained sections from Mmp8-/- mice (b) compared with Mmp8+ mice (e). Increased tartrate-resistant acid phosphatase (TRAP)-positive multinucleated osteoclast-like cells (arrows) were observed at sites of bone erosions on sections stained for TRAP activity from Mmp8-deficient mice (c) compared with control mice (f).
As shown in Figures 2 and 3c,f, bone erosion was more marked in Mmp8-/- mice than in wildtype mice (P = 0.04 by Mann-Whitney U test). Furthermore, staining sections for TRAP activity revealed a significantly increase of TRAP-positive multinucleated cells in Mmp8-/- mice compared with Mmp8+ mice (P = 0.025 by Mann-Whitney U test). These cells were observed at sites of bone erosion in both groups of mice (Figure 3c,f).
A trend to higher cartilage damage in Mmp8-/- mice than control Mmp8+ mice was detected (Figures 3 and 4b,e), although the difference was not significant (P = 0.11 by Mann-Whitney U test). Significant correlations between synovial inflammation, cartilage damage, bone erosion and TRAP staining with clinical scores were observed (RS >0.64 and P < 0.017).
Figure 4.
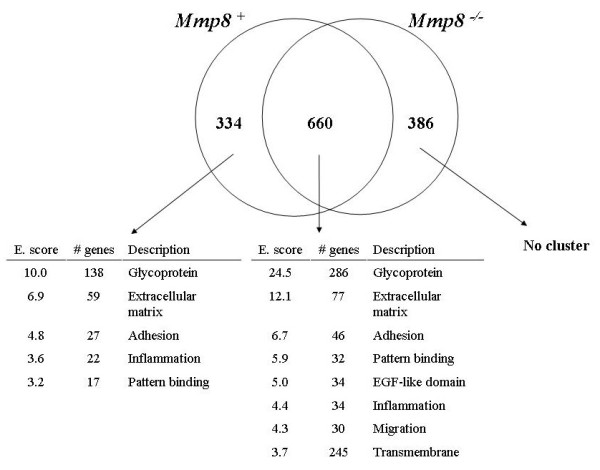
Clustering of differentially expressed genes. The DAVID utility [24,25] was used to cluster differentially expressed genes according to a false discovery rate 0.01 threshold, in both Mmp8+/+ and Mmp8-/-arthritic mice, exclusive arthritic Mmp8+/+ mice and exclusive arthritic Mmp8-/- mice. The functional description and the number of genes with enrichment score (E. score) >3.0 are shown.
Overall these results suggest that MMP-8 plays a protective role in inflammatory arthritis.
Microarray analysis
To explore the mechanisms underlying the increased arthritis severity in Mmp8-deficient mice, we used a genome-wide microarray analysis including probes for more than 28,000 mouse transcripts. Ankle joints from three mice from each of the following groups were studied: Mmp8+/+ and Mmp8-/- mice with and without arthritis. Mice with arthritis were injected intraperitoneally on days 0 and 2 with K/BxN mice serum, and joints were taken 7 days after injection. Comparison of expression levels between arthritic and nonarthritic control mice yielded a list of about 3,200 genes that were differentially expressed according to an FDR of 5% (2,996 genes in the comparison among Mmp8+/+ mice and 3,407 genes in the Mmp8-/- comparison), or about 1,000 genes according to the more stringent FDR 1% threshold (Table 2). These lists were largely concordant in the two independent comparisons, as assessed by the fact that most genes differently expressed in Mmp8+/+ mice were also differently expressed in Mmp8-/- mice. In fact, direct comparison of arthritic Mmp8+/+ mice with arthritic Mmp8-/- mice did not show any significant difference.
Table 2.
Number of differentially expressed genes
| Mice | Control versus rheumatoid arthritis | ||
|---|---|---|---|
| FDR <0.05 | FDR <0.01 | P corr <0.05 | |
| Mmp8+/+ | 2,996 | 994 | 77 |
| Mmp8-/- | 3,407 | 1,046 | 52 |
| Mmp8+/+ and Mmp8-/- | 2,136 | 660 | 40 |
| Mmp8+/+ exclusive | 860 | 334 | 37 |
| Mmp8-/- exclusive | 1,271 | 386 | 12 |
No differences were found between arthritic Mmp8+/+ and arthritic Mmp8-/- mice. FDR, false discovery rate.
We therefore conducted other types of analyses. First, we compared the functional groups of differentially expressed genes modified in both groups of arthritic mice, only in arthritic Mmp8+/+ mice and only in Mmp8-/- mice. The 660 genes that were modified both in Mmp8+/+ mice and Mmp8-/- mice, according to a FDR 0.01 threshold, could be grouped into eight clusters with an enrichment score over 3.0 (Figure 4). These clusters included some that are more structurally defined and others that are more related with cellular or biological pathways. The same type of analysis was also carried out for the sets of genes that were different only in the Mmp8+/+ arthritic mice according to the same FDR 0.01 criteria. There were 334 genes in this class and they were grouped into five clusters of annotations with an enrichment score over 3.0. These five clusters were a subgroup of the eight clusters that were modified in both groups of arthritic mice. The only three clusters missing here were the cluster of epidermal growth factor-like domain proteins and the two last clusters: the one grouping cell migration and motility genes, and the one containing transmembrane proteins. The genes whose expression was significantly modified in arthritic Mmp8+/+ mice and not in arthritic Mmp8-/- mice were therefore largely from the same functional classes as the genes that were modified in both types of mice.
A similar analysis with the 386 genes that were modified only in arthritic Mmp8-/-mice gave very different results. No single cluster of genes showed an enrichment score over 3.0, and only two clusters showed a score over 2.0. This indicates that the modified genes specific of arthritis in the Mmp8-/- mice are very varied and difficult to group. The pattern of genes that were differentially regulated in Mmp8+/+ mice and Mmp8-/- mice are therefore very different: the genes regulated specifically in arthritic Mmp8-/- mice are similar in number but much more diverse functionally.
The lack of any clearly defined functional class of genes specifically modified in arthritic Mmp8-/- mice made it impossible to focus on them to try to discern important factors in the differential arthritis phenotype. We decided to concentrate instead on the genes that, having a most clearly changed expression with arthritis, were also most differentially affected in Mmp8+/+ and Mmp8-/- mice. We selected the 86 nonredundant genes that were different between arthritic and control mice in the comparison of either Mmp8+/+ mice or Mmp8-/- mice according to the very conservative Bonferroni-corrected threshold of P = 0.05. We obtained the fold change ratios between their respective comparisons. Genes with fold change ratios higher than 1.35 and lower than 0.75 were considered interesting (Table 3). That is, differences between arthritic Mmp8-/- mice and their controls were compared with differences between arthritic Mmp8+/+ mice and their controls, and the most extreme fold change ratios were selected. Seven out of 29 genes were chosen for confirmatory real-time PCR experiments given their interest in inflammation, autoimmunity or arthritis.
Table 3.
Genes with the most discordant changes in expression in Mmp8+/+ and Mmp8-/-arthritic mice
| Gene | Fold change: CRL Mmp8 -/- vs. RA Mmp8 -/- (A) | Fold change: CRL Mmp8 +/+ vs. RA Mmp8 +/+ (B) | Ratio A/B |
|---|---|---|---|
| Anpep | 2.107 | 3.766 | 0.559 |
| Arsi | 3.440 | 4.685 | 0.734 |
| Aspa | 0.289 | 0.195 | 1.481 |
| C1qtnf3 | 5.773 | 8.349 | 0.691 |
| Capn6 | 5.503 | 11.324 | 0.485 |
| Ces3 | 0.050 | 0.074 | 0.682 |
| Csgalnact1 | 4.318 | 5.906 | 0.731 |
| Ddefl1 | 1.411 | 2.010 | 0.702 |
| Fbln2 | 2.254 | 3.128 | 0.720 |
| Gltd5d1 | 1.411 | 2.158 | 0.654 |
| Grb10 | 1.639 | 2.421 | 0.677 |
| H19 | 3.154 | 5.834 | 0.541 |
| Ifitm1 | 2.100 | 4.232 | 0.496 |
| Il1 β | 5.014 | 3.711 | 1.351 |
| Itga5 | 2.184 | 3.079 | 0.709 |
| Mmp3 | 27.264 | 41.104 | 0.663 |
| Nt5dc2 | 3.532 | 4.891 | 0.722 |
| P4ha3 | 6.826 | 9.504 | 0.718 |
| Postn | 2.949 | 4.571 | 0.645 |
| Prokr2 | 4.791 | 3.527 | 1.358 |
| Ptgfrn | 1.705 | 2.407 | 0.708 |
| Ptx3 | 6.117 | 4.493 | 1.361 |
| Rentla | 0.031 | 0.043 | 0.709 |
| Sfrp1 | 8.278 | 11.900 | 0.696 |
| Slc39a14 | 5.882 | 7.949 | 0.739 |
| Sult1a1 | 0.251 | 0.181 | 1.379 |
| Syne2 | 0.590 | 0.335 | 1.759 |
| TenascinN | 3.653 | 5.793 | 0.631 |
| Tpd52 | 2.410 | 1.781 | 1.353 |
Genes with the most discordant changes (<0.75 or >1.35) in expression in Mmp8+/+ and Mmp8-/-arthritic mice in relation to their respective nonarthritic controls (selected from arthritis-associated genes according to Bonferroni-corrected P < 0.01). Bold genes were chosen for confirmatory real-time PCR. CRL, control; RA, rheumatoid arthritis.
The data discussed in this publication have been deposited in NCBI'S Gene Expression Omnibus [26] and are accessible through the GEO Series accession number [GEO:GSE22971] [27].
Induction of IL-1β, PROKR2 and PTX3 in arthritic Mmp8-deficient mice
To corroborate the results obtained by the microarray analysis, real-time RT-PCR experiments were performed in arthritic joints from six other Mmp8-deficient mice and six wildtype mice treated in the same way. Increased mRNA expression of IL-1β, PROKR2 and pentraxin-3 (PTX3) was found in arthritic Mmp8-/- mice compared with wildtype mice (P = 0.035, P = 0.032 and P = 0.028, respectively; Figure 5a). Real-time PCR did not, however, confirm the expression changes observed in CALPAIN 6, MMP-3, C1QTNF3 and TenascinW in Mmp8-deficient mice compared with wildtype mice (data not shown).
Figure 5.
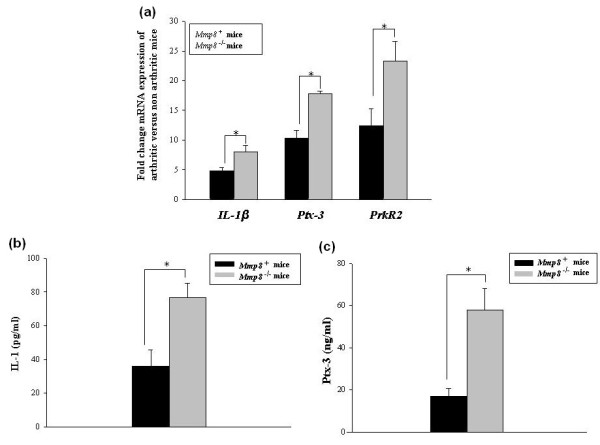
Increased IL-1β, PTX-3 and PROKR2 mRNA and protein levels in mice lacking Mmp8. (a) IL-1β, pentraxin-3 (PTX-3) and prokineticin receptor 2 (PROKR2) mRNA levels were measured by quantitative real-time PCR in arthritic joints of Mmp8-/- mice (n = 6) and Mmp8+/+ mice (n = 6) at day 7 after intraperitoneal serum injection. Levels of IL-1β (b) and PTX-3 (c) proteins were measured by ELISA in extracts from arthritic joints of mice at day 7 after intraperitoneal serum injection. Values expressed as mean ± standard error of the mean; *P < 0.05, by two-sided Mann-Whitney U test.
Increased production of IL-1β and PTX3 was verified by ELISA assay (Figure 5b,c), and results showed a significant increase of both proteins in joints from Mmp8-/- mice compared with Mmp8+/+ mice (P = 0.031 and P = 0.017, respectively). PROKR2 production was assessed by western blot and is shown in Figure 6a,b. As expected, PROKR2 levels were significantly higher in joints from Mmp8-deficient than in control male mice (P = 0.031).
Figure 6.
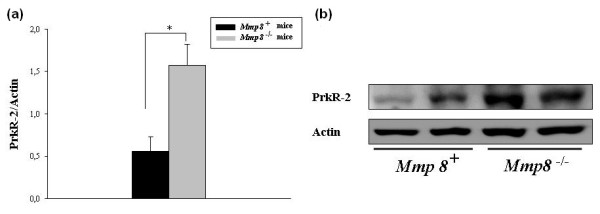
Increased PROKR2 expression in Mmp8-deficient mice. Protein expression of prokineticin receptor 2 (PROKR2) was determined by western blot in arthritic joints of six Mmp8-/- mice and six Mmp8+/+ mice at 7 days after arthritis induction. Densitometric analysis of PROKR2 and that normalized with the intensity of actin is shown. (a) Data represent mean ± standard error of the mean; *P < 0.05, by Mann-Whitney U test. (b) Representative blot.
Discussion
Accumulated evidence indicates that MMPs are involved in the cartilage destruction observed in RA [1-4,7]; MMP inhibitors are thereby of special interest for the treatment of RA. Results from clinical trials of MMP inhibitors in RA have not been encouraging, however, probably due to lack of specificity of such inhibitors [28,29]. In fact, analyses of several MMPs in animal models have shown either [9-11] exacerbation or reduction of arthritis severity depending on the MMP analyzed. This indicates that specific MMPs could have either a promoting or a protective role in arthritis pathogenesis. Knowledge of the role of specific MMPs in the pathogenesis of arthritis therefore seems pivotal to obtain successful inhibitors for treatment. In the present study, we have investigated the impact of lack of Mmp8 in the K/BxN serum-transfer arthritis model. The advantages of this transfer model with respect to other arthritis models is its 100% penetrance, early onset, rapid development of osteolytic lesions and its MHC independence. Clinical features and histopathology are very similar to human RA. Another characteristic of the K/BxN transfer model is that it allows us to focus on the effector phase mechanisms of arthritis that are dependent on neutrophils, macrophages, mast cells, and inflammatory mediators, especially IL-1β, but independent of T cells and B cells.
We have found that the absence of Mmp8 increased the severity of arthritis without noticeably affecting its time course, either at its onset or at its spontaneous remission. The aggravated clinical course of arthritis was accompanied by exacerbated synovial inflammation and bone erosion. These effects were associated with modified expression of a varied array of genes, including overexpression of IL-1β, PTX3 and PROKR2 in arthritic joints. Surprisingly, despite the known collagenolytic activity of Mmp-8, its absence did not protect from cartilage damage but a trend to increased damage was observed compared with Mmp8 wildtype mice. This finding may indicate that other Mmps could compensate for its absence. These data indicate that Mmp-8 plays a protector role against arthritis in this model. This effect is consistent with the previously reported effect of Mmp-8 absence in other inflammation models such as OVA-induced airway inflammation [17], chemically-induced skin carcinomas [19] and skin wound healing [16], in which the absence of Mmp-8 increased the severity of these pathologies and delayed wound healing. In these studies, disease aggravation was linked to increased neutrophil accumulation in the mice lacking Mmp8. In our work, we did not observe differences in cellular infiltrate composition between Mmp8 control and deficient mice, suggesting that mechanisms involved in the Mmp-8 regulation of inflammation are complex and include its effect in other aspects of inflammation as shown by our expression studies. It is possible that differences between models depend on the nature of the inflammatory stimulus or of the tissue affected.
To elucidate the mechanisms behind arthritis aggravation in Mmp8-/- mice, we have investigated the gene expression profile in Mmp8-sufficient and Mmp8-deficient mice with and without arthritis using microarray technology. There was a wide array of genes that changed expression in arthritic mice. Most were coincident in Mmp8-sufficient and Mmp8-deficient mice, and they can be grouped in functional categories that are congruent with current knowledge of arthritis mechanisms. The functional spread of the genes whose expression was only modified in the arthritic Mmp8-/- mice contrasted with the clustering in five functional categories of the genes significantly modified only in the arthritic Mmp8 wildtype mice, despite being similar in number. This result is consistent with the lack of any clearly different phenotype in the histological analysis and has been taken into consideration to interpret the analyses of individual genes. To select genes for detailed analysis, we decided to focus on the genes that with high likelihood were differentially expressed with respect to arthritis and the presence of Mmp8. After selection of a group of seven genes, we found an increased expression of IL-1β, PTX3 and PROKR2 in arthritic joints from Mmp8-deficient mice compared with wildtype mice that were confirmed by real-time PCR assays. The corresponding increase in protein expression was validated by ELISA and western blot.
IL-1β is highly expressed in the synovium of RA patients and plays a crucial role in production of inflammatory mediators and articular damage [2,30]. This cytokine's functional relevance has been demonstrated in several animal models, including the K/BxN model [30-36]. Results of these studies indicate that the increased IL-1β expression observed in Mmp8-deficient mice can contribute to the higher clinical score, synovial inflammation, osteoclast activity and bone erosion found in these mice.
PTX3 is the prototypic member of the long pentraxin family of acute phase reactants. PTX3 rapidly increases in serum during endotoxic shock, inflammation and infections [37]. A possible role of this protein in potentiating inflammation has been reported in a model of intestinal injury by ischemia/reperfusion in which PTX3 transgenic mice showed exacerbated inflammatory response and increased lethality [38]. Also, mice lacking PTX3 displayed reduced tissue inflammation and increased survival rates [39]. Our results showed an increased PTX3 expression in mice lacking Mmp8 compared with wildtype mice, where it was also increased, indicating that PTX3 upregulation could have contributed to the higher arthritis severity in the knockout mice. This result suggests that the accumulation of PTX3 in the synovial fluid of RA patients after being produced by synoviocytes and synovial endothelial cells [40] can be also a contributor to the inflammation process.
PROKR2 is a seven-transmembrane coupled G-protein receptor that binds prokineticin-2. PROKR2 is highly expressed in the bone marrow, and in neutrophils, monocytes and dendritic cells [41]. Signaling through this receptor induces survival, differentiation and activation of granulocytic and monocytic lineages [42]. The higher expression of PROKR2 found in the arthritic joints from Mmp8-deficient mice could therefore have contributed to the increased inflammatory infiltration observed in them.
Changes in the expression of these three genes exemplify different ways in which the lack of MMP-8 led to an aggravation of arthritis: promotion of inflammation by IL-1β and other molecules like PTX3, induction of maturation and activation of osteoclasts by IL-1β and PROKR2, and enhanced inflammatory infiltrate by IL-1β and possibly PROKR2 - however, other contributing mechanisms are possible as only a fraction of the genes with possible differential expression were analyzed. Similar analysis in other models of inflammation will help to unravel the many ways in which MMP-8 seems to protect against inflammation.
Conclusions
The present study indicates that MMP-8 protects against inflammatory synovitis and bone erosion in the K/BxN serum-transfer arthritis model. Expression analysis indicates that protection is due to changes in multiple genes belonging to different functional categories. Three of these genes have been validated exemplifying the involved pathways: IL-1β, PTX3 and PROKR2.
Abbreviations
ANCOVA: analysis of covariance; MMP: matrix metalloproteinases; ELISA: enzyme-linked immunosorbent assay; FDR: false discovery rate; IL: interleukin; PBS: phosphate-buffered saline; PCR: polymerase chain reaction; RA: rheumatoid arthritis; PTX3: pentraxin-3; PROKR2: prokineticin receptor 2; TRAP: tartrate-resistant acid phosphatase; C1QTNF3: C1q and TNF-related protein; TNF: tumor necrosis factor.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SG carried out the experiments and participated in the analysis of data and in drafting the manuscript. JF carried out the histological scoring. CL-O participated in design of the study and revision of the manuscript. JJG-R participated in design and coordination of the study, and revision of the manuscript. AG performed the statistical analysis, and participated in design of the study, interpretation of data and drafting the manuscript. CC conceived of the study, and participated in its design, in coordination and interpretation of the data and in drafting the manuscript. All authors read and approved the final manuscript.
Contributor Information
Samuel García, Email: samugp13@yahoo.es.
Jerónimo Forteza, Email: Jeronimo.Forteza.Vila@sergas.es.
Carlos López-Otin, Email: clo@uniovi.es.
Juan J Gómez-Reino, Email: Juan.Jesus.Gomez-Reino.Carnota@sergas.es.
Antonio González, Email: Antonio.Gonzalez.Martinez-Pedrayo@sergas.es.
Carmen Conde, Email: Carmen.Conde.Muro@sergas.es.
Acknowledgements
The authors thank Dr D Mathis, Dr C Benoist and Dr H Illges for providing us with KRN mice, and A Bodaño for help with the arthritis clinical score. The present work was supported by grants PI04/0783, PI08/0038, RETICS Program, RD08/0075 (RIER), all from Fondo de Investigación Sanitaria, Instituto de Salud Carlos III within the VI PN de I+D+I 2008-2011 with participation of FEDER funds (European Union). SG is supported by Xunta de Galicia.
References
- Sweeney SE, Firestein GS. Rheumatoid arthritis: regulation of synovial inflammation. Int J Biochem Cell Biol. 2004;36:372–378. doi: 10.1016/S1357-2725(03)00259-0. [DOI] [PubMed] [Google Scholar]
- McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- Takaishi H, Kimura T, Dalal S, Okada Y, D'Armiento J. Joint diseases and matrix metalloproteinases: a role for MMP-13. Curr Pharm Biotechnol. 2008;9:47–54. doi: 10.2174/138920108783497659. [DOI] [PubMed] [Google Scholar]
- Rengel Y, Ospelt C, Gay S. Proteinases in the joint: clinical relevance of proteinases in joint destruction. Arthritis Res Ther. 2007;9:221. doi: 10.1186/ar2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara Y, Nakamura H, Obata K, Yamada H, Hayakawa T, Fujikawa K, Okada Y. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in synovial fluids from patients with rheumatoid artrhritis or osteoarthritis. Ann Rheum Dis. 2000;59:455–461. doi: 10.1136/ard.59.6.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchetverikov I, Lard LR, DeGroot J, Verzijl N, TeKoppele JM, Breedveld FC, Huizinga TW, Hanemaaijer R. Matrix metalloproteinases-3, -8, -9 as markers of disease activity and joint damage progression in early rheumatoid arthritis. Ann Rheum Dis. 2003;62:1094–1099. doi: 10.1136/ard.62.11.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolboom TC, Pieterman E, van der Laan WH, Toes RE, Huidekoper AL, Nelissen RG, Breedveld FC, Huizinga TW. Invasive properties of fibroblast-like synoviocytes: correlation with growth characteristics and expression of MMP-1, MMP-3, and MMP-10. Ann Rheum Dis. 2002;61:975–980. doi: 10.1136/ard.61.11.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MJ, Gough AK, Devlin J, Smith J, astin P, Taylor D, Emery P. Serum MMP-3 and MMP-1 and progression of joint damage in early rheumatoid arthritis. Rheumatology. 2003;42:83–88. doi: 10.1093/rheumatology/keg037. [DOI] [PubMed] [Google Scholar]
- Itoh T, matsuda H, Tanioka M, Kuwabara K, Itohara S, Suzuki R. The role of matrix metalloproteinase-2 and matrix metalloproteinase-9 in antibody-induced arthritis. J Immunol. 2002;169:2643–2647. doi: 10.4049/jimmunol.169.5.2643. [DOI] [PubMed] [Google Scholar]
- van Meurs J, Van Lent P, Stoop R, Holthuysen A, Singer I, Bayne E, Mudgett J, Poole R, Billinghurst C, Van der Kraan P, Burna P, Van den Berg W. Cleavage of aggrecan at the Asn341-Phe342 site coincides with the initiation of collagen damage in murine antigen-induced arthritis: a pivotal role for stromelysin 1 in matrix metalloproteinase activity. Arthritis Rheum. 1999;42:2074–2084. doi: 10.1002/1529-0131(199910)42:10<2074::AID-ANR7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Mudgett JS, Hutchinson NI, Chartrain NA, Forsyth AJ, Mcdonnell J, Sdinger II, Bayne EK, Flanagan J, Kawka D, Shen CF, Stevens K, Chen H, Trumbauer M, Visco DM. Susceptibility of stromelysin-1-deficient mice to collagen-induced arthritis and cartilage destruction. Arthritis Rheum. 1998;41:110–121. doi: 10.1002/1529-0131(199801)41:1<110::AID-ART14>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Cole AA, Chubinskaya S, Schumacher B, Huch K, Szabo G, Yao J, Mikecz K, Hasty KA, Kuettner KE. Chondrocyte matrix metalloproteinase-8. Human articular chondrocytes express neutrophil collagenase. J Biol Chem. 1996;271:11023–11026. doi: 10.1074/jbc.271.18.11023. [DOI] [PubMed] [Google Scholar]
- Hanemaaijer R, Sorsa T, Konttinen YT, Ding Y, Sutinen M, Visser H, van Hinsbergh VW, Helaakoski T, Kainulainen T, Rönkä H, Tschesche H, Salo T. Matrix metalloproteinase-8 is expressed in rheumatoid synovial fibroblasts and endothelial cells. Regulation by tumor necrosis factor-alpha and doxycycline. J Biol Chem. 1997;272:31504–31509. doi: 10.1074/jbc.272.50.31504. [DOI] [PubMed] [Google Scholar]
- van Lint P, Wielockx B, Puimège L, Nöel A, López-Otin C, Libert C. Resistance of collagenase-2 (matrix metalloproteinase-8)-deficient mice to TNF-induced lethal hepatitis. J Immunol. 2005;175:7642–7649. doi: 10.4049/jimmunol.175.11.7642. [DOI] [PubMed] [Google Scholar]
- Tester AM, Cox JH, Connor AR, Starr AE, Dean RA, Puente XS, López-Otin C, Overall CM. LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PloS ONE. 2007;2:e312. doi: 10.1371/journal.pone.0000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez-Fernández A, Inada M, Balbín M, Fueyo A, Pitiot AS, Astudillo A, Hirose K, Hirata M, Shapiro SD, Noël A, Werb Z, Krane SM, López-Otin C, Puente XS. Increased inflammation delays wound healing in mice deficient in collagenase-2 (MMP-8) FASEB J. 2007;21:2580–2591. doi: 10.1096/fj.06-7860com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueders MM, Balbin M, Rocks N, Foidart JM, Gosset P, Louis R, Shapiro S, Lopez-Otin C, Noël A, Cataldo DD. Matrix metalloproteinase-8 deficiency promotes granulocytic allergen-induced airway inflammation. J Immunol. 2005;175:2589–2597. doi: 10.4049/jimmunol.175.4.2589. [DOI] [PubMed] [Google Scholar]
- van den Steen PE, Husson SJ, Proost P, Van Damme J, Opdenakker G. Carboxyterminal cleavage of the chemokines MIG and IP-10 by gelatinase B and neutrophil collagenase. Biochem Biophys Res Commun. 2003;310:889–896. doi: 10.1016/j.bbrc.2003.09.098. [DOI] [PubMed] [Google Scholar]
- Balbín M, Fueyo A, Tester AM, Pendás AM, Pitiot AS, Astudillo A, Overall CM, Shapiro SD, López-Otín C. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003;35:252–257. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/S0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- García S, Bodaño A, González A, Forteza J, Gomez-Reino JJ, Conde C. Partial protection against collagen antibody-induced arthritis in PARP-1 deficient mice. Arthritis Res Ther. 2006;8:R14. doi: 10.1186/ar1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scatizzi JC, Bickel E, Hutcheson J, Haines GK III, Perlman H. Bim deficiency leads to exacerbation and prolongation of joint inflammation in experimental arthritis. Arthritis Rheum. 2006;54:3182–3193. doi: 10.1002/art.22133. [DOI] [PubMed] [Google Scholar]
- Pettit AR, Ji H, von Stechow D, Müller R, Goldring SR, Choi Y, Benoist C, Gravallese EM. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001;159:1689–1699. doi: 10.1016/S0002-9440(10)63016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acid Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Expression Omnibus series accession number. http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE22971
- Close DR. Matrix metalloproteinase inhibitors in rheumatic diseases. Ann Rheum Dis. 2001;60:iii62–iii67. doi: 10.1136/ard.60.90003.iii62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IM, Parker AE. Metalloproteinases: their role in arthritis and potential as therapeutic targets. Expert Opin Ther Targets. 2003;7:19–34. doi: 10.1517/14728222.7.1.19. [DOI] [PubMed] [Google Scholar]
- Brennan FM, McInnes IB. Evidence that cytokines plays a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–3545. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, Ikuse T, Asano M, Iwakura Y. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000;191:313–320. doi: 10.1084/jem.191.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata H, Sakaguchi N, Yoshitomi H, Iwakura Y, Sekikawa K, Azuma Y, Kanai C, Moriizumi E, Nomura T, Nakamura T, Sakaguchi S. Distinct contribution of IL-6, TNF-α, IL-1, and IL-10 to T cell-mediated spontaneous autoimmune arthritis in mice. J Clin Invest. 2004;114:582–588. doi: 10.1172/JCI21795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Pettit A, Ohmura K, Ortiz-Lopez A, Duchatelle V, Degott C, Gravallese E, Mathis D, Benoist C. Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. J Exp Med. 2002;196:77–85. doi: 10.1084/jem.20020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe JY, Crain B, Wu SR, Corr M. Interleukin 1 receptor dependence of serum transferred arthritis can be circumvented by toll-like receptor 4 signaling. J Exp Med. 2003;197:537–542. doi: 10.1084/jem.20021850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 mediated TNF-induced osteoclastogenesis. J Clin Invest. 2005;115:282–290. doi: 10.1172/JCI23394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwerina J, Redlich K, Polzer K, Joosten L, Krönke G, Distler J, Hess A, Pundt N, Pap T, Hoffmann O, Gasser J, Scheinecker C, Smolen JS, van den Berg W, Schett G. TNF-induced structural joint damage is mediated by IL-1. Proc Natl Acad Sci USA. 2007;104:11742–11747. doi: 10.1073/pnas.0610812104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi AA, Rovere-Querini P, Bottazzi B, Garlanda C, Mantovani A. Pentraxins, humoral innate immunity and tissue injury. Curr Opin Immunol. 2008;20:538–544. doi: 10.1016/j.coi.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Luchetti MM, Piccinini G, Mantovani A, Peri G, Mattencci C, Pomponio G, Fratini M, Fraticelli P, Sambo P, DiLoreto C, Introna M, Gabrielli A. Expression and production of the long pentraxin PTX3 in rheumatoid arthritis (RA) Clin Exp Immunol. 2000;119:196–202. doi: 10.1046/j.1365-2249.2000.01110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza DG, Soares AC, Pinho V, Torloni H, Reis LFL, Martins MT, Dias AAM. Increased mortality and inflammation in tumor necrosis factor-stimulated gene-14 transgenic mice after ischemia and reperfusion injury. Am J Pathol. 2002;160:1755–1765. doi: 10.1016/s0002-9440(10)61122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deban L, Russo RC, Sironi M, Moalli F, Scanziani M, Zambelli V, Cuccovillo I, Bastone A, Gobbi M, Valentino S, Doni A, Garlanda C, Danese S, Salvatori G, Sassano M, Evangelista V, Rossi B, Zenaro E, Constantin G, Laudanna C, Bottazzi B, Mantovani A. Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol. 2010;11:328–334. doi: 10.1038/ni.1854. [DOI] [PubMed] [Google Scholar]
- LeCouter J, Zlot C, Tejada M, Peale F, Ferrara N. Bv8 and endocrine gland-derived vascular endothelial growth factor stimulate hematopoiesis and hematopoietic cell mobilization. Proc Natl Acad Sci USA. 2004;101:16813–16818. doi: 10.1073/pnas.0407697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier J, Samson M. Cytokine properties of prokineticins. FEBS J. 2008;275:4014–4021. doi: 10.1111/j.1742-4658.2008.06559.x. [DOI] [PubMed] [Google Scholar]