

Abstract
Advances in protein chemistry and molecular and structural biology have empowered modern chemists to build complex biological architectures using a “first principles” approach, which is known as de novo protein design. In this Perspective we demonstrate how simple three-stranded α-helical constructs can be prepared by the sole consideration of the primary amino acid sequence of a peptide. With these well defined systems, we then demonstrate that metal binding cavities can be carved out of the hydrophobic cores of these aggregates in order to bind metal ions such as cadmium with well defined coordination geometries. Examples will be given of homoleptic CdS3 complexes, CdS3O sites and proteins which contain equilibrium mixtures of these two species. We will provide a description of a strategy that allows us to build heterochromic peptides (small proteins that complex two metals in nearly identical environments but which result in different physical properties and allow for metal site selectivity). We conclude with a new class of designed peptides, diastereopeptides, which can exploit changes in amino acid chirality to control metal ion coordination number and lead to an alternative path towards heterochromic systems. The constructs described herein represent the initial steps of preparing protein structures that may simultaneous contain structural and catalytic metal binding centers. These studies inform the community on a developing field, which promises new opportunities for the study of bioinorganic chemistry.
Introduction
Today we take for granted many biochemical and biophysical techniques that only 25 years ago were considered major accomplishments. The structural determination of proteins through either X-ray crystallography or solution NMR as well as the modification of specific amino acid residues through site-directed mutagenesis are now common place. Scientists have amassed mountains of structural information and made significant advances in understanding protein folding, protein–protein interactions, electronic structure and function for biological molecules, revolutionizing the way we now think about such systems. As we look forward, we are ideally placed to use de novo designed peptides to understand biological systems on a molecular and even atomic level. Great advances have also been made in the field of peptide synthesis and the prediction of protein structure from primary amino acid sequences, ultimately generating the field of de novo design in which a linear sequence can be designed to give a well defined secondary and tertiary protein structure.1–6 These systems offer a simplified construct, by removing a significant amount of the large biological structure, while maintaining the core features with which to work. Ultimately, this allows one to develop structure–function relationships through systematically correlating chemical observations with three-dimensional structure.
The field of de novo peptide design has targeted a number of structural motifs commonly found in biological systems, such as β-sheets,7–9 mixed α/β-motifs7,10 and most importantly α-helices and bundles thereof. A number of different α-helical aggregates have been reported, ranging from two-, three- and four-stranded coiled coils,11–13 as well as single peptides that fold to form a bundle of α-helices.14,15 Further variations have looked at both homo vs. hetero-aggregates,16 as well as parallel and anti-parallel orientation of helices.17 The subgroup of coiled coils typically result from sequences containing a seven amino acid heptad repeat, where the position in the sequence is defined as abcdefg, respectively. An example of this being the TRI family, on which the work in this perspective is based, with the sequence Ac-G(LaKbAcLdEeEfKg)4G-NH2, see Table 1.18,19 These peptides form α-helices in solution in which all of the hydrophobic “a” and “d” residues are on one face of the helix, and all the hydrophilic residues are on the opposite face, see Fig. 1A. This results in an amphipatic helix, which aggregates in aqueous solution to form a coiled coil with a hydrophobic interior. At low pH (<5) these form two-stranded coiled coils, but as the pH is raised (>7) and the Glu side chains are deprotonated, these form three-stranded coiled coils due to stabilizing salt bridges between the “e” and “g” residues of adjacent helixes, see Fig. 1B. Thus, the TRI family of peptides form parallel left-handed coiled coil structures comprised of three amphipatic α-helices as the primary and secondary structure elements. Furthermore, the overall stability of the coiled coil is affected by both chain length and amino acid compositions. BABY, which consists of one fewer heptad repeat, Ac-G(LKALEEK)3G-NH2, is less stable than TRI and in a similar fashion GRAND, Ac-G(LKALEEK)5G-NH2, which consists of an additional heptad, is more stable (each additional heptad stabilizing the peptide by ~9 kcal mol−1).20,21 Substitutions made to the leucine interior of the coiled coil scaffold result in an overall destabilization.22,23
Table 1.
Derivatives of peptides
| Peptide | Sequencea |
|---|---|
| TRI | Ac-G LKALEEK LKALEEK LKALEEK LKALEEK G-NH2 |
| TRIL9C | Ac-G LKALEEK CKALEEK LKALEEK LKALEEK G-NH2 |
| TRIL19C | Ac-G LKALEEK LKALEEK LKACEEK LKALEEK G-NH2 |
| TRIL9CL19C | Ac-G LKALEEK CKALEEK LKACEEK LKALEEK G-NH2 |
| TRIL12AL16C | Ac-G LKALEEK LKAAEEK CKALEEK LKALEEK G-NH2 |
| TRIL16X | Ac-G LKALEEK LKALEEK XKALEEK LKALEEK G-NH2 |
| BABY | Ac-G LKALEEK LKALEEK LKALEEK G-NH2 |
| GRAND | Ac-G LKALEEK LKALEEK LKALEEK LKALEEK LKALEEK G-NH2 |
| GRANDL16XL26AL30C | Ac-G LKALEEK LKALEEK XKALEEK LKAAEEK CKALEEK G-NH2 |
| TRIL12LDL16C | Ac-G LKALEEK LKALD EEK CKALEEK LKALEEK G-NH2 |
| GRANDL12LDL16CL26AL30C | Ac-G LKALEEK LKALD EEK CKALEEK LKAAEEK CKALEEK G-NH2 |
| CoilSer | Ac-E WEALEKK LAALESK LQALEKK LEALEHG-NH2 |
X = Penicillamine. LD = D-leucine. Residues in bold and italics indicate substitutions.
Fig. 1.

(A) Amphipathic α-helix placing hydrophobic residues (“a” and “d”) on one face, and hydrophilic residues on the other. (B) Helical wheel diagrams showing the hydrophobic (“a” and “d”) and electrostatic (“e” and “g”) interactions in parallel two and three-stranded coiled coil peptides.
Scaffolds such as these can be employed in the sub-field of de novo designed metallopeptides. The fact that a third of all proteins contain metal ion sites provides a strong motivation for such studies.24 Metalloprotein design requires an additional level of complexity as one must perturb the original bioorganic design in order to construct an environment amenable to a specific metal in a defined oxidation state. There are many fascinating questions that we and others are keen to answer. Among those points we will address in this article are: how does one define the coordination number of a metal in a protein by controlling first coordination sphere ligands and modifying the second coordination environment? Can one harness steric effects associated with the spatial orientation of side chains by incorporating D-amino acids? Can one engineer proteins containing two nearly identical sites, which discriminate metal binding based on coordination number? Can one prepare proteins that have different metal affinity while retaining identical first coordination ligands? Can we isolate proteins which contain the same metal, in the same oxidation state with dramatically different physical properties? Our group and others in the de novo metallopeptide design community have made significant advances in recent years towards understanding the mechanism of metal insertion into well defined biological constructs,25,26,30,31 rates of insertion,25,30 and selectivity.27–29
To address questions such as these, metal binding sites can be designed or engineered into the hydrophobic interior of coiled coils, for example by substitution of an interior Leu layer with an amino acid capable of metal binding. The use of the TRI peptide family allows us to harness the stability provided by a protein’s tertiary structure to control the coordination chemistry of a metal ion site. Our work has focused on the generation of thiol-rich sites (by incorporation of a Cys residue) which have a high affinity for heavy, and often toxic, metals such as Hg(II), Cd(II), Bi(III), Pb(II) and As(III).18,19,30–34 This Perspective focuses on how we have been able to utilize these TRI peptides to influence and control how cadmium binds in unfavourable, but biologically relevant, trigonal thiolate coordination geometries in aqueous solution. In particular, we have been able to achieve specific coordination environments by controlling the steric constraints within the metal ion binding pocket, by making modifications to either the first or second coordination sphere residues. In this way we have prepared peptides capable of binding cadmium as distorted tetrahedral CdS3O, CdS4 and trigonal CdS3. We will illustrate how the fundamental principles identified from these studies lead to selective binding of cadmium into sites that confer different physical properties on the metal.
In order to characterize these sites, we have employed two important and complementary techniques with which to probe cadmium species, 113Cd NMR and 111mCd perturbed angular correlation (PAC) spectroscopy. Most commonly used is 113Cd NMR spectroscopy, as the chemical shift is highly sensitive to both the nature of the coordinating ligands as well as the metal ion coordination geometry, which is reflected by a large chemical shift range.35 In addition, the detection of multiple cadmium binding sites in a biological system is possible if the metal does not move between sites faster than the NMR timescale (0.01–10 ms). Cadmium was first used to probe the Zn(II) site in carbonic anhydrase and alkaline phosphatase using 113Cd NMR in 1976.36 The use of 111mCd PAC is a complimentary technique due to the very different (nanosecond) timescale, and involves the successive emission of two γ rays from the radioactive 111mCd isotope.35 Subsequently five parameters can be extracted from the PAC spectrum, which provide important information about the electronic environment, the symmetry of the metal ion and the dynamics of the site. This technique has proved successful in establishing the structure and dynamics of metal ion sites in biomolecules.37,38
Cadmium binding to TRI peptides
“a” vs. “d” sites
Our aim was to develop a thiolate-rich environment by replacing one of the hydrophobic interior Leu layers with Cys residues capable of metal binding. However, there are two different possibilities within the heptad repeat, as both the “a” and the “d” residues are found in the interior of the coiled coil. Due to the different position in the heptad sequence, substituting an “a” or “d” Leu for a Cys results in two sites with subtly different orientation of the thiolate ligands.18 Energy minimized models are shown in Fig. 2A and 2B, respectively, and illustrate how in an “a” site the sulfur atoms point towards the interior of the coiled coil and appear to be pre-organized for metal binding, whereas in a “d” site, which occurs slightly further down the α-helix, the Cys are orientated such that the sulfurs point away from the interior towards the helical interfaces. As well as displaying a larger cavity size (distance of the sulfur atom from the central axis 3.9 for “d” sites compared to 2.1 Å for “a” sites), the “d” site appears less pre-organized, presumably requiring energetically unfavourable reorganization on metal binding.18 These observations have been confirmed by crystallography.39,40 The different conformations of the Cys ligands raises the question as to how this will influence metal binding affinity and selectivity.
Fig. 2.

MOLSCRIPT figure showing the energy minimized orientations of Cys residues (ball and stick) in an (A) “a” and (B) “d” site model. Additional residues have been omitted for clarity. Reprinted from ref. 18 with permission from Elsevier copyright 1998.
The binding of cadmium to three thiolates can be monitored by a characteristic ligand-to-metal charge-transfer (LMCT) band at around 230 nm, which is similar for both “a” and “d” substituted peptides (Table 2 and Fig. 3). However, one is generally required to go to more basic pH to ensure that all three cysteines bind cadmium in a “d” site, with “a” sites fully formed at lower pH (Table 3).41 Metal binding constants were obtained for the different sites and cadmium was found to bind to “a” sites more strongly, by an order of magnitude, than the analogous “d” sites.41 Taken together, it seems likely that the energy penalty for re-orientation of the Cys ligand in a “d” site disfavours cadmium binding and reduces the affinity for this site. Intriguingly, the cadmium region of the difference circular dichroism (CD) spectra for cadmium bound to “a” and “d” site peptides were very different, with similar LMCT transitions but with opposite polarity.41 Clearly cadmium bound to “a” or “d” substituted peptides are spectroscopically similar, yet distinct.
Table 2.
Spectroscopic values for CdS complexes
Fig. 3.

Typical UV-vis difference titration of cadmium into Cys containing peptides, here that for TRIL16C is shown. The change in absorption at 235 nm is plotted against equivalents of peptide added (inset), illustrating that three helices bind per cadmium at pH 8.5.
Table 3.
Physical properties of cadmium bound to TRI peptides
| Peptide | Apparent pKa2a | 113Cd NMRb | 111mCd PACc |
|---|---|---|---|
| TRIL9C | 13.4 | 615 | — |
| TRIL19C | 14.3 | 627 | — |
| TRIL9CL19C | — | 615 and 623 | — |
| TRIL16C | 13.4 | 625 | 60% CdS3 O, 40% CdS3 |
| TRIL12AL16C | 12.2 | 574 | 100% CdS3 O |
| TRIL16Xd | 15.8 | 684 | 100% CdS3 |
| TRIL12LD L16Cd | 15.1 | 697 | 100% CdS3 |
Mixed 3- and 4-coordinate cadmium41
A significant amount of effort has been expended using small molecule complexes to understand Cd–S chemistry. Table 2 provides a comparison of the 113Cd NMR, UV spectral features and metrical parameters for CdS3, CdS4 and the TRIL16C and TRIL12C designed peptides. 113Cd bound to “a” (TRIL16C) and “d” (TRIL12C) substituted peptides, resulted in chemical shifts of 625 and 619 ppm, respectively. These resonances fall within the acceptable chemical shift range for cadmium bound to three thiolates, four thiolates or CdS3X (where X = O, N).41 Extended X-ray absorption fine structure (EXAFS) spectroscopy also could not distinguish between these three alternatives since the average Cd–S bond distances of 2.49 Å41 are intermediate between purely trigonal CdS3 (2.45 Å)42,43 or tetrahedral CdS4 (2.54 Å)44–47 species (Table 2). Similarly, both the energies and extinction coefficients of the electronic transitions provided no basis for discrimination of metal site structure. We next turned to 111mCd PAC spectroscopy to study the cadmium coordination geometry.37,38 Most notably PAC spectroscopy is capable of probing both the electronic environment around the cadmium ion and the symmetry of the site, on a very fast timescale (0.1–100 ns). The key result is that the cadmium ion is actually bound as a dynamic mixture of two species, trigonal CdS3 (40% ωo = 0.44 rad ns−1) and tetrahedral CdS3O (60% ω0 = 0.34 rad ns−1) (where the O corresponds to an exogenous water molecule), see Fig. 4A.41 The interchange between these two species is rapid on the NMR timescale and as such we observe a single coalesced peak. However, the two independent species can be detected on the faster 111mCd PAC timescale, further highlighting the combined strength of these techniques.
Fig. 4.

111mCd PAC spectra showing a mixture of 2 species for (A) Cd(TRIL16C)3 −, which correspond to the single species formed by (B) Cd(TRIL12AL16C)3− and (C) Cd(TRIL16Pen)3−, respectively.
We can conclude that cadmium binds to our single Cys designed coiled coils as a mixture of 3- and 4-coordinate cadmium. However, nature is capable of binding metals in one well defined coordination geometry and so our aim was to sequester cadmium as either exclusively 4- or 3-coordinate CdS3O and CdS3, respectively. The following section describes how we were able to obtain either fully tetrahedral CdS3O or trigonal CdS3 by systematically making single alterations to the parent peptide sequence, TRIL16C, and how this subsequently resulted in systems with very different physical properties.
4-Coordinate cadmium49
The preparation of 4-coordinate CdS3O peptides with a small exogenous ligand are vital towards understanding the origin behind “active site water” and substrate binding in designed systems. It seemed logical that by altering the steric constraints in the metal binding pocket we should be able to perturb the cadmium speciation to affect a fully 4-coordinate site. We reasoned that by removing steric bulk above the metal binding site we would provide a water “pocket” or “hole”, as illustrated by Hodges et al.48 and that this would lead to fully CdS3O coordination. The strategy was to replace the Leu layer at the 12 site directly above the Cys (a 2nd coordination sphere residue) by Ala. In order to assess the impact of such modifications, it is useful to build models using known crystallographic structures. Fortunately, we previously reported the X-ray crystal structure of an arsenated Cys derivative of CoilSer, As(CSL9C)3, which forms a parallel three-stranded coiled coil.39 Therefore, this scaffold can be used to build the metal binding sites of various peptides by modelling substitutions made to residues in the hydrophobic interior. The model for the metal binding site of TRIL12AL16C, see Fig. 5A, shows the cavity produced on substituting a Leu with an Ala directly above the plane of the Cys, when compared to the parent construct based on TRIL16C (Fig. 5B). Indeed, this construct provides a fully hydrated metal binding cavity, resulting in a 100% 4-coordinate CdS3O species as determined by 111mCd PAC analysis of 111mCd(TRIL12AL16C)3− (100% ωo = 0.34 rad ns−1), see Fig. 4B.49,50 The 113Cd NMR chemical shift of this purely CdS3O complex is 574 ppm (Fig. 6A). Because the 625 ppm signal for 113Cd bound to TRIL16C corresponds to a coalescence resonance of CdS3O and CdS3, the observation that CdS3O has values around 574 ppm indicates that pure CdS3 species should be found downfield of 625 ppm.
Fig. 5.
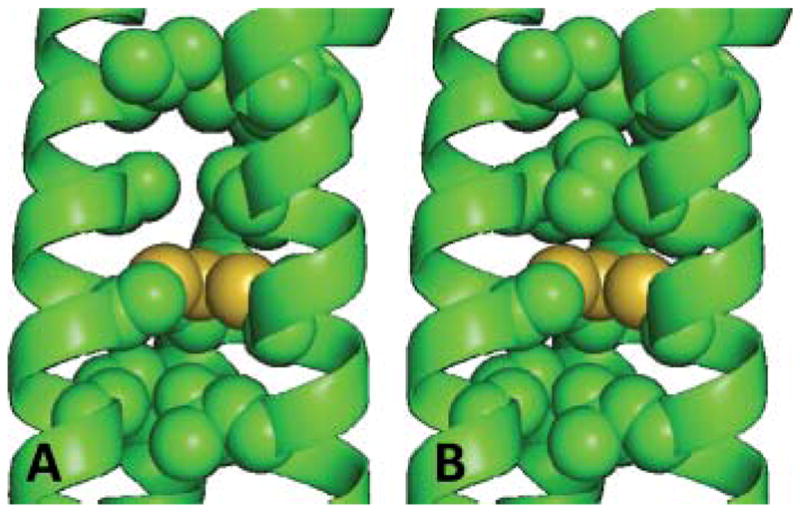
Pymol representations of the cavity formed by replacing a leucine at position 12 with an alanine in (A) L12AL16C compared to the peptide (B) L16C based on the X-ray crystal structure of As(CS L9C)3.39 Interior layer (9, 12, 16 and 19) residues have been shown as spheres, carbons (green) and sulfur (yellow). Additional residues side chains have been omitted for clarity.
Fig. 6.

113Cd NMR spectra for (A) Cd(TRIL12AL16C)3−, (B) Cd(TRIL16Pen)3 −, (C) Cd2 (GRANDL16PenL26AL30C)32− and (D) Cd2 (GRANDL12LD L16CL26AL30C)32−.
Somewhat surprisingly, placement of these “holes” at positions in the coiled coil further away from the metal site, separated by an intervening Leu layer, still influences the CdS3/CdS3O speciation. For example, a “hole” two layers above the metal site, such as in TRIL9AL16C, resulted in a fully 4-coordinate species (100% ωo = 0.33 rad ns−1) with a lower chemical shift (583 ppm) than the parent peptide, TRIL16C (625 ppm).51 However, in an analogous peptide, TRIL16CL23A, placing the “hole” two layers below the metal site actually results in a more 3-coordinate CdS3 species (45% ωo = 0.45 rad ns−1, 643 ppm).51 This is another example of how non-coordinating amino acids may dramatically alter the resultant coordination environment of a metal ion in a protein.49 These peptides with remote alanine substitution illustrate that the metal coordination sphere is directly influenced by the packing of leucine above and below the cysteine plane.
3-Coordinate cadmium50
One might reason that if removing steric bulk above the plane of the Cys led to fully CdS3O then it would be likely that the addition of steric bulk could push the equilibrium towards CdS3. Unfortunately, it is difficult to obtain a much more sterically demanding naturally occurring amino acid than Leu; however, one of the benefits of de novo design is that we are not limited to the 20 commonly found amino acids, and we can readily incorporate non-natural amino acids into our sequence. Our first approach was to replace the Leu layer at position 12 with amino acids containing more bulky side chains, such as the non-natural hexafluoroleucine. This, however, resulted in a shift in the 113Cd NMR from 625 to 607 ppm, suggesting the formation of a more CdS3O species.50 The second approach was to increase the steric bulk directly around the metal ion binding pocket. Therefore, the non-natural amino acid penicillamine (Pen), which had previously been used in de novo peptide design,52 was incorporated. Pen can be considered either the bulkier analogue of Cys with methyl groups in place of the β-methylene hydrogens, or as a thiol containing analogue of Val, see Fig. 7. Viewed as a bulky analogue of cysteine, one might consider the methyl groups as excluding water access to the metal just as a sterically bulky small molecule ligand can decrease the coordination number of a metal.53 However, when viewed as a valine analogue, the additional methyl groups may prevent water from reaching the active site by blocking access either at helical interfaces, restricting flexibility of the coordinating ligand, or through improved packing of Leu residues about the metal ion binding site. Whatever the effective mechanism may be, a fully CdS3 coordination was confirmed for Cd(TRIL16Pen)3− by 111mCd PAC spectroscopy (100% ωo = 0.45 rad ns−1) (Fig. 4C).50 As predicated, the 684 ppm resonance for this complex is significantly downfield of the signal for Cd(TRIL16C)3− (Fig. 6B).50 Subsequently, we have shown that replacement of Cys by Pen in “a” or “d” sites can yield trigonal planar CdS3 structures.
Fig. 7.

Representation of penicillamine as being either the bulkier analogue if cysteine containing methyl groups in place of the β-methylene hydrogens; or the thiol analogue of valine.
As shown above, the 113Cd NMR chemical shift is different for cadmium bound to TRIL12AL16C as fully 4-coordinate CdS3O (574 ppm), compared to cadmium bound to TRIL16Pen as fully 3-coordinate CdS3 (684 ppm). In fact, these sites differ from one another to an even greater extent. For example the cadmium region of the difference CD spectra for cadmium bound to TRIL16Pen and TRIL12AL16C are distinct, as are their UV spectra, λ227 (28 900 M−1 cm−1) and λ230 (21 800 M−1 cm−1), respectively.28
One of the most notable physical differences between CdS3O and CdS3 centers, is the pH at which these peptides sequester cadmium. Peptides such as TRIL12AL16C that bind cadmium as tetrahedral CdS3O sequester the metal at more acidic pH with a pKa2, for the simultaneous release of two protons, of 12.2.28 In contrast, the pKa2 for the binding of cadmium to TRIL16Pen, which forms the trigonal CdS3 site, is significantly higher at 15.8.51 Peptides that bind cadmium as a mixture of 3- and 4-coordinate species such as TRIL16C, 40% CdS3 and 60% CdS3O, have intermediate pKa2 values (13.4).34,40 The peptide TRIL12AL16Pen was synthesized and found to bind cadmium as fully 4-coordinate CdS3O species, as confirmed by both 111mCd PAC (ωo = 0.34 rad ns−1) and 113Cd NMR (583 ppm) spectroscopy.51 Furthermore this peptide sequesters cadmium with a pKa2 of 12.7, which is consistent with a fully 4-coordinate CdS3O site.51 This result confirms that these physical properties are due to the 4-coordinate CdS3O structure and not a consequence of inductive effects associated with Pen coordinating to the cadmium rather than Cys.
A direct correlation exists between the 113Cd NMR chemical shift and the percentage of 3- and 4-coordinate, CdS3 and CdS3O, species based on 111mCd PAC spectroscopy.51 In fact, based on this correlation, we predict that the 113Cd NMR chemical shift of a perfectly trigonal 100% CdS3 site within a coiled coil, would be 702 ppm and that of a 100% CdS3O site 579 ppm.50,51 Using the 113Cd NMR range (579–702 ppm) allows us to predict the percentage speciation between the 3- and 4-coordinate cadmium species. This correlation has already proved to be a valuable tool in the de novo design of our peptides, especially as 111mCd PAC spectroscopy is less readily available than 113Cd NMR. This correlation may be more widely applicable to metal binding in natural biological systems, in particular cadmium substituted thiolate-rich zinc proteins.
Recently, the structure of cadmium bound to CmtR, one of the ArsR-SmtB type DNA binding repressors found in Mycobacterium tuberculosis, was solved by 2D NMR. The authors propose the first biological three coordinate CdS3 site.54 Since our mercury peptide constructs provided such good water soluble models for the protein MerR,19,32,55 it is of interest to compare the spectroscopic data for our first water soluble model of CdS3 in a biological construct with that of CmtR. It is intriguing, therefore, that the 113Cd NMR chemical shift of 480 ppm has been reported for cadmium bound to CmtR,56 which is in contrast to the values (675–700 ppm) we have observed in our systems. Thus, it is possible that under certain conditions CdS3O or CdS2O2 may be formed. The more common CdS3O site, also found in cadmium substituted δ-aminolevulinic acid dehydratase (ALAD), should be modelled well by our CdS3O 4-coordinate complexes. Finally, the 113Cd NMR chemical shift of 622 ppm reported for cadmium bound to the toxic metal-sensing transcriptional repressor CadC, is consistent with a mixture of CdS3/CdS3X species interconverting rapidly on the NMR timescale.57 Studies with these aqueous peptide models are extremely powerful for interpreting metal coordination in sulfur containing biomolecules.
Disubstituted TRI peptides with no intervening leucine layers45
Incorporating two cysteine layers near one another within the same sequence offers the potential to generate new metal ion sites, with higher thiol coordination numbers. The use of a Cys-X-X-Cys motif has previously been described for constructs which are similar, but form exclusively two-stranded coiled coils.58,59 One of these peptides is unstructured in the absence of metal ions; however, becomes structured on binding cadmium as CdS4.59 We designed sequences that incorporate the common Cysa-X-X-Cysd binding motif and less common Cysd-X-X-X-Cysa sequence into the TRI parent peptide in order to address if similar coordination geometries are achieved on binding cadmium. This resulted in peptides that contain adjacent Cys sites on the hydrophobic face of the α-helix, TRIL9CL12C and TRIL12CL16C, with no intervening leucine layers. However, the chemistry of these constructs differ from one another in part due to the different orientation of cysteine side chains in “a” vs. “d” sites. Furthermore, cadmium coordination to these constructs has been complicated by the formation of both two- and three-stranded coiled coils in solution. We have confirmed by 113Cd NMR and 111mCd PAC spectroscopy that cadmium binds to TRIL12CL16C as a mixture of species, but to TRIL9CL12C as a distorted tetrahedral CdS4 complex in two stranded and three stranded bundles (100% ωo = 0.14 rad ns−1; 650 and 680 ppm).45 This illustrates how we can achieve higher coordination number species utilizing our TRI scaffold. Furthermore, the coordination chemistry of these systems allows one the rare opportunity to compare directly metal binding behaviour between Cys-X-X-Cys and Cys-X-X-X-Cys in a similar environment.
Selective metal sequestration in disubstituted TRI peptides29
Having achieved coordination number control of cadmium in these thiol rich environments within the interior of our coiled coils, we decided to tackle the more challenging obstacle which is to design multiple independent metal binding sites into the same short peptide sequence. The two sites can be made to behave independent from one another by introducing intervening leucine layers between the two Cys sites. Such a complex would be the first step towards preparing designed metalloproteins, which contain both a stabilizing, structural site and a separate catalytic metal center. Two metals have been previously incorporated into heme maquettes or as binuclear metal centers within the hydrophobic interior of similar constructs.60,61 Heterometal binding to the same peptide has also been achieved, by utilizing the different affinity of metals for chemically different sites.62 We decided to address the more subtle and challenging issue of homometal binding to two independent but very similar sites, utilizing the different affinities of cadmium for “a” and “d” substituted peptides. The peptide sequence utilized was TRIL9CL19C which contains both an “a” (9) and a “d” (19) Cys site, separated by a layer (16) of intervening leucine residues so as to prevent interaction between the two metal centers.
TRIL9CL19C shows sequential and selective binding of cadmium to first the “a” and then the “d” site, as confirmed by 113Cd NMR, 2D NOESY and CD spectroscopy.29 The first equivalent of 113Cd binds exclusively to the “a” or 9 site as can be seen by the growth of a peak at 615 ppm in the NMR. Only on further addition of 113Cd does population of the “d” or 19 site occur, as revealed by the appearance of a peak at 623 ppm, see Fig. 8.29 2D NOESY spectra were recorded and changes in the Cys side chain resonances for the “a” and “d” sites were monitored on addition of cadmium. These observations support a model in which the first equivalent of cadmium binds exclusively to the “a” site and the second equivalent to the “d” site. The order of metallation was further confirmed by monitoring the cadmium region of the difference CD spectra, as cadmium bound to the “a” and “d” sites produce signals with opposite intensity (vide supra) and can, therefore, be used to determine to which site the cadmium is binding first. The observed selectivity is thought to be due to the different binding strengths, which is most likely a result of the distinct orientation of the Cys ligands in the two similar, yet distinct, sites (see Fig. 2). As alluded to earlier, Cys in an “a” site would not require significant rearrangement on binding cadmium, in contrast to the sulfur ligands in a “d” site. Clearly, very conservative modifications to a peptide sequence, such as the position of a substitution, can result in significantly different metal binding and subsequent discrimination.
Fig. 8.

113Cd NMR spectra for the titration of 113CdCl2 into TRIL9CL19C, at pH 8.5. Reprinted from ref. 29. Copyright 2005 American Chemical Society.
The two resonances in the 113Cd NMR spectrum for the fully loaded peptide, Cd2(TRIL9CL19CL)32−, at 615 and 623 ppm (Fig. 8), are similar to those of their monosubstituted analogues suggesting that the two sites behave independently, with no significant metal exchange between the two on the NMR timescale. This is noteworthy as these sights are very close to one another (~15–16 Å) and have been separated by only one intervening layer of Leu (16) residues. Notably, site selectivity for the binding of the same metal ion to very similar thiolatesites, which behave independently, has been achieved in a relatively short peptide (30 amino acids). The success of this simple system demonstrates that large extensive biological architectures are not necessarily required to obtain this type and degree of selectivity.
Heterochromic peptides: 3- and 4-coordinate cadmium28
At this point, we wanted to combine our two significant achievements (binding cadmium exclusively as tetrahedral CdS3O or trigonal CdS3 and selective binding of two cadmium ions into two distinct, independent sites) into a single peptide construct. Peptide sequences capable of binding two equivalents of the same metal ion in separate sites with different coordination geometries more closely approaches our goal of preparing analogues for metalloproteins with both structural and catalytic sites. The heme biosynthesis enzyme δ-amino levulinic acid dehydratase is an example of such a system as it contains a 5-coordinate Zn(II) structural center and a 4-coordinate Zn(II) catalytic site. Carrying out such studies with designed peptides raises the questions whether these sites can maintain their separate and distinct physical properties in a short sequence, if the metallation of one site affects the affinity and physical properties of the second, and if metal ions will show selectivity for one site over the other.
Initial attempts to incorporate both a 3- and 4-coordinate site into the short TRI peptide sequence were unsuccessful. For example, cadmium binding to TRIL12AL16CL23Pen resulted in both sites being predominantly 4-coordinate.28 The use of the GRAND family of peptides, consisting of an additional heptad repeat (Table 1), which provides higher aggregate stability and enhanced packing near the Pen site, was required to achieve a fully 3-coordinate site. The resultant peptide GRANDL16PenL26AL30C has been shown to bind cadmium as 100% 3-coordinate CdS3 and 100% 4-coordinate CdS3O in the 16 and 30 sites, respectively, see Fig. 6C.28 In addition, the pH profile for sequestering two equivalents of cadmium shows two well defined regions, consistent with those for the analogous mono-substituted peptides, GRANDL16Pen and GRANDL26AL30C, see Fig. 9. Not only is the first equivalent of cadmium sequestered to the 4-coordinate site at low pH due to the lower pKa2 associated with binding, but at pH 9.6 when theoretically both sites are capable of binding the cadmium, the 4-coordinate site fills first, as indicated by a peak with 113Cd NMR chemical shift of 588 ppm. Only on further addition of 113Cd do we observe formation of a second peak at 687 pm, which is consistent with the 3-coordinate site. This corresponds to the first example of a single, short peptide capable of binding two equivalents of the same metal ions close to one another in the same coiled coil, with similar ligand sets yet distinct and different coordination geometries and physical properties. We designated these “heterochromic” peptides as you can liken it to the condition where a person has two eyes (metal ions), but with different coloured irises (coordination geometries).
Fig. 9.

UV-vis pH titrations of cadmium loading. pH dependence of 2 equiv. of cadmium binding to GRANDL16PenL26AL30C (20 μM trimer; ●), of 1 equiv. cadmium binding to GRANDL16Pen (20 μM trimer; ■), and of 1 equiv. cadmium binding to GRANDL26AL30C (20 μM trimer; ▲). Reprinted from ref. 28 Wiley-VCH Verlag GmbH & Co. KGaA copyright 2007.
Diastereopeptides: D-amino acids63
While a great success, the generation of a heterochromic peptide using penicillamine was not fully satisfying as the ultimate objective is to prepare heterochromic peptides using identical first coordination sphere ligands. Thus we began to explore ways to modify the environment adjacent to the metal ligands. The type and position of second sphere residues is often crucial and can play an important role in achieving the correct biological activity. We have previously shown that changing the chemical nature of a second coordination sphere ligand (Leu → Ala) can significantly perturb the ultimate metal coordination geometry. Unfortunately, increasing the steric bulk using hexafluoroleucine led to unsatisfactory results, so an alternative strategy was needed. We reasoned that by changing the spatial orientation of a second coordination sphere ligand by altering the chirality at a single carbon backbone atom, one could achieve the desired steric constraints.
In order to assess the impact of changing the chirality of the internal hydrophobic residues, we again prepared models using the known crystallographic structure of As(CSL9C)3. This structure illustrates that all of the leucine sidechains point towards the N-terminus.39 We anticipated that replacing the Leu in the 12 position by its D-amino acid analogue would cause the isopropyl moeity to point towards the C-terminus. This re-orientation of the leucine moves the hydrophobic side chain 2–3 Å towards the metal ion binding site (modelled in Fig. 10). We predicted that this would subsequently exclude any exogenous water, leading to a fully trigonal CdS3 site. We refer to this class of peptides as “diastereopeptides”, as we have incorporated a single D-amino acid into what is otherwise an all L-amino acid α-helix. Though D-amino acids have previously been successfully incorporated into predominantly L-amino acid α-helices, to the best of our knowledge this was the first example where such a modification is made in the interior of a three-stranded coiled coil. Based on the correlations established previously, we would anticipate a 113Cd chemical shift of 702 ppm for a 100% fully trigonal CdS3 species, and this is indeed very close to the value of 697 ppm obtained for Cd(TRIL12LDL16C)3−. In addition the pKa2 for cadmium binding (15.1) is close to that of 15.8 obtained for Cd(TRIL16Pen)3−. Finally 111mCd PAC confirmed that cadmium was bound to the Cys as a 100% trigonal CdS3 species (100% ωo = 0.46 rad ns−1).63
Fig. 10.
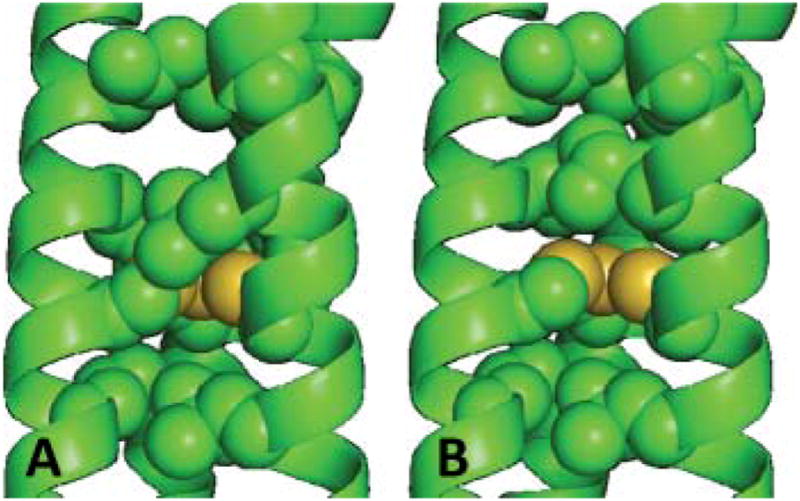
Pymol representation, based on the X-ray crystal structure of As(CS L9C)3, 39 of the L- vs. D-Leu substitution at position 12, for (A) L12LD L16C and (B) L16C. Interior layer (9, 12, 16 and 19) residues have been shown as spheres, carbons (green) and sulfur (yellow). Additional residues side chains have been omitted for clarity.
We next came full circle and prepared the heterochromic diastereopeptide GRANDL12LDL16CL26AL30C. This was found to be capable of binding two equivalents of cadmium ions to the Cys residues, but one as a fully trigonal 3-coordinate CdS3 and the other as a tetrahedral 4-coordinate CdS3O, as indicated by two distinct peaks at 690 and 589 ppm in the 113Cd NMR spectrum, Fig. 6D.63 At pH 9.6, where both sites are capable of binding cadmium, we again observe selectivity for the 4-coordinate site, illustrating that this preference is due to the coordination number of the cadmium and not the nature of the coordinating ligand (Cys vs. Pen). This peptide represents the first example of a sequence capable of binding two equivalents of the same metal ion to two identical sites (three Cys ligands) that differ only in the nature of a non-coordinating second coordination sphere ligand (Ala vs. D-Leu), yet which displays different coordination geometries, specificity and physical properties.
It should be noted that proteins do not have access to D-amino acids within their sequence. Our assemblies are by design parallel constructs; however, helical bundles in biological systems consist of anti-parallel helices. Thus, one can envision that the orientation of these D-Leu residues in our constructs, may resemble the orientation of side chains on an anti-parallel helix.
Future directions
25 years ago chemists would have attempted to interpret large complex biological systems solely by synthesizing small molecule analogues. Today because of technological advances, we are ideally positioned to start addressing important questions and challenging issues concerning metals in biology using peptide design. Recently, scientists have revolutionized their thinking by considering the possibilities made available by nanomaterials that have new and unique properties based on the fact that they are neither small molecules or larger composites. De novo designed proteins are the biological equivalents, providing a more simplified architecture or scaffold allowing us to answer challenging questions that may be too complicated to address using the natural protein system, yet may require a degree of secondary or tertiary structure elements not present in small molecule models. The benefit of de novo designed peptides is that we can choose to adopt certain secondary and tertiary structure elements of biological molecules, and yet maintain the ability to directly correlate chemical observation with chemical structure on an atomic level, in a fashion similar to small molecules, Fig. 11.
Fig. 11.

De novo designed metallopeptides represent the intermediary ground between small molecule model complexes and the larger complex natural biological systems.
While we have illustrated in this perspective how one can achieve a high degree of control in designing cadmium binding sites, we could have illustrated these points with other metals such as mercury or lead.18–20,25,30–32,34,41,45,55,64,65 We have also shown that what initially appear to be very similar constructs, notably the concept of “a” vs. “d” sites, are in fact different (see Fig. 2), binding metals with different affinities and, ultimately, with different physical properties and spectroscopic signatures. Though we have successfully designed sites that bind cadmium as structures with exclusively 3- or 4-coordination geometry, and determined the resulting properties of these sights, new questions arise regarding metalloprotein chemistry. For example, what do we mean by a “3-or 4-coordinate site”? How do the properties of a 4-coordinate site buried in the center of a helix differ from one found at the end of a helical bundle? How may they differ, despite both being found at a similar location in a helix, when one is prepared in a “d” site and the other in an “a” site? We have learned from crystallography that metal ions are capable of binding as either endo or exo configurations, above or below the Cys coordinating ligands (see Fig. 12A and 12B, respectively).39 Does this orientation change exogenous ligand access or metal affinity for a site? Can one distinguish such structures with physical methods, and if there is a preference for one site over another for a metal that is capable of adopting both configurations, is there a difference in the physical properties? The development of dual substituted peptides capable of sequestering multiple equivalents of metal ions (see Fig. 12C), will allow us to address issues of cooperative binding. An advantage of de novo design is that we can measure the thermodynamic preferences and kinetics associated with metal ion binding to these different sites in water and with real biologically relevant ligands. This will allow us to unravel the complexities associated with what leads to the various site preferences of different metal ions in biomolecules. Answering questions such as these is something that can not readily be achieved, or would be more difficult to answer, using small molecule systems, but which can be tackled using the approach of de novo metalloprotein design.65,66
Fig. 12.

The variety of different “types” of metal ion coordination geometries vary from (A) endo (below plane) vs. (B) exo (above plane) coordination modes. Further variations result from (C) varying positions within a coiled coil bundle.
Acknowledgments
V. L. P. thanks the National Institute of Health for support of this research (R01 ES0 12236) and O. I. thanks the Margaret and Herman Sokol Foundation for a Postdoctoral Award.
Biographies

Anna Peacock obtained her MChem at The University of York in 2003, and her PhD at The University of Edinburgh in 2007 on the design of osmium arene anticancer complexes in Professor Peter Sadler’s group. For her post-doctoral work, she joined Professor Vincent Pecoraro’s group at the University of Michigan, working of the design of de novo metallopeptides. She has been appointed as lecturer at the University of Birmingham (UK). Her research interests are centred on the chemistry of metals in biology and medicine.

Olga Iranzo obtained her B.S. in Chemistry (Universidad de Barcelona) in 1990. After working as a research scientist in Rhône-Poulenc, she went on to do her M.S. in Biotechnology. She received her PhD (2003) at The University of Buffalo under supervision of Professors J. R. Morrow and J. P. Richard, on Zn(II) catalysts for RNA cleavage. Her postdoctoral work was at the University of Michigan with Professor Pecoraro and she became an AHA postdoctoral fellow (2006). She is currently at the Instituto de Tecnologia Química e Biológica at Oeiras (Portugal). Her interests include bioinorganic chemistry and metalloprotein design and engineering.

Vincent L. Pecoraro received his B.S. degree in biochemistry from UCLA, (1977) and his Ph.D. in chemistry (1981) at UC Berkeley (advisor: Prof. Kenneth N. Raymond). After an NIH postdoctoral fellowship with Prof. W. W. Cleland in Biochemistry (University of Wisconsin), he joined the University of Michigan (1984; promoted to associate professor in 1989; professor in 1992; and John T. Groves Professor in 2005). He researches the role of metals in biology (the biological chemistry of manganese, vanadium, heavy metals and de novo metalloprotein design) and metallamacrocyclic chemistry. He has received numerous awards and has served as an Associate Editor of Inorganic Chemistry since 1994.
References
- 1.Regan L, DeGrado WF. Science. 1988;241:976–978. doi: 10.1126/science.3043666. [DOI] [PubMed] [Google Scholar]
- 2.DeGrado WF, Wasserman ZR, Lear JD. Science. 1989;243:622–628. doi: 10.1126/science.2464850. [DOI] [PubMed] [Google Scholar]
- 3.Lau SY, Taneja AK, Hodges RS. J Biol Chem. 1984;259:13253–13261. [PubMed] [Google Scholar]
- 4.Monera OD, Zhou NE, Kay CM, Hodges RS. J Biol Chem. 1993;268:19218–19227. [PubMed] [Google Scholar]
- 5.Kaiser ET, Kdzdy FJ. Proc Natl Acad Sci U S A. 1983;80:1137–1143. doi: 10.1073/pnas.80.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaiser ET, Kdzdy FJ. Science. 1984;223:249–255. doi: 10.1126/science.6322295. [DOI] [PubMed] [Google Scholar]
- 7.DeGrado WF, Summa C, Pavone V, Nastri F, Lombardi A. Annu Rev Biochem. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 8.Hecht MH. Proc Natl Acad Sci U S A. 1994;91:8729–8730. doi: 10.1073/pnas.91.19.8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramirez-Alvarado M, Blanco FJ, Serrano L. Nat Struct Biol. 1996;3:604–612. doi: 10.1038/nsb0796-604. [DOI] [PubMed] [Google Scholar]
- 10.Dahiyat BI, Mayo SL. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 11.Tripet B, Wagschal K, Lavigne P, Mant CT, Hodges RS. J Mol Biol. 2000;300:377–402. doi: 10.1006/jmbi.2000.3866. [DOI] [PubMed] [Google Scholar]
- 12.Harbury PB, Zhang T, Kim PS, Albert T. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 13.Lovejoy B, Choe S, Cascio D, McRorie DK, DeGrado WF, Eisenberg D. Science. 1993;259:1288–1293. doi: 10.1126/science.8446897. [DOI] [PubMed] [Google Scholar]
- 14.Walsh ST, Cheng H, Bryson JW, Roder H, DeGrado WF. Proc Natl Acad Sci U S A. 1999;96:5486–5491. doi: 10.1073/pnas.96.10.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho SP, DeGrado WF. J Am Chem Soc. 1987;109:6751–6758. [Google Scholar]
- 16.O’Shea EK, Lumb KJ, Kim PS. Curr Biol. 1993;3:658–667. doi: 10.1016/0960-9822(93)90063-t. [DOI] [PubMed] [Google Scholar]
- 17.Monera OD, Kay CM, Hodges RS. Biochemistry. 1994;33:3862–3871. doi: 10.1021/bi00179a010. [DOI] [PubMed] [Google Scholar]
- 18.Dieckmann GR, McRorie DK, Lear JD, Sharp KA, DeGrado WF, Pecoraro VL. J Mol Biol. 1998;280:897–912. doi: 10.1006/jmbi.1998.1891. [DOI] [PubMed] [Google Scholar]
- 19.Dieckmann GR, McRorie DK, Tierney DL, Utschig LM, Singer CP, O’Halloran TV, Penner-Hahn JE, DeGrado WF, Pecoraro VL. J Am Chem Soc. 1997;119:6195–6196. [Google Scholar]
- 20.Ghosh D, Lee K-H, Demeler B, Pecoraro VL. Biochemistry. 2005;44:10732–10740. doi: 10.1021/bi0506674. [DOI] [PubMed] [Google Scholar]
- 21.Su JY, Hodges RS, Kay CM. Biochemistry. 1994;33:15501–15510. doi: 10.1021/bi00255a032. [DOI] [PubMed] [Google Scholar]
- 22.Moitra J, Szilák L, Krylov D, Vinson C. Biochemistry. 1997;36:12567–12573. doi: 10.1021/bi971424h. [DOI] [PubMed] [Google Scholar]
- 23.Wagschal K, Tripet B, Hodges RS. J Mol Biol. 1999;285:785–803. doi: 10.1006/jmbi.1998.2284. [DOI] [PubMed] [Google Scholar]
- 24.Holm RH, Kennepohl P, Solomon EI. Chem Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh D, Pecoraro VL. unpublished work. [Google Scholar]
- 26.Li X, Suzuki K, Kanaori K, Tajima K, Kashiwada A, Hiroaki H, Kohda D, Tanaka T. Prot Sci. 2000;9:1327–1333. doi: 10.1110/ps.9.7.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiyokawa T, Kanaori K, Tajima K, Koike M, Mizuno T, Oku J-I, Tanaka T. J Pep Res. 2004;63:347–353. doi: 10.1111/j.1399-3011.2004.00109.x. [DOI] [PubMed] [Google Scholar]
- 28.Iranzo O, Cabello C, Pecoraro VL. Angew Chem, Int Ed. 2007;46:6688–6691. doi: 10.1002/anie.200701729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matzapetakis M, Pecoraro VL. J Am Chem Soc. 2005;127:18229–18233. doi: 10.1021/ja055433m. [DOI] [PubMed] [Google Scholar]
- 30.Farrer BT, Pecoraro VL. Proc Natl Acad Sci U S A. 2003;100:3760–3765. doi: 10.1073/pnas.0336055100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farrer BT, Harris NP, Balchus KE, Pecoraro VL. Biochemistry. 2001;40:14696–14705. doi: 10.1021/bi015649a. [DOI] [PubMed] [Google Scholar]
- 32.Iranzo O, Thulstrup PW, Ryu S, Hemmingsen L, Pecoraro VL. Chem–Eur J. 2007;13:9178–9190. doi: 10.1002/chem.200701208. [DOI] [PubMed] [Google Scholar]
- 33.Farrer BT, McClure CP, Penner-Hahn JE, Pecoraro VL. Inorg Chem. 2000;39:5422–5423. doi: 10.1021/ic0010149. [DOI] [PubMed] [Google Scholar]
- 34.Matzapetakis M, Ghosh D, Weng T-C, Penner-Hahn JE, Pecoraro VL. JBIC, J Biol Inorg Chem. 2006;11:876–890. doi: 10.1007/s00775-006-0140-7. [DOI] [PubMed] [Google Scholar]
- 35.Hemmingsen L, Olsen L, Antony J, Sauer SPA. JBIC, J Biol Inorg Chem. 2004;9:591–599. doi: 10.1007/s00775-004-0553-0. [DOI] [PubMed] [Google Scholar]
- 36.Armitage IM, Pajer RT, Uiterkamp AJMS, Chlebowski JF, Coleman JE. J Am Chem Soc. 1976;98:5710–5712. doi: 10.1021/ja00434a058. [DOI] [PubMed] [Google Scholar]
- 37.Hemmingsen L, Sas KN, Danielsen E. Chem Rev. 2004;104:4027–4062. doi: 10.1021/cr030030v. [DOI] [PubMed] [Google Scholar]
- 38.Bauer R. Q Rev Biophys. 1985;18:1–64. doi: 10.1017/s0033583500004972. [DOI] [PubMed] [Google Scholar]
- 39.Touw DS, Nordman CE, Stuckey JA, Pecoraro VL. Proc Natl Acad Sci U S A. 2007;104:11969–11974. doi: 10.1073/pnas.0701979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Touw D. PhD Thesis. University of Michigan; 2007. [Google Scholar]
- 41.Matzapetakis M, Farrer BT, Weng T-C, Hemmingsen L, Penner-Hahn JE, Pecoraro VL. J Am Chem Soc. 2002;124:8042–8054. doi: 10.1021/ja017520u. [DOI] [PubMed] [Google Scholar]
- 42.Gruff ES, Koch SA. J Am Chem Soc. 1990;112:1245–1247. [Google Scholar]
- 43.Duhme A-K, Strasdeit H. Z Anorg Allg Chem. 1999;625:6–8. [Google Scholar]
- 44.Swenson D, Baenziger NC, Coucouvanis D. J Am Chem Soc. 1978;100:1932–1934. [Google Scholar]
- 45.Luczkowski M, Stachura M, Schirf V, Demeler B, Hemmingsen L, Pecoraro VL. Inorg Chem. 2008;47:10875–10888. doi: 10.1021/ic8009817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kharenko OA, Ogawa MY. J Inorg Biochem. 2004;98:1971–1974. doi: 10.1016/j.jinorgbio.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 47.Henehan CJ, Pountney DL, Zerbe O, Vasak M. Prot Sci. 1993;2:1756–1764. doi: 10.1002/pro.5560021019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Monera OD, Sönnichsen FD, Hicks L, Kay CM, Hodges RS. Protein Eng. 1996;9:353–363. doi: 10.1093/protein/9.4.353. [DOI] [PubMed] [Google Scholar]
- 49.Lee K-H, Matzapetakis M, Mitra S, Marsh ENG, Pecoraro VL. J Am Chem Soc. 2004;126:9178–9179. doi: 10.1021/ja048839s. [DOI] [PubMed] [Google Scholar]
- 50.Lee K-H, Cabello C, Hemmingsen L, Marsh ENG, Pecoraro VL. Angew Chem, Int Ed. 2006;45:2864–2868. doi: 10.1002/anie.200504548. [DOI] [PubMed] [Google Scholar]
- 51.Iranzo O, Jakusch T, Lee K-H, Hemmingsen L, Pecoraro VL. Chem–Eur J. 2009 doi: 10.1002/chem.200802105. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petros AK, Shaner SE, Costello AL, Tierney DL, Gibney BR. Inorg Chem. 2004;43:4793–4795. doi: 10.1021/ic0497679. [DOI] [PubMed] [Google Scholar]
- 53.Gorrell IB, Looney A, Parkin G, Rheingold AL. J Am Chem Soc. 1990;112:4068–4069. [Google Scholar]
- 54.Banci L, Bertini I, Cantini F, Ciofi-Baffoni S, Cavet JS, Dennison C, Graham AI, Harvie DR, Robinson NJ. J Biol Chem. 2007;282:30181–30188. doi: 10.1074/jbc.M701119200. [DOI] [PubMed] [Google Scholar]
- 55.Pecoraro VL, Peacock AFA, Iranzo O, Luczkowsk M. Advances in Inorganic Biochemistry: From Synthetic Models to Cellular Systems. In: Baldwin MJ, Long EC, editors. ACS Symposium Series. in press. [Google Scholar]
- 56.Wang Y, Hemmingsen L, Giedroc DP. Biochemistry. 2005;44:8976–8988. doi: 10.1021/bi050094v. [DOI] [PubMed] [Google Scholar]
- 57.Busenlehner LS, Cosper NJ, Scott RA, Rosen BP, Wong MD, Giedroc DP. Biochemistry. 2001;40:4426–4436. doi: 10.1021/bi010006g. [DOI] [PubMed] [Google Scholar]
- 58.Mukherjee M, Zhu X, Ogawa MY. Inorg Chem. 2008;47:4430–4432. doi: 10.1021/ic702370k. [DOI] [PubMed] [Google Scholar]
- 59.Kharenko OA, Ogawa MY. J Inorg Biochem. 2004;98:1971–1974. doi: 10.1016/j.jinorgbio.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 60.Lombardi A, Summa CM, Geremia S, Randaccio L, Pavone V, DeGrado WF. Proc Natl Acad Sci U S A. 2000;97:6298–6305. doi: 10.1073/pnas.97.12.6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Summa CM, Rosenblatt MM, Hong J-K, Lear JD, DeGrado WF. J Mol Biol. 2002;321:923–938. doi: 10.1016/s0022-2836(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 62.Tanaka T, Mizuno T, Fukui S, Hiroaki H, Oku J, Kanaori K, Tajima K, Shirakawa M. J Am Chem Soc. 2004;126:14023–14028. doi: 10.1021/ja047945r. [DOI] [PubMed] [Google Scholar]
- 63.Peacock AFA, Hemmingsen L, Pecoraro VL. Proc Natl Acad Sci U S A. 2008;105:16566–16571. doi: 10.1073/pnas.0806792105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iranzo O, Ghosh D, Pecoraro VL. Inorg Chem. 2006;45:9959–9973. doi: 10.1021/ic061183e. [DOI] [PubMed] [Google Scholar]
- 65.Ghosh D, Pecoraro VL. Inorg Chem. 2004;43:7902–7915. doi: 10.1021/ic048939z. [DOI] [PubMed] [Google Scholar]
- 66.Summers MF. Coord Chem Rev. 1988;86:43–134. [Google Scholar]