

Abstract
Phosphorylation of the cardiac ryanodine receptor (RyR2) is thought to be important not only for normal cardiac excitation-contraction coupling but also in exacerbating abnormalities in Ca2+ homeostasis in heart failure. Linking phosphorylation to specific changes in the single-channel function of RyR2 has proved very difficult, yielding much controversy within the field. We therefore investigated the mechanistic changes that take place at the single-channel level after phosphorylating RyR2 and, in particular, the idea that PKA-dependent phosphorylation increases RyR2 sensitivity to cytosolic Ca2+. We show that hyperphosphorylation by exogenous PKA increases open probability (P o) but, crucially, RyR2 becomes uncoupled from the influence of cytosolic Ca2+; lowering [Ca2+] to subactivating levels no longer closes the channels. Phosphatase (PP1) treatment reverses these gating changes, returning the channels to a Ca2+-sensitive mode of gating. We additionally found that cytosolic incubation with Mg2+/ATP in the absence of exogenously added kinase could phosphorylate RyR2 in approximately 50% of channels, thereby indicating that an endogenous kinase incorporates into the bilayer together with RyR2. Channels activated by the endogenous kinase exhibited identical changes in gating behavior to those activated by exogenous PKA, including uncoupling from the influence of cytosolic Ca2+. We show that the endogenous kinase is both Ca2+-dependent and sensitive to inhibitors of PKC. Moreover, the Ca2+-dependent, endogenous kinase–induced changes in RyR2 gating do not appear to be related to phosphorylation of serine-2809. Further work is required to investigate the identity and physiological role of this Ca2+-dependent endogenous kinase that can uncouple RyR2 gating from direct cytosolic Ca2+ regulation.
Keywords: Ca2+-release channel, Ion channel, Excitation-contraction coupling, Ryanodine receptor, Sarcoplasmic reticulum, Cardiac
Introduction
Phosphorylation of the cardiac ryanodine receptor (RyR2) is suggested to play a key role not only in physiological regulation of sarcoplasmic reticulum (SR) Ca2+-release (Marx et al. 2000; Wehrens et al. 2003) but also in the damaging changes to Ca2+ homeostasis that are seen in heart failure (Marx et al. 2000; Belevych et al. 2007). RyR2 can be phosphorylated by both protein kinase A (PKA) and calmodulin-dependent protein kinase II (CaMKII) (Witcher et al. 1991; Marx et al. 2000; Rodriguez et al. 2003), and to date, three phosphorylation sites on RyR2 have been identified: serine-2809, serine-2815 and serine-2030 (Marx et al. 2000). Although there is not absolute agreement about the specificity of PKA and CaMKII for these phosphorylation sites, there is a generally held view that serine-2030 is a PKA site, serine-2815 is a CaMKII site and serine-2809 can be phosphorylated by either PKA or CaMKII (Wehrens et al. 2004; Rodriguez et al. 2003; Marx et al. 2000; Xiao et al. 2005; Huke and Bers 2008). The presence of three phosphorylation sites that can be phosphorylated by different kinases potentially introduces a highly flexible but complex system for RyR2 channel regulation. Indeed, given the large size of RyR2, it is not unreasonable to assume there may be other, as yet unidentified phosphorylation sites.
What, then, are the mechanisms by which phosphorylation of RyR2 alters channel function? Not surprisingly, given the complexity of the situation, a clear grasp of the mechanisms underlying phosphorylation-dependent changes in RyR2 activity is lacking. It has been reported that both PKA-dependent and CaMKII-dependent phosphorylation of RyR2 can give rise to an increase in open probability (P o) (Marx et al. 2000; Carter et al. 2006; Wehrens et al. 2004). On the other hand, we, and others, have shown that dephosphorylating RyR2 can also increase P o, indicating that there must be an inhibitory component to phosphorylation (Carter et al. 2006; Terentyev et al. 2003; Lokuta et al. 1995). The aim of this study was, therefore, to examine the underlying changes in RyR2 single-channel gating that occur following phosphorylation of RyR2. Since it has been suggested (Marx et al. 2000) that PKA-dependent phosphorylation of RyR2 increases the sensitivity of the channel to cytosolic [Ca2+], we specifically addressed this hypothesis.
Our results demonstrate that approximately 50% of native sheep RyR2 reconstituted into bilayers can be activated by incubation with cytosolic Mg2+/ATP, suggesting that they are associated with a kinase endogenous to the sarcoplasmic reticulum (SR). The unknown kinase displays an inhibitor sensitivity profile compatible with PKC enzymes. Kinetic analysis of channel gating reveals (1) that the unknown endogenous kinase is Ca2+-dependent, (2) a distinctive yet common mechanism of channel activation by the unknown endogenous kinase and by exogenous PKA and (3) that both types of phosphorylation effectively uncouple channel gating from the influence of cytosolic [Ca2+], allowing substantial channel openings at nanomolar [Ca2+].
Materials and Methods
Materials
The catalytic subunits of PKA and ATP were obtained from Sigma-Aldrich (Dorset, UK). PKI was obtained from Alta Biosciences (Birmingham, UK). AIPII and chelerythrine chloride were obtained from EMD Biosciences (San Diego, CA). Other chemicals were Analar or the best equivalent grade from BDH (Poole, UK) or Sigma-Aldrich. All solutions were made up in deionized water, and those for use in bilayer experiments were filtered through a Millipore (Bedford, MA) membrane filter (0.45 μm pore).
SR Vesicle Preparation and Planar Phospholipid Bilayer Techniques
Sheep hearts were obtained from a local abattoir, and heavy SR (HSR) membrane vesicles were prepared and fused with planar phosphatidylethanolamine lipid bilayers, as described previously (Sitsapesan et al. 1991). SR vesicles fused in a fixed orientation such that the cis chamber corresponded to the cytosolic space and the trans chamber to the SR lumen. The trans chamber was held at ground and the cis chamber at potentials relative to ground. After fusion, the cis chamber was perfused with 250 mM HEPES, 80 mM Tris and 50 μM free Ca2+ (pH 7.2), unless stated otherwise. The trans chamber was perfused with 250 mM glutamic acid and 10 mM HEPES (pH to 7.2) with Ca(OH)2 (free [Ca2+], approximately 50 mM). Experiments were carried out at room temperature (22 ± 2°C). The free [Ca2+] and pH of the solutions were determined at the relevant temperatures using a Ca2+ electrode (Orion 93-20; Orion, Boston, MA) and a Ross-type pH electrode (Orion 81-55) as previously described (Sitsapesan et al. 1991). In the presence of 10 mM EGTA, the free [Ca2+] was calculated using the maxchelator program (http://maxchelator.stanford.edu).
Single-Channel Data Acquisition and Analysis
Channel recordings were displayed on an oscilloscope and recorded on digital audiotape. Current recordings were filtered at 1 kHz (–3 dB) and digitized at 20 kHz using Pulse (Heka, Lambrecht, Germany). P o was determined over 3 min of continuous recording, unless otherwise stated, using the method of 50% threshold analysis (Colquhoun and Sigworth 1983). Lifetime analysis was carried out only when a single channel was incorporated into the bilayer. Events <1 ms in duration were not fully resolved and were excluded from lifetime analysis. Individual lifetimes were fitted to a probability density function (pdf) by the method of maximum likelihood according to the equation
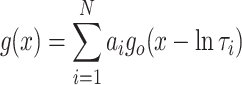 |
where ln τi is the logarithm of the ith time constant and a i is the fraction of the total events represented by that component (Sigworth and Sine 1987).
PKA-Dependent Phosphorylation Experiments
Control-channel P o was determined in the presence of 50 μM cytosolic Ca2+. Ten units of the catalytic subunit of PKA, 5 mM Mg2+ and 1 mM ATP were then added to the cytosolic face of the channel and incubated for 5 min. At the end of the incubation, the cytosolic face of the channel was washed back to control conditions.
Mg2+/ATP Treatment Experiments
Control-channel P o was determined in the presence of 50 μM cytosolic Ca2+. We then added 5 mM Mg2+ and 1 mM ATP to the cytosolic face of the channel and incubated for 5 min. At the end of the incubation, the cytosolic face of the channel was washed back to control conditions. For kinase inhibitor experiments the relevant inhibitor was added to the cytosolic face of the channel, incubated for 3 min prior to the addition of Mg2+ and ATP and maintained throughout the period of exposure to Mg/ATP.
Western Blotting
HSR proteins were size-fractionated by SDS-PAGE on a 6% polyacrylamide gel for resolving RyR2 or a 10% polyacrylamide gel for PKC isoforms (Laemmli 1970). Following separation, proteins were transferred to nitrocellulose membranes and nonspecific binding sites were blocked for 1.5–2 h at room temperature using 5% dried milk and Tris-buffered saline (pH 7.4), 0.1 % Tween 20. Depending on the target protein, the blots were then treated as follows:
To detect the phosphorylation state of RyR2, membranes were probed overnight at 4°C with a primary antibody (1:5,000) specific for RyR dephosphorylated at serine-2809 (RYR2-2809deP) or phosphorylated at serine-2809 (RyR2-PS2809) (Rodriguez et al. 2003; Carter et al. 2006) in 5% dried milk and Tris-buffered saline (pH 7.4), 0.1% Tween 20.
To identify PKC isoforms, membranes were probed with antibodies for various PKC isoforms (Santa Cruz Biotechnology, Santa Cruz, CA) for 90 min at room temperature in 5% dried milk and Tris-buffered saline (pH 7.4), 0.1% Tween 20. A secondary horseradish peroxidase–linked anti-rabbit IgG (Amersham Biosciences, Aylesbury, UK) or horseradish peroxidase–linked anti-mouse IgG (Santa Cruz Biotechnology) was used in combination with an enhanced chemiluminescent detection system (Amersham Biosciences) to visualize the primary antibodies.
Endogenous Kinase–Dependent Phosphorylation of RyR2
Cardiac SR vesicles were incubated in a solution containing 50 mM HEPES, 16 mM Tris, 5 mM Mg2+ and 5 mM NaF (pH 7.2) with various other additions as described in the individual figure legends. At the end of the relevant incubation time, 1 ml of ice-cold buffer containing 250 mM HEPES, 80 mM Tris and 5 mM NaF (pH 7.2) was added to each sample. Samples were centrifuged at 9,720×g for 15 min at 4°C, the supernatant was removed and samples were resuspended in 50 μl of Laemmeli sample buffer and incubated at 45°C for 30 min.
Determination of the Phosphorylation State of RyR at Serine-2809
For PKA treatment, cardiac SR vesicles were incubated in a solution containing 50 mM HEPES, 16 mM Tris and 5 mM NaF (pH 7.2) for 5 min at 37°C in the presence of 1 U of the catalytic subunit of PKA per microgram of protein. RyR2-P2809 staining of control and PKA-treated samples was quantified by densitometry. For PP1 dephosphorylation of RyR2, SR vesicles were incubated with 0.1 U PP1 per microgram of protein for 5 min at 37°C. As previously described (Carter et al. 2006), PKA treatment results in complete phosphorylation at S2809, as demonstrated by the increase in staining with the RyR2-P2809 antibody and the absence of signal from the RyR2-2809deP antibody. PP1 treatment results in complete dephosphorylation at serine-2809, as demonstrated by the increase in staining with RyR2-2809deP and the absence of RyR2-P2809 signal (Carter et al. 2006). Comparing the RyR2-2809deP staining of a control sample with the PP1-treated sample staining gives the percentage of dephosphorylated monomers in the sample and the basal level of phosphorylation, which we find to be 75% of the maximum (Carter et al. 2006). For simplicity, we have expressed all antibody signals as a percentage of the control (basal) levels. In all cases control and treated samples were always quantified from the same Western blot. Densitometry was carried out using Scion Image (Scion, Frederick, MD) and the level of staining did not saturate the measurement by the densitometry software in any blot quantified.
Statistics
All values are means ± SEM. Where appropriate, a Mann-Whitney U-test was used to assess the difference between the mean values. A P value of 0.05 was taken as a significant difference. An F-test was additionally used to determine the variance within groups of data.
Results
We previously reported that PKA-dependent phosphorylation of RyR2 above background levels results in a sustained increase in P o (Carter et al. 2006). In these experiments, Mg2+ and ATP were present throughout and we could not specifically determine whether phosphorylation affected channel regulation by cytosolic Ca2+ (Carter et al. 2006). We therefore investigated how phosphorylation affects the response of the channel to cytosolic Ca2+ by applying Mg2+/ATP only during the kinase incubation time. Figure 1a shows a representative experiment demonstrating the effect of PKA-dependent phosphorylation on the gating of channels activated solely by cytosolic Ca2+. The top trace shows channel activity under control conditions of 50 μM cytosolic Ca2+. The channel was then treated with ten units of PKA catalytic subunit, 1 mM ATP and 5 mM Mg2+ for 5 min before washout to control conditions (lower trace). A large increase in P o following PKA incubation was observed. This effect was extremely robust, occurring in 100% of experiments (n = 9). The importance of this consistency became apparent with subsequent experiments. Control experiments demonstrated that none of the individual components of the incubation solutions was capable of producing a sustained increase in P o. ATP (10 mM) increased P o from 0.090 ± 0.024 to 0.65 ± 0.062 (n = 14), but after washout, P o returned to 0.076 ± 0.025 (n = 12). Incubation with kinase alone without ATP/Mg2+ caused no change in P o (0.141 ± 0.054 before and 0.128 ± 0.84 [n = 5] after kinase incubation). Mg2+ (5 mM) produced only a reversible decrease in P o (from 0.132 ± 0.070 to 0.006 ± 0.005 [n = 4]).
Fig. 1.

Effects of PKA on RyR2 channel activity. a A representative single RyR2 channel gating in the presence of 50 μM cytosolic Ca2+ before (top) and after (bottom) PKA treatment is shown. Arrows and dotted lines indicate closed and open channel levels, respectively. b Western blot analysis showing the phosphorylation of RyR2 at serine-2809 that occurs at 50 μM cytosolic Ca2+ for the indicated conditions. For both gels, cardiac HSR was incubated with 5 mM Mg2+ (lane 1), 5 mM Mg2+ + 1 mM ATP (lane 2) and 5 mM Mg2+ + 1 mM ATP + 1 U of PKA per microgram of protein (lane 3). All lanes were loaded at 30 μg total protein. Western blots were probed with antibodies specific for RyR2 dephosphorylated at serine-2809 (RyR2-2809deP, left gel) or for RyR2 phosphorylated at serine-2809 (RyR2-PS2809, right gel). Size markers are indicated in kilodaltons. The histogram shows the average percentage change in phosphorylation levels with respect to the control (5 mM Mg2+ only) by probing the blot with either RyR2-2809deP or RyR2-PS2809. Data are expressed as mean ± SD (n = 4)
Similar PKA treatment of isolated SR vesicles was performed to investigate changes in phosphorylation at serine-2809 (Fig. 1b). Two identical Western blots were produced and probed with antibodies specific for RyR2, dephosphorylated or phosphorylated at serine-2809. Lane 1 shows the background level of phosphorylation at serine-2809 for HSR vesicles incubated in the presence of Mg2+ alone. Lane 3 demonstrates the increase in the level of phosphorylation at S2809 in response to PKA-dependent phosphorylation. The increase in signal from the phosphorylated serine-2809-specific antibody coupled with loss of the dephosphorylated state–specific antibody staining confirms the increase in phosphorylation state. Important to notice is the apparent change in phosphorylation state at serine-2809 when PKA was omitted from the incubation medium (lane 2). Mg2+/ATP incubation alone appeared to increase phosphorylation at serine-2809, although this effect was much lower than that observed when PKA was included in the incubation medium. In light of this change, we then examined if Mg2+/ATP incubation in the absence of exogenously added kinase could affect RyR2 single-channel function.
Figure 2 shows that transient Mg2+/ATP treatment can significantly activate RyR2 channels incorporated into bilayers beyond the period of exposure to Mg2+/ATP. Cytosolic incubation with Mg2+/ATP (5 min) was followed by washout to control solutions (50 μM cytosolic free Ca2+). Interestingly, some channels appeared not to be affected by this treatment, whereas other channels were obviously activated. We therefore grouped the channels according to whether Mg2+/ATP treatment had no apparent lasting effect following washout of Mg2+/ATP or whether they were irreversibly activated by Mg2+/ATP treatment. Figure 2a shows an example of an experiment where Mg2+/ATP treatment did not increase P o. The mean data are shown in Fig. 2b. Likewise, Fig. 2c shows an example of an experiment where Mg2+/ATP treatment did increase P o (Fig. 2d shows the mean data). Figure 2e combines all the data, and the individual experiments are imposed on the histogram, demonstrating the heterogeneous response to Mg2+/ATP treatment. This demonstrates an important difference between these Mg2+/ATP only treated channels and the PKA-treated channels: Mg2+/ATP treatment activates only a subpopulation of channels, whereas PKA treatment activates all channels.
Fig. 2.

Effects of Mg2+/ATP treatment on RyR2 channel gating. a Representative current fluctuations through a single RyR2 channel that demonstrated no increase in P o after Mg2+/ATP treatment. b Mean P o from experiments where P o was equivalent or reduced after Mg2+/ATP treatment. Mean ± SEM (n = 7). c Representative current fluctuations through a single RyR2 channel where P o increased after Mg2+/ATP treatment. d Mean channel P o from experiments where P o increased after Mg2+/ATP treatment. Mean ± SEM (n = 10). e Histogram showing the mean channel P o of all experiments (regardless of whether P o increased or decreased) in the presence of 50 μM cytosolic Ca2+, before and after Mg2+/ATP treatment. Open circles show channel P o for the individual experiments. Mean ± SEM (n = 17, **P < 0.01). f Histogram illustrating the effect of PP1 treatment on RyR2 channels that displayed increased P o after Mg2+/ATP treatment. Control P o was determined in the presence of 50 μM cytosolic Ca2+ alone. The cytosolic face of the channel was then treated for 5 min in the presence of 1 mM ATP and 5 mM Mg2+ and washed back to control conditions. The cytosolic face of each channel was then incubated in the presence of a maximum of 12.5 units of PP1, for 10 min, before being washed out to control conditions. Mean ± SD (n = 3)
The increase in P o observed following Mg2+/ATP activation of the channels (this occurred in 10 out of 17 channels) could not be reversed by washout of Mg2+/ATP, thus demonstrating that activation was not due to direct effects of Mg2+ or ATP on gating. P o was reversed by the phosphatase PP1 (Fig. 2f), thus demonstrating that the changes in gating were most likely caused by channel phosphorylation by an endogenous kinase incorporating into the bilayer along with RyR2. The heterogeneity observed with the Mg2+/ATP-treated channels possibly is a result of the kinase not being associated with all RyR2 channels that incorporate into the bilayer.
The above results indicate that phosphorylation of RyR2 occurs by two distinct mechanisms: (1) activation of an endogenous kinase by treatment with Mg2+/ATP, causing activation of only a subpopulation of channels, and (2) activation by exogenous PKA incubation, an effect observed in all treated channels. The change in channel gating, however, after Mg2+/ATP treatment (Fig. 2c) closely resembles that observed after PKA treatment (Fig. 1a). Figure 3 illustrates the open and closed lifetime distributions of the Mg2+/ATP-treated and PKA-treated channels. The figure demonstrates that control-channel events were best fit by two open and three closed states, as expected for a channel activated by 50 μM cytosolic Ca2+ as the sole channel activator (and Ca2+ as permeant ion) (Ashley and Williams 1990; Stewart et al. 2008). Channel activity was characterized by short openings and long closings. After Mg2+/ATP treatment (Fig. 3a) or PKA treatment (Fig. 3b), the closed lifetimes remained best fit by three exponentials but the duration of each closed time constant was reduced substantially, demonstrating that an increase in the frequency of channel opening had occurred. Much more profound, however, was the effect on open lifetime durations. Before treatment, most of the openings (>80%) occurred to the shortest time constant and were close to 1 ms in duration. After Mg2+/ATP treatment (Fig. 3a) or PKA treatment (Fig. 3b), two extra, long open time constants (four in total) could be detected and the distributions of the events were shifted so that a much lower proportion of events occurred to the shortest time constant. These gating changes were highly reproducible (n = 8) for activation by PKA or Mg2+/ATP (see Table 1 for detailed lifetime data).
Fig. 3.

Effect of RyR2 phosphorylation on channel gating. a Open and closed lifetime distributions and pdfs for a typical single RyR2 that displayed increased P o after Mg2+/ATP treatment. b Open and closed lifetime distributions and pdfs for a typical single RyR2 before and after PKA treatment
Table 1.
Comparison of the changes in RyR2 channel gating in response to PKA-dependent phosphorylation or ATP/Mg2+ treatment
| τ1 (ms) | Area (%) | τ2 (ms) | Area (%) | τ3 (ms) | Area (%) | τ4 (ms) | Area (%) | |
|---|---|---|---|---|---|---|---|---|
| Open lifetime parameters | ||||||||
| Control | 0.9 ± 0.1 | 88.3 ± 3.8 | 5.6 ± 0.6 | 12.0 ± 3.8 | ||||
| PKA-treated | 1.3 ± 0.2 | 41.0 ± 10.0 | 8.0 ± 2.6 | 32.0 ± 4.4 | 44.2 ± 17.7 | 12.8 ± 2.1 | 194.6 ± 44.8 | 14.0 ± 12.7 |
| Closed lifetime parameters | ||||||||
| Control | 2.6 ± 0.6 | 38.6 ± 8.3 | 20.7 ± 5.9 | 43.4 ± 5.0 | 94.4 ± 34.9 | 18.0 ± 4.6 | ||
| PKA-treated | 1.0 ± 0.2 | 65.8 ± 6.3 | 4.5 ± 1.6 | 29.5 ± 4.1 | 22.0 ± 11.1 | 4.8 ± 2.4 | ||
| τ1 (ms) | Area (%) | τ2 (ms) | Area (%) | τ3 (ms) | Area (%) | τ4 (ms) | Area (%) | |
|---|---|---|---|---|---|---|---|---|
| Open lifetime parameters | ||||||||
| Control | 0.7 ± 0.04 | 94.3 ± 1.6 | 4.0 ± 0.5 | 5.8 ± 1.5 | ||||
| ATP/Mg2+-treated | 0.9 ± 0.1 | 59.0 ± 9.5 | 3.6 ± 0.6 | 29.8 ± 5.6 | 15.9 ± 2.4 | 9.8 ± 3.4 | 68.4 ± 16.0 | 1.7 ± 0.6 |
| Closed lifetime parameters | ||||||||
| Control | 3.1 ± 0.6 | 32.5 ± 4.6 | 23.1 ± 8.6 | 44.8 ± 5.9 | 184.6 ± 119.1 | 22.8 ± 2.4 | ||
| ATP/Mg2+-treated | 1.1 ± 0.2 | 67.0 ± 6.2 | 5.1 ± 1.4 | 27.9 ± 10.3 | 27.9 ± 10.3 | 4.5 ± 1.0 | ||
Upper table shows open and closed lifetime parameters in the presence of 50 μM cytosolic Ca2+, before and after 5-min cytosolic incubation with 1 U PKA, 5 mM Mg2+ and 1 mM ATP. Mean ± SEM (n = 4). Lower table shows open and closed lifetime parameters in the presence of 50 μM cytosolic Ca2+, before and after 5-min cytosolic with 5 mM Mg2+ and 1 mM ATP. Mean ± SEM (n = 4)
We then examined the interplay between phosphorylation status and regulatory cytosolic [Ca2+]. Figures 4 and 5 provide typical examples of RyR2 gating for channels activated either by the endogenous Mg2+/ATP-activated kinase or by PKA-dependent phosphorylation. In Fig. 4 the top traces show channel activity under control conditions in the presence of 50 μM cytosolic Ca2+ (i). Increased activity and altered channel gating are observed after the 5-min Mg2+/ATP treatment and subsequent washout back to 50 μM cytosolic Ca2+ (ii). In (iii), subsequent addition of 10 mM EGTA to the cytosolic face of the channel reduces the free Ca2+ concentration to <1 nM. This is a subactivating cytosolic [Ca2+], and the expected P o would therefore be zero (Stewart et al. 2008). There is, however, very little change in steady-state P o but an obvious change in the type of gating, now characterized by long openings and long closings. The channel is now gating in a Ca2+-independent manner since there is no cytosolic Ca2+ available. Treatment with the phosphatase, PP1 (iv), dephosphorylates the channel (Carter et al. 2006) and brings P o to zero, thus reinstating the normal Ca2+ sensitivity of the channel. After 3 min, the cytosolic solutions were perfused back to the original control conditions of 50 μM cytosolic Ca2+ (v) and brief channel openings return with P o slightly higher than the initial conditions (i), in line with our previous report (Carter et al. 2006).
Fig. 4.

Effect of Mg2+/ATP treatment on the Ca2+ sensitivity of RyR2. a Channel activity (i) in the presence of 50 μM cytosolic Ca2+ alone, (ii) after Mg2+/ATP treatment and washout to control conditions, (iii) after 10 mM cytosolic EGTA addition, (iv) after subsequent addition of 12.5 units cytosolic PP1 and (v) after washout of EGTA and PP1 back to control conditions. b Open and closed lifetime distributions and pdfs of the Mg2+/ATP-treated RyR2 channel shown in a in the absence and presence of 10 mM cytosolic EGTA
Fig. 5.

Effect of PKA-dependent phosphorylation on RyR2 Ca2+ sensitivity. Channel activity (i) in the presence of 50 μM cytosolic Ca2+ alone, (ii) after addition of 10 mM EGTA to the cytosolic chamber, (iii) after washout of EGTA and PKA treatment and (iv) after 10 mM EGTA was again added to the cytosolic chamber
The addition of EGTA provides evidence of the mechanisms by which the channel gates following phosphorylation by Mg2+/ATP treatment. Lifetime histograms of channel openings and closings before and after EGTA addition are shown in Fig 4b. The open and closed lifetime distributions of the Mg2+/ATP-treated channel are best described by at least four open and three closed exponentials (as expected following phosphorylation, see Fig. 3). After the addition of 10 mM EGTA, the events occurring to the shortest time constant are lost and the durations of the three remaining time constants are greatly increased. The closed lifetime durations are also markedly increased, shifting from three closed states to four. The data suggest that following RyR2 phosphorylation above background levels, only one, minor component of the channel openings (openings occurring to the shortest time constant) is dependent on cytosolic Ca2+. The majority of channel openings occur in a Ca2+-independent manner; essentially, the channel has become uncoupled from ligand regulation. Dephosphorylation of the channel by PP1 (as demonstrated in Fig. 4a iv) switches the channel gating back into a Ca2+-regulated mode, closing the channel down until activating [Ca2+] levels are restored (Fig. 4a v).
Figure 5 demonstrates that exactly the same mechanisms are responsible for the gating changes in the PKA-phosphorylated channels. RyR2 activated by 50 μM cytosolic Ca2+ as the sole channel activator exhibits a wide range of P o values (Sitsapesan and Williams 1994a; Copello et al. 1997). For this figure, we have chosen a channel that gates in control conditions with a relatively high P o in order to demonstrate the robustness of Ca2+ sensitivity. Even when the channel gates with high P o at background levels of phosphorylation, lowering the cytosolic [Ca2+] to subactivating levels (<1 nM) by the addition of 10 mM EGTA (ii) still completely shuts the channel. Subsequent PKA treatment followed by perfusion to 50 μM Ca2+ (iii) increased P o to high levels, but then lowering the [Ca2+] to subactivating levels again (iv) revealed the characteristic long open and closed events of a channel gating in a Ca2+-independent manner. It is clear from Figs. 4 and 5 that phosphorylation by PKA treatment or Mg2+/ATP treatment causes a huge increase in P o by an identical kinetic mechanism. There is still a small Ca2+-sensitive component to the gating of the hyperphosphorylated channel, but the use of EGTA reveals that the overriding mechanism increasing P o can be attributed to channel openings that occur independently of the cytosolic [Ca2+].
Since PKA is reported to be closely associated with RyR2 (Marx et al. 2000) and since Mg2+/ATP-treated and PKA-treated channels exhibit virtually identical gating, we investigated whether an endogenous PKA was responsible for the Mg2+/ATP-dependent effect by using the specific PKA inhibitor PKI. We have previously shown that a protein kinase similar to PKA copurifies with cardiac SR (canine) but that 1 μM PKI completely inhibits this enzyme with respect to phosphorylation of phospholamban (Jackson and Colyer 1996). Figure 6a shows that Mg2+/ATP continues to activate some channels even in the presence of inhibitory concentrations of PKI (10 μM). In other words, as in Fig. 2, there is still a subpopulation of channels that can be activated by Mg2+/ATP treatment in the presence of PKI. The failure of PKI to prevent the Mg2+/ATP-induced increase in P o suggests that Mg2+/ATP treatment activates a kinase other than PKA. Likewise, in the presence of PKI (10 μM), although PKA no longer activates 100% of channels (see inset to Fig. 6), a proportion of channels are still activated, presumably because Mg2+/ATP in the incubation medium can activate some channels via the endogenous kinase.
Fig. 6.

Effect of Mg2+/ATP treatment in the presence of various kinase inhibitors. a All data are shown under the same conditions of 50 μM cytosolic Ca2+ alone before (pooled data) and after Mg2+/ATP treatment. Data from individual experiments are shown as open circles. Mean ± SEM (n ≥ 4). Inset Histogram showing mean channel P o in the presence of 50 μM cytosolic Ca2+, before and after PKA treatment in the presence of 10 μM PKI. Open circles show channel P o for the individual experiments. Mean ± SEM (n = 4). b Identification of PKC isoforms in the HSR preparation. Cardiac HSR vesicle samples were probed with antibodies for different isoforms of PKC. Mouse brain lysate was included as a positive control
Figure 6a additionally demonstrates that the CaMKII inhibitor AIPII (50 nM) did not inhibit the effects of the endogenous kinase on RyR2 P o, suggesting that CaMKII is not involved. At this concentration (50 nM), AIPII specifically blocks the action of CaMKII (Ishida et al. 1998). High concentrations of AIPII (1 μM) also inhibits PKC (Ishida et al. 1998) and, as the histogram shows, abolishes the effect of Mg2+/ATP treatment. These data suggest that PKC may be the kinase responsible for the Mg2+/ATP-induced phosphorylation effects. To explore this further, we examined the effect of specific PKC inhibition by chelerythrine (Herbert et al. 1990). In the presence of chelerythrine (see histogram), Mg2+/ATP treatment could not activate the channels. Together, the AIPII (1 μM) and chelerythrine data strongly suggest that the Mg2+/ATP-induced gating changes, seen in a subpopulation of RyR2, could be caused by endogenous PKC. Control experiments show that none of the inhibitors, alone, has any effect on RyR2 P o (P o was 0.12 ± 0.039 before and 0.203 ± 0.089 [n = 4] after 1 μM chelerythrine treatment, P o was 0.056 ± 0.021 before and 0.072 ± 0.023 [n = 4] after 10 μM PKI treatment, P o was 0.038 ± 0.033 before and 0.048 ± 0.038 [n = 4] after 1 μM AIPII treatment).
If PKC is the endogenous kinase responsible for the Mg2+/ATP-induced phosphorylation effects, there should be evidence that PKC is present in our cardiac HSR membrane fraction. Figure 6b demonstrates that there are significant levels of the β and ε PKC isoforms but also some evidence for the α and ζ isoforms. Although not proof of the mechanism, these results add support to our suggestion that Mg2+/ATP could be activating a PKC closely associated with RyR2 channels incorporated into bilayers.
To further characterize the activity of the endogenous kinase, we tested the Ca2+ dependence of this enzyme. Figure 7a shows that lowering the cytosolic [Ca2+] from 10 μM (P o = 0.05 ± 0.03, n = 4) to a subactivating level (<1 nM) by addition of EGTA completely closes RyR2 (P o = 0, n = 4, as expected [Stewart et al. 2008]). Under these conditions, incubation with 5 mM Mg2+ and 1 mM ATP (Mg2+/ATP) for 6 min has no effect on RyR2 gating and the channel remains closed. In contrast to the effects of Mg2+/ATP in the presence of 50 μM cytosolic Ca2+ (see Fig. 4a), which lead to the irreversible increases in RyR2 P o, washout of Mg2+/ATP back to control conditions (50 μM Ca2+) shows that no irreversible changes in RyR2 gating have taken place (P o = 0.004 ± 0.001, n = 4). Readdition of EGTA completely closes the channel again (P o = 0, n = 4), suggesting that no long-term modification, such as channel phosphorylation, could have occurred. These data provide strong evidence that the endogenous kinase is Ca2+-dependent.
Fig. 7.

Effects of Mg2+/ATP treatment at subactivating cytosolic [Ca2+]. a A representative experiment showing a single RyR2 channel gating in the presence of 50 μM cytosolic free Ca2+. Addition of 1 mM cytosolic EGTA to lower cytosolic [Ca2+] to <1 nM completely closes the channel. Subsequent addition of 5 mM Mg2+ and 1 mM ATP has no effect on the RyR2 channel. Cytosolic washout back to control solutions (50 μM free Ca2+) causes the channel to reopen, showing the reversible nature of the Mg2+/ATP treatment. Readdition of 1 mM cytosolic EGTA is able to close the channel again, showing no long-term modification of the channel by Mg2+/ATP. Arrows and dotted lines indicate closed and open channel levels, respectively. b Western blot analysis showing the phosphorylation of RyR2 at serine-2809 that occurs at nanomolar cytosolic [Ca2+] (<1 nM) for the indicated conditions. EGTA (10 mM) was present throughout all incubations. For both gels, cardiac HSR was incubated with 5 mM Mg2+ (lane 1), 5 mM Mg2+ + 1 mM ATP (lane 2) and 5 mM Mg2+ + 1 mM ATP + 1 U of PKA per microgram of protein (lane 3). All lanes were loaded at 30 μg total protein. Western blots were probed with antibodies specific for RyR2 dephosphorylated at serine-2809 (RyR2-2809deP, left gel) or for RyR2 phosphorylated at serine-2809 (RyR2-PS2809, right gel). Size markers are indicated in kilodaltons. The histogram shows the average percentage change in phosphorylation levels with respect to the control (5 mM Mg2+ only) by probing the blot with either RyR2-2809deP or RyR2-PS2809. Data are expressed as mean ± SD (n = 3)
In Fig. 7b, conditions comparable to those in Fig. 7a were used (<1 nM Ca2+), but it can be seen that Mg2+/ATP treatment (Fig. 7b, lane 2 both gels) and PKA treatment (Fig. 7b, lane 3 both gels) produce approximately the same degree of phosphorylation as in the presence of 50 μM free Ca2+ (compare with Fig. 1b). Thus, phosphorylation of serine-2809 by endogenous kinases or by exogenously added PKA is not a Ca2+-dependent process. We therefore conclude that the Ca2+-dependent increase in P o caused by the endogenous kinase is not due to phosphorylation at serine-2809.
Discussion
Our study demonstrates that RyR2 hyperphosphorylation, both by exogenous PKA and by Mg2+/ATP activation of an endogenous kinase, produces a dominant change in RyR2 gating that overrides the usual influence of ligand regulation and maintains P o at extremely high levels. Lifetime analysis highlights the unique, distinctive gating kinetics of a highly phosphorylated channel (Fig. 3, Table 1). The channel becomes so dominated by the influence of phosphorylation that the regulatory effects of cytosolic Ca2+ become trivial. When activating levels of cytosolic Ca2+ are removed, the highly phosphorylated channel still gates with high P o; it has effectively become uncoupled from the influence of cytosolic Ca2+.
Mechanism of Phosphorylation-Induced Gating Changes
It has previously been assumed that phosphorylation-induced increases in RyR2 P o result from an increase in cytosolic Ca2+ sensitivity (Marx et al. 2000), but our results point to a more fundamental change in the mechanism of channel activation. Cytosolic Ca2+ activates RyR2 in a particularly characteristic manner, namely by increasing the frequency, not the duration, of channel openings (Ashley and Williams 1990; Sitsapesan and Williams 1994a; Carter et al. 2006; Stewart et al. 2008). As a result, P o values are rarely raised above 0.5 with cytosolic Ca2+ as sole activator. The large increases in open lifetime duration observed here suggest that the phosphorylation-dependent changes are not primarily due to increased Ca2+ sensitivity (although the slight increase in opening frequency may represent a minor Ca2+-dependent component). The dominant RyR2-gating component is Ca2+-independent and the contribution to overall P o made by Ca2+-dependent gating is trivial in comparison. This conclusion is supported by our observation that hyperphosphorylated channels are active in the presence or absence of cytosolic Ca2+ and the level of activity is similar in both situations. As lifetime analysis demonstrates (Fig. 4b), removal of activating cytosolic Ca2+ almost eliminates brief openings; but since these events are so short, they do not markedly reduce P o.
This Ca2+-independent gating behavior is very different from that displayed by RyR2 phosphorylated to lower levels (basal/background phosphorylation states and fully dephosphorylated states), where cytosolic Ca2+ is an essential ligand required for channel opening. We have previously demonstrated that the background level of phosphorylation at serine-2809 in our SR vesicles is 75% of the maximum (Carter et al. 2006). In the absence of cytosolic Ca2+, channels phosphorylated to background or lower levels remain shut (P o = 0; Figs. 4, 5) (Sitsapesan and Williams 1989, 1994b; Stewart et al. 2008). Ca2+-independent activity is seen only when channels are hyperphosphorylated above this level and is reversed by PP1-catalyzed dephosphorylation. Phosphorylating RyR2 above background levels therefore switches the channel from a ligand-dependent to a ligand-independent mode of gating.
Nonlinearity of the RyR2 Activity–Phosphorylation Relationship
Although RyR2 P o is very variable (Saftenku et al. 2001; Copello et al. 1997; Laver et al. 1995), we have previously demonstrated (Carter et al. 2006) that channels phosphorylated at background levels gate with low P o when activated solely by cytosolic Ca2+ and that complete dephosphorylation of RyR2 with PP1 slightly, but significantly, increases P o in a manner consistent with increased cytosolic Ca2+ sensitivity (increased frequency, but not duration, of channel opening [Ashley and Williams 1990; Sitsapesan and Williams 1994a; Carter et al. 2006; Stewart et al. 2008]). This observation is confirmed in the present study (e.g., compare channel activities in Fig. 4, traces i and v, and see Fig. 2f). The PP1 experiments demonstrate that phosphorylation of RyR2 is complex and that P o does not increase linearly with increasing levels of phosphorylation. Increasing phosphorylation above background levels causes huge increases in P o characterized by long open times and Ca2+-independent gating. Reducing phosphorylation below background levels by dephosphorylating with PP1 also increases P o but by increasing the frequency of channel opening, thus highlighting the fact that certain phosphorylation events are inhibitory. Figure 8 shows a simple model to explain modulation of RyR2 gating by phosphorylation. At basal phosphorylation levels, RyR2 gates in a Ca2+-sensitive manner, and hence, P o can be regulated by cytosolic Ca2+ and other ligands. Therefore, lowering the free cytosolic [Ca2+] to <1 nM reduces P o to zero. An example of this type of behavior is seen in Fig. 7a (first and second traces). When RyR2 is fully dephosphorylated, RyR2 P o is higher than is observed at basal levels of phosphorylation but remains sensitive to cytosolic Ca2+; lowering or raising the free cytosolic Ca2+ reversibly alters P o (as in Fig. 4, traces iv and v). However, when the channel is hyperphosphorylated, by either PKA or the proposed endogenous PKC, exceptionally long open states lead to very high P o. In this conformation, RyR2 is uncoupled from the usual regulatory effects of cytosolic Ca2+ and cannot be closed by lowering cytosolic [Ca2+] to below diastolic levels (<1 nM Ca2+).
Fig. 8.

Model summarizing the relationship between the phosphorylation level of RyR2 and channel gating behavior. Large vertical arrow on the left indicates increasing P o, and large horizontal arrow indicates increasing RyR2 phosphorylation levels. At the basal levels of phosphorylation, channel P o is maintained at a low level and the channel gates in a Ca2+-sensitive manner, fully responsive to the levels of other channel regulators. Lowering the free [Ca2+] to subactivating levels (<1 nM) reduces P o to zero. The fully dephosphorylated channel (dephosphorylated state) gates with a higher P o than the channel phosphorylated at basal levels but remains sensitive to cytosolic Ca2+ (again, lowering the free [Ca2+] to subactivating levels [<1 nM] reduces P o to zero). When the channel is maximally phosphorylated (hyperphosphorylated state), exceptionally long open states result, leading to high P o values. In this conformation, RyR2 is uncoupled from the usual regulatory effects of cytosolic Ca2+ and cannot be closed by lowering cytosolic [Ca2+] below activating levels. P and de-P indicate phosphorylation and dephosphorylation, respectively
A Ca2+-Dependent Endogenous Kinase Triggers Ca2+-Independent Channel Opening
A number of kinases are known to phosphorylate RyR2 including PKA, CaMKII and PKC (Wehrens et al. 2004; Rodriguez et al. 2003; Marx et al. 2000; Xiao et al. 2005; Takasago et al. 1991). In the present study we demonstrate that the endogenous kinase is totally Ca2+-dependent since Mg2+/ATP treatment cannot activate RyR2 at subactivating cytosolic [Ca2+]. Yet, once phosphorylated by the endogenous kinase, RyR2 gating becomes independent of the cytosolic [Ca2+] and is characterized by high P o levels and long open states. Using a range of kinase inhibitors, we identified PKC as a likely candidate for the endogenous kinase as the alteration in RyR2 function was blocked by two distinct PKC inhibitors (chelerythrine and AIPII [1 μM]). Although novel, this is not an outlandish hypothesis. Figure 6b demonstrates that several isoforms of PKC (PKCα, PKCβ1, PKCε and PKCζ) are present in our SR vesicles and two of these isoforms are known to be Ca2+-sensitive (PKCα and PKCβ1) (Newton 2003). It is interesting to note that Bowling et al. (1999) show that there is an increased level of expression of the Ca2+-sensitive PKCβ1 in failing human hearts. Mayrleitner et al. (1995), measuring incorporation of [γ-32P]phosphate, demonstrated that RyR1 can be phosphorylated by PKC. Phosphorylation of terminal cisternae (TC) vesicles by PKC also resulted in a fall in the Ca2+-loading rate of the TC vesicles. This was attributed to an increase in the rate of Ca2+ leak from the vesicles through RyR1, suggesting an increase in P o after PKC phosphorylation. Although these effects were observed in RyR1, a second group (Takasago et al. 1991) reported phosphorylation of RyR2 by PKC and a related increase in [3H]ryanodine binding, again suggesting an increase in the P o of PKC-treated channels. Futhermore, in failing human hearts, PKC-β1 and -β2 expression and contribution to total PKC activity are significantly increased (Bowling et al. 1999), thus highlighting a possible role for Ca2+-sensitive PKC isoforms in the changes in SR Ca2+ release that occur in heart failure.
Physiological and Pathophysiological Implications
Marks and colleagues (2000) hypothesized that RyR2 hyperphosphorylation led to increases in P o that shaped the progression of heart failure, producing changes to SR Ca2+ release that favored propagation of delayed after depolarizations (DADs) and fatal cardiac arrhythmias. The present study provides further dramatic evidence of RyR2 regulation (or dysregulation) as a consequence of enhanced phosphorylation. Heightened RyR2 phosphorylation, by either PKA or an endogenous PKC associated with cardiac SR, largely eliminates the requirement for cytosolic Ca2+ as a trigger for channel opening (Ca2+-induced Ca2+-release). The endogenous kinase, of course, is dependent on cytosolic Ca2+ levels; but once the kinase becomes activated during systole and phosphorylates RyR2, without an efficient dephosphorylation system, RyR2 will remain activated during diastole because its gating has effectively become uncoupled from the influence of cytosolic Ca2+. This new status of RyR2 activity is incompatible with our current understanding of excitation–contraction coupling in the heart and would lead to RyR2 opening at high frequency at any point in the contractile cycle of a myocyte under any circumstances where this level of phosphorylation of RyR2 arises. This would be expected to be a destabilizing event leading to an increased likelihood of generating arrhythmic DADs. Further research will be needed to understand how the fundamental behavior of RyR2 channels described in this study is accommodated in cardiac myocytes. It will be important to establish whether and how this behavior is masked in the normal course of contraction in healthy cells, perhaps by the aggressive activity of phosphatases, such gating changes play a role in arrhythmia generation and cardiovascular disease.
Acknowledgement
This work was supported by the British Heart Foundation. J. C. benefits from the commercial exploitation of antibodies used in this study. No promotion of this activity is made in this article.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Ashley RH, Williams AJ. Divalent cation activation and inhibition of single calcium release channels from sheep cardiac sarcoplasmic reticulum. J Gen Physiol. 1990;95:981–1005. doi: 10.1085/jgp.95.5.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Gyorke S. Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J. 2007;93:4083–4092. doi: 10.1529/biophysj.107.114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling N, Walsh RA, Song GJ, Estridge T, Sandusky GE, Fouts RL, Mintze K, Pickard T, Roden R, Bristow MR, Sabbah HN, Mizrahi JL, Gromo G, King GL, Vlahos CJ. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- Carter S, Colyer J, Sitsapesan R. Maximum phosphorylation of the cardiac ryanodine receptor at serine-2809 by protein kinase A produces unique modifications to channel gating and conductance not observed at lower levels of phosphorylation. Circ Res. 2006;98:1506–1513. doi: 10.1161/01.RES.0000227506.43292.df. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Sigworth FJ. Fitting and statistical analysis of single-channel recording. In: Sakmann B, Neher E, editors. Single-channel recording. New York: Plenum; 1983. pp. 191–263. [Google Scholar]
- Copello JA, Barg S, Onoue H, Fleischer S. Heterogeneity of Ca2+ gating of skeletal muscle and cardiac ryanodine receptors. Biophys J. 1997;73:141–156. doi: 10.1016/S0006-3495(97)78055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1990;172:993–999. doi: 10.1016/0006-291X(90)91544-3. [DOI] [PubMed] [Google Scholar]
- Huke S, Bers DM. Ryanodine receptor phosphorylation at serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem Biophys Res Commun. 2008;376:80–85. doi: 10.1016/j.bbrc.2008.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida A, Shigeri Y, Tatsu Y, Uegaki K, Kameshita I, Okuno S, Kitani T, Yumoto N, Fujisawa H. Critical amino acid residues of AIP, a highly specific inhibitory peptide of calmodulin-dependent protein kinase II. FEBS Lett. 1998;427:115–118. doi: 10.1016/S0014-5793(98)00405-0. [DOI] [PubMed] [Google Scholar]
- Jackson WA, Colyer J. Translation of Ser16 and Thr17 phosphorylation of phospholamban into Ca2+-pump stimulation. Biochem J. 1996;316(Pt 1):201–207. doi: 10.1042/bj3160201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Laver DR, Roden LD, Ahern GP, Eager KR, Junankar PR, Dulhunty AF. Cytoplasmic Ca2+ inhibits the ryanodine receptor from cardiac muscle. J Membr Biol. 1995;147:7–22. doi: 10.1007/BF00235394. [DOI] [PubMed] [Google Scholar]
- Lokuta AJ, Rogers TB, Lederer WJ, Valdivia HH. Modulation of cardiac ryanodine receptors of swine and rabbit by a phosphorylation–dephosphorylation mechanism. J Physiol. 1995;487:609–622. doi: 10.1113/jphysiol.1995.sp020904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/S0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Mayrleitner M, Chandler R, Schindler H, Fleischer S. Phosphorylation with protein kinases modulates calcium loading of terminal cisternae of sarcoplasmic reticulum from skeletal muscle. Cell Calcium. 1995;18:197–206. doi: 10.1016/0143-4160(95)90064-0. [DOI] [PubMed] [Google Scholar]
- Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez P, Bhogal MS, Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem. 2003;278:38593–38600. doi: 10.1074/jbc.C301180200. [DOI] [PubMed] [Google Scholar]
- Saftenku E, Williams AJ, Sitsapesan R. Markovian models of low and high activity levels of cardiac ryanodine receptors. Biophys J. 2001;80:2727–2741. doi: 10.1016/S0006-3495(01)76241-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth FJ, Sine SM. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsapesan R, Williams AJ. Caffeine activation of single calcium-release channels from sheep cardiac sarcoplasmic reticulum [abstract] J Physiol. 1989;418:26P. doi: 10.1113/jphysiol.1990.sp018031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsapesan R, Williams AJ. Gating of the native and purified cardiac SR Ca2+-release channel with monovalent cations as permeant species. Biophys J. 1994;67:1484–1494. doi: 10.1016/S0006-3495(94)80622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsapesan R, Williams AJ. Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca2+-release channel by luminal Ca2+ J Membr Biol. 1994;137:215–226. doi: 10.1007/BF00232590. [DOI] [PubMed] [Google Scholar]
- Sitsapesan R, Montgomery RAP, MacLeod KT, Williams AJ. Sheep cardiac sarcoplasmic reticulum calcium release channels: modification of conductance and gating by temperature. J Physiol. 1991;434:469–488. doi: 10.1113/jphysiol.1991.sp018481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart R, Song L, Carter SM, Sigalas C, Zaccai NR, Kanamarlapudi V, Bhat MB, Takeshima H, Sitsapesan R. Single-channel characterization of the rabbit recombinant RyR2 reveals a novel inactivation property of physiological concentrations of ATP. J Membr Biol. 2008;222:65–77. doi: 10.1007/s00232-008-9102-z. [DOI] [PubMed] [Google Scholar]
- Takasago T, Imagawa T, Furukawa K-I, Ogurusu T, Shigekawa M. Regulation of the cardiac ryanodine receptor by protein kinase–dependent phosphorylation. J Biochem. 1991;109:163–170. doi: 10.1093/oxfordjournals.jbchem.a123339. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Gyorke S. Protein phosphatases decrease sarcoplasmic reticulum calcium content by stimulating calcium release in cardiac myocytes. J Physiol. 2003;552:109–118. doi: 10.1113/jphysiol.2003.046367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D’Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/S0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- Witcher DR, Kovacs RJ, Schulman H, Cefali DC, Jones LR. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem. 1991;266:11144–11152. [PubMed] [Google Scholar]
- Xiao B, Jiang MT, Zhao M, Yang D, Sutherland C, Lai FA, Walsh MP, Warltier DC, Cheng H, Chen SR. Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ Res. 2005;96:847–855. doi: 10.1161/01.RES.0000163276.26083.e8. [DOI] [PubMed] [Google Scholar]