

Small self-cleaving ribozymes such as the hammerhead ribozyme (HHR) have been instrumental as model systems for RNA catalysis.1–3 Recently, an extended HHR structure was determined by X-ray crystallography at 2.0-Å resolution,4 in which a divalent metal ion was observed near the active site. Subsequent computer simulations lend credence to the possibility that this metal ion may play an active role in catalysis,4,5 although free energy profiles to elucidate specific pathways have not yet been reported.
We report results from combined quantum mechanical/molecular mechanical (QM/MM) free energy simulations to explore metalassisted phosphoryl transfer and general acid catalysis in the extended HHR. The mechanisms considered here assumes that the 2′OH group of C17 has already been activated (i.e., is deprotonated) and acts as a nucleophile to go on an in-line attack to the adjacent scissile phosphate, passing through a pentavalent phosphorane intermediate/transition state, followed by acid-catalyzed departure of the O5′ leaving group of C1.1. The general acid is assumed to be the 2′OH group of G8.6–8
A series of six 2D potential of mean force (PMF) profiles are herein reported, requiring an aggregate of over 100 ns of QM/MM simulation. Simulations were based on the extended HHR solvent structure (PDB: 2OEU)4 solvated by over 10K water molecules. Active site residues (G8, A9, C1.1, C17, and an Mg2+ ion with coordinated water molecules shown in Figure 3) were treated quantum mechanically (81 atoms total) using the AM1/d-PhoT quantum model9 with AM1/d model for Mg2+.10 We used the all-atom AMBER parmbsc0 force field,11 to describe the HHR outside of the active site, along with the TIP4P-Ewald water model12 and the consistent set of monovalent ion parameters.13 Multidimensional PMF profiles were generated along reaction coordinates corresponding to phosphoryl transfer, proton transfer from the general acid to the leaving group, and divalent metal ion binding mode. Complete details are given in the Supporting Information.
Figure 3.
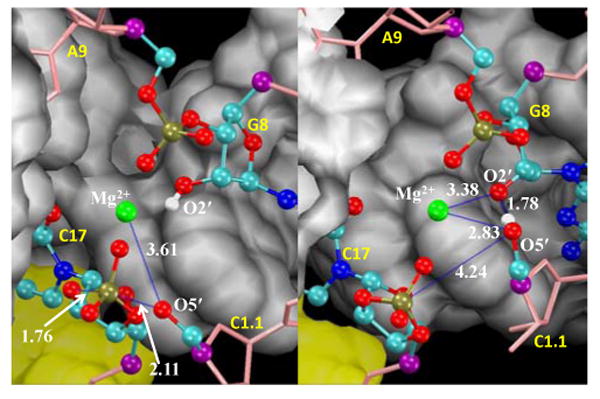
Snapshots of the active site at the transition states for phosphoryl transfer (left) and general acid catalysis (right) with average distances labeled.
Phosphoryl transfer and general acid steps are stepwise, and sensitive to Mg2+ binding mode
Our initial attempts to study the chemical steps of the HHR reaction from 2D PMF profiles using phosphoryl transfer and proton transfer reaction coordinates, but not considering a reaction coordinate associated with Mg2+ ion binding mode, led to free energy barriers that were unexpectedly high (∼37 kcal/mol) compared to an estimated barrier of ∼20 kcal/mol derived from the experimental rate of one turnover per minute in HHR catalysis.14 We extended the calculations so as to include a 3D-PMF profiles with a course-grained reaction coordinate associated with the Mg2+ binding mode, and confirmed the sensitivity of the barriers to the Mg2+ ion position along the reaction coordinate. A common feature of the reaction mechanism derived from the 3D profile was that the phosphoryl transfer and general acid steps were stepwise (e.g., Fig. 2a), allowing these steps to be decoupled. Both the phosphoryl transfer and general acid steps of the reaction were coupled with Mg2+ binding mode, and hence separate 2D profiles were generated for each step with a reaction coordinate corresponding to the Mg2+ binding mode as a second dimension. Table 1 summarizes key average geometrical parameters, and free energy values for stationary points along the reaction. The corresponding reaction rate constants are depicted in Fig. 1
Figure 2.

(A) Selected 2D surface, harmonically restrained along the course-grained metal ion binding coordinate at d(Mg2+,G8:O2′) = 2.5Å, where z1 = d(O5′,P) - d(P,O2′), z2 = d(G8:O2′,H) - d(H,O5′). (B) 2D PMF for Mg2+ binding mode in phosphoryl transfer step, where z4 = d(Mg2+,O5′) + d(Mg2+,G8:O2′). (C) 2D PMF for Mg2+ binding mode in general acid step, where z5 = d(Mg2+,O5′) - d(Mg2+,G8:O2′). d(x,y) denotes distance between x and y. TS is the acronym of transition state.
Table 1.
Relative free energies and internuclear distances at various states of RNA self-cleavage catalysis in hammerhead ribozymesa
| React. | TS1 | Int. | TS2 | Prod. | |
|---|---|---|---|---|---|
| Nu-P | 3.50(04) | 1.76(05) | 1.66(03) | 1.67(03) | 1.68(03) |
| P-Lea | 1.65(03) | 2.11(05) | 4.51(04) | 4.24(48) | 3.63(23) |
| gA-H | 0.96(00) | 0.96(00) | 0.96(00) | 1.78(04) | 3.75(04) |
| H-Lea | 2.57(51) | 4.07(47) | 4.13(73) | 1.04(03) | 1.00(03) |
| Mg2+-Lea | 3.99(18) | 3.61(17) | 2.02(05) | 2.83(86) | 4.48(05) |
| Mg2+-gA | 4.56(18) | 4.03(18) | 4.33(06) | 3.38(86) | 2.03(05) |
| ΔG | 0.0(4) | 24.4(6) | -6.7(3) | 13.7(7) | -13.6(9) |
Free energies (ΔG) are in kcal/mol, which were extracted from 1D PMF profiles along the minimum free-energy path through the 2D profiles. Average distances (X-Y) are in Å. Standard deviations are listed in parentheses divided by the decimal precision of the average values. The abbreviations “React”, “TS”, “Int”, and “Prod” signify reactant, transition, intermediate, and product states, respectively, and for the distance metrics, “Nu”, “Lea”, “gA” and “H” refer to the O2′ nucleophile, O5′ leaving group, general acid residues G8:O2′ and H2′, respectively.
Figure 1.

Schematic diagram for relative free-energy barriers and the corresponding reaction rate constants.
Phosphoryl transfer is rate limiting and facilitated by electrostatic stabilization by Mg2+
The phosphoryl transfer step is rate controlling, having a free energy barrier of approximately 24.4 kcal/mol. The position of the Mg2+ ion follows the negative charge along the phosphoryl transfer reaction coordinate in order to provide electrostatic stabilization. The change in the Mg2+ position is continuous and monotonic throughout the phosphoryl transfer step (Fig. 2b), and is most pronounced in the initial and final stages when the nucleophile and leaving group have the greatest negative charge. The transition state is late (Fig. 3), having a P-O5′ distance of 2.11 Å. As the P-O5′ bond breaks, the Mg2+ ion forms an inner-sphere coordination, leading to a Mg2+-bound O5′ alkoxide intermediate.
General acid catalysis is concerted with changes in Mg2+ binding mode
The general acid step considered here assumes the 2′OH of G8 acts as a general acid catalyst to transfer a proton to the O5′ leaving group. Examination in Fig. 2c indicates that proton transfer occurs after formation of the Mg2+-coordinated cleaved intermediate, and is concerted with changes in Mg2+ binding mode. Unlike the phosphoryl transfer step, participation of the Mg2+ along the reaction coordinate is most pronounced not at the endpoints of the step, but near the midpoint where the proton transfer occurs.
The free energy barrier for the transition state of the general acid step (Fig. 3) is 13.7 kcal/mol with respect to the activated precursor state. The intermediate is only 6.7 kcal/mol lower in free energy than the activated precursor, and has a 20.4 kcal/mol barrier to breakdown into the product state with a proton fully transferred to the O5′ leaving group.
Relation with experiment
The present work explores a specific mechanistic scenario, departing from the activated precursor state, that assumes the catalytic metal ion is in a position bridging the A9 and scissile phosphates, and the 2′OH of G8 acts as a general acid catalyst. The former metal ion binding mode has not yet been observed experimentally, but has been inferred from biochemical experiments on the minimal15 and extended16 HHR, and predicted by molecular simulations.5,17,18 The latter role of G8 is consistent from structural4 and biochemical data.6–8 There has been seminal work in the study of metal ion interactions for the minimal HHR.19, 20 Time-resolved NMR experiments suggest that there is a dynamic equilibrium between energetically similar conformations in the minimal HHR that are sensitive to Mg2+ binding,21 and it has been suggested that the minimal and extended HHR may utilize a similar dynamic reaction mechanism for catalysis.22 In the present study, we provide computational support for the plausibility of a cleavage mechanism where phosphoryl transfer and general acid catalysis are stepwise, and that the catalytic divalent metal ion plays an active role in the chemical steps of catalysis. It is the hope that this work, together with experimental work that probes the nature of metal ion interactions at the active site, will provide deeper insight into the underpinnings of chemical catalysis in the HHR.
Supplementary Material
Acknowledgments
The authors are grateful for support from the National Institutes of Health (GM62248 to DY) and the Minnesota Supercomputing Institute (MSI). We thank Professor Victoria J. DeRose for useful comments on the manuscript.
Footnotes
Supporting Information Available: Additional computational details. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Strobel SA, Cochrane JC. Curr Opin Chem Biol. 2007;11:636–643. doi: 10.1016/j.cbpa.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scott WG. Curr Opin Struct Biol. 2007;17:280–286. doi: 10.1016/j.sbi.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Leclerc F. Molecules. 2010;15:5389–5407. doi: 10.3390/molecules15085389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martick M, Lee TS, York DM, Scott WG. Chem Biol. 2008;15:332–342. doi: 10.1016/j.chembiol.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee TS, Silva Lopez C, Giambaşu GM, Martick M, Scott WG, York DM. J Am Chem Soc. 2008;130:3053–3064. doi: 10.1021/ja076529e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blount KF, Uhlenbeck OC. Annu Rev Biophys Biomol Struct. 2005;34:415–440. doi: 10.1146/annurev.biophys.34.122004.184428. [DOI] [PubMed] [Google Scholar]
- 7.Nelson JA, Uhlenbeck OC. RNA. 2008;14:605–615. doi: 10.1261/rna.912608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas JM, Perrin DM. J Am Chem Soc. 2009;131:1135–1143. doi: 10.1021/ja807790e. [DOI] [PubMed] [Google Scholar]
- 9.Nam K, Cui Q, Gao J, York DM. J Chem Theory Comput. 2007;3:486–504. doi: 10.1021/ct6002466. [DOI] [PubMed] [Google Scholar]
- 10.Imhof P, Noé F, Fischer S, Smith JC. J Chem Theory Comput. 2006;2:1050–1056. doi: 10.1021/ct600092c. [DOI] [PubMed] [Google Scholar]
- 11.Pérez Alberto, Marchán Iván, Svozil D, Sponer J, C TE, III, Laughton CA, Orozco M. Biophys J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horn HW, Swope WC, Pitera JW, Madura JD, Dick TJ, Hura GL, Head-Gordon T. J Chem Phys. 2004;120:9665–9678. doi: 10.1063/1.1683075. [DOI] [PubMed] [Google Scholar]
- 13.Joung IS, Cheatham TE., III J Phys Chem B. 2008;112:9020–9041. doi: 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott WG. What can the New Hammerhead Ribozyme Structures Teach us About Design? In: Erdmann V, Barciszewski J, editors. RNA Technologies and Their Applications. Springer-Verlag; 2010. [Google Scholar]
- 15.Wang S, Karbstein K, Peracchi A, Beigelman L, Herschlag D. Biochemistry. 1999;38:14363–14378. doi: 10.1021/bi9913202. [DOI] [PubMed] [Google Scholar]
- 16.Osborne EM, Schaak JE, Derose VJ. RNA. 2005;11:187–196. doi: 10.1261/rna.7950605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee TS, Silva-Lopez C, Martick M, Scott WG, York DM. J Chem Theory Comput. 2007;3:325–327. doi: 10.1021/ct6003142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee TS, Giambaşu GM, Sosa CP, Martick M, Scott WG, York DM. J Mol Biol. 2009;388:195–206. doi: 10.1016/j.jmb.2009.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogt M, Lahiri S, Hoogstraten CG, Britt DR, DeRose VJ. J Am Chem Soc. 2006;128:16764–16770. doi: 10.1021/ja057035p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Osborne EM, Ward WL, Ruehle MZ, DeRose VJ. Biochemistry. 2009;48:10654–10664. doi: 10.1021/bi900614v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fürtig B, Richter C, Schell P, Wenter P, Pitsch S, Schwalbe H. RNA Biol. 2008;5:41–48. doi: 10.4161/rna.5.1.5704. [DOI] [PubMed] [Google Scholar]
- 22.Nelson JA, Uhlenbeck OC. RNA. 2008;14:43–54. doi: 10.1261/rna.717908. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.