

Abstract
Gram-negative bacteria, such as Escherichia coli, frequently utilize tripartite efflux complexes in the resistance-nodulation-cell division (RND) family to expel diverse toxic compounds from the cell. These efflux systems span the entire cell envelope to mediate the phenomenon of bacterial multidrug resistance. The three parts of the efflux complexes are: (1) a membrane fusion protein (MFP) connecting (2) a substrate-binding inner membrane transporter to (3) an outer membrane-anchored channel in the periplasmic space. One such efflux system CusCBA is responsible for extruding biocidal Cu(I) and Ag(I) ions. We recently determined the crystal structures of both the inner membrane transporter CusA and MFP CusB of the CusCBA tripartite efflux system from E. coli. These are the first structures of the heavy-metal efflux (HME) subfamily of the RND efflux pumps. Here, we summarize the structural information of these two efflux proteins and present the accumulated evidence that this efflux system utilizes methionine residues to bind and export Cu(I)/Ag(I). Genetic and structural analyses suggest that the CusA pump is capable of picking up the metal ions from both the periplasm and cytoplasm. We propose a stepwise shuttle mechanism for this pump to extrude metal ions from the cell.
Keywords: CusCBA efflux complex, heavy-metal efflux, resistance-nodulation-cell division, multidrug resistance, membrane fusion protein, outer membrane channel
Introduction
Silver is a heavy metal with a relatively high toxicity in prokaryotes. Ionic silver exhibits antimicrobial activity against a broad range of microorganisms and has long been used as an effective broad antimicrobial agent against pathogens.1,2 Copper, although required in trace amounts for bacterial growth, is highly toxic even at low concentrations.3 Thus, both silver and copper are well-known bactericides, and their biocidal effects have been used for centuries. In Gram-negative bacteria, efflux systems of the resistance-nodulation-cell division (RND) superfamily play major roles in the intrinsic and acquired tolerance of antibiotics and toxic compounds, including silver and copper ions.4,5 The Gram-negative bacterium Escherichia coli harbors seven different RND efflux transporters. These transporters can be categorized into two distinct sub-families, the hydrophobic and amphiphilic efflux RND (HAE-RND) and heavy-metal efflux RND (HME-RND) families.4,5 Six of these transporters, such as AcrB,6–16 AcrD,7,17,18 AcrF,7,19,20 MdtB,7,21,22 MdtC,7,21,22 and YhiV,7,23,24 are multidrug efflux pumps (Table I), which belong to the HAE-RND protein family.4 In addition to these multidrug efflux pumps, E. coli contains one HME-RND efflux transporter, CusA (Table I), which specifically recognizes and confers resistance to Ag(I) and Cu(I) ions.25,26 The two sub-families of these RND transporters share relatively low protein sequence homology. For example, sequence alignment shows that CusA and AcrB have only 19% identity.
Table I.
The RND Efflux Pumps of Escherichia coli
| RND protein | Subfamily | Function | MFP | OMC | References |
|---|---|---|---|---|---|
| AcrB | HAE | Multi-drug efflux pump (conferring resistance to chloramphenicol, tetracycline, minocycline, erythromycin, nalidixic acid, norfloxacin, enoxacin, doxorubicin, novobiocin, rifampin, trimethoprim, acriflavine, crystal violet, ethidium bromide, rhodamine 6G, tetraphenylphosphonium bromide, benzalkonium, Triton X-100, sodium dodecyl sulfate, deoxycholate, taurocholate, etc.) | AcrA | TolC | 6–16 |
| AcrD | HAE | Multi-drug efflux pump (conferring resistance to aminoglycosides, novobiocin, sodium dodecyl sulfate, and deoxycholate) | AcrA | TolC | 7,17,18 |
| AcrF | HAE | Multi-drug efflux pump (conferring resistance to doxorubicin, acriflavine, ethidium bromide, rhodamine 6G, sodium dodecyl sulfate, deoxycholate; important in the maintenance of cell division) | AcrE | TolC | 7,19,20 |
| MdtB | HAE | Multi-drug efflux pump (conferring resistance to nalidixic acid, norfloxacin, fosfomycin, novobiocin, benzalkonium, sodium dodecyl sulfate, and deoxycholate) | MdtA | TolC | 7,21,22 |
| MdtC | HAE | Multi-drug efflux pump (conferring resistance to nalidixic acid, norfloxacin, fosfomycin, novobiocin, benzalkonium, sodium dodecyl sulfate, and deoxycholate) | MdtA | TolC | 7,21,22 |
| YhiV | HAE | Multi-drug efflux pump (conferring resistance to erythromycin, doxorubicin, crystal violet, ethidium bromide, rhodamine 6G, tetraphenylphosphonium bromide, benzalkonium, sodium dodecyl sulfate, and deoxycholate) | YhiU | TolC | 7,23,24 |
| CusA | HME | Heavy-metal efflux pump (conferring resistance to Cu(I) and Ag(I) ions) | CusB | CusC | 7,25,26 |
As an RND transporter, CusA works in conjunction with a periplasmic component, belonging to the membrane fusion protein (MFP) family, and an outer membrane channel (OMC) to form a functional protein complex. CusA is a large proton motive force-dependent inner membrane efflux pump comprised of 1047 amino acids.25,26 CusC, however, is a 457 amino acid protein that forms an OMC.25,26 The MFP CusB (379 amino acids) contacts both the inner membrane CusA and outer membrane CusC proteins.25,26 Presumably, the three components of this HME-RND system form a tripartite efflux complex CusCBA, which spans both the inner and outer membranes of E. coli to export Ag(I) and Cu(I) directly out of the cell. HME by the CusCBA complex is driven by proton import, and this process is catalyzed through the inner membrane transporter CusA.
Between the cusC and cusB genes, there is a small chromosomal gene that produces a periplasmic protein CusF.26 CusF is also involved in Cu(I)/Ag(I) resistance. It functions as a chaperone that carries Cu(I)/Ag(I) to the CusCBA tripartite efflux pump.27,28
Among those transporters belonging to the RND superfamily, the structural and functional relationships of the HAE-RND pumps have been elucidated extensively. Currently, two crystal structures of the HAE-RND-type efflux pumps, the E. coli AcrB8–16 and Pseudomonas aeruginosa MexB29 multidrug transporters, have been determined. Their structures suggest that both AcrB and MexB span the entire width of the inner membrane and protrude approximately 70 Å into the periplasm. Along with the models of these two HAE-RND transporters, the crystal structures of the other components of these tripartite complex systems have also been determined. These include the OMCs, E. coli TolC30 and P. aeruginosa OprM,31 as well as the periplasmic MFPs, E. coli AcrA32 and P. aeruginosa MexA.33–35
In contrast to the HAE-RND pumps, transporters belonging to the HME-RND sub-family have not yet been studied in detail. Different from the HAE-RND family, members of the HME-RND family are highly substrate-specific, with the ability to differentiate between monovalent and divalent ions. To elucidate the mechanism used by the CusCBA efflux system for Cu(I)/Ag(I) recognition and extrusion, we have recently determined the crystal structures of the CusB MFP36 and the CusA inner membrane efflux pump,37 both in the absence and presence of Cu(I) or Ag(I). In this review, we summarize the structural information of these two efflux proteins. Based on the new findings of this efflux system, we put forward our opinion on how these protein machineries facilitate metal transport. The structures strongly suggest that the CusCBA efflux system relies upon methionine residues to bind and export the metal ions.
Crystal structure of the CusB membrane fusion protein
The crystal structure of CusB (pdb code: 3H9I), comprising ∼80% of the protein (residues 89–385), was originally determined to a resolution of 3.40 Å.36 The structure revealed that the asymmetric unit of the crystal consists of two protomers. These two CusB molecules were found to interact with each other. However, the functional oligomerization state of this protein is still not clear. Each CusB molecule is folded into an elongated polypeptide of ∼120 Å long and ∼40 Å wide. Intriguingly, this MFP can be divided into four different domains (Fig. 1). The first three domains of the protein are mostly β-strands. However, the fourth domain forms an all α-helical domain, which is folded into a three-helix bundle secondary structure.
Figure 1.
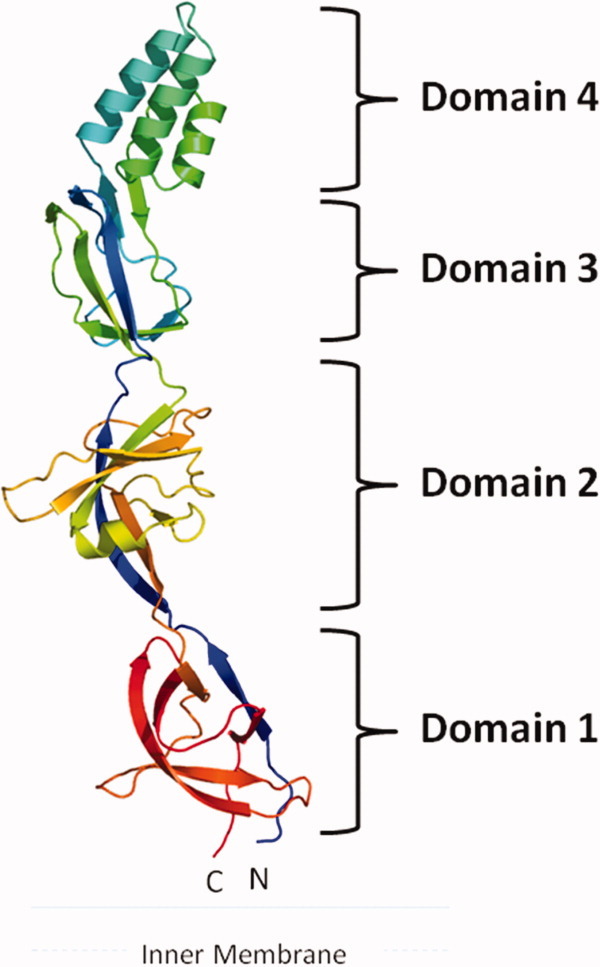
Crystal structure of the CusB membrane fusion protein. The structure can be divided into four distinct domains. Domain 1 is formed by the N and C-termini and is located above the inner membrane. The loops between Domains 2 and 3 appear to form an effective hinge to allow the molecule to shift from an open conformation to a more compact structure. Domain 4 is folded into an antiparallel, three-helix bundle, which is thought to be located near the outer membrane.
The first β-domain (Domain 1) of CusB is formed by the N and C-terminal ends of the polypeptide (residues 89–102 and 324–385). This domain is located directly above the outer-leaflet of the inner membrane. It has later been found that this domain specifically interacts with the periplasmic domain of the CusA efflux pump.36 Overall, Domain 1 is a β-barrel domain, consisting of six β-strands, with the N-terminal end forming one of the β-strands while the C-terminus of the protein constitutes the remaining five strands (Fig. 1).
The second β-domain (Domain 2) of CusB is formed by residues 105–115 and 243–320. This domain comprises six β-strands and one short α-helix. Again, the N-terminal residues form one of the β-strands that is incorporated into this domain. The C-terminal residues contribute a β-strand, an α-helix and four antiparallel β-sheets. Interestingly, in an asymmetric unit of the crystal, the two molecules of CusB contact one another in an antiparallel fashion, with Domains 1 and 2 of one molecule interact closely with Domains 2 and 1 of the other molecule, respectively.36 It is suspected that these two domains may form an interaction site for CusB oligomerization.
Domain 3 is another globular β-domain adjacent to the second domain of CusB. This domain consists of residues 121–154 and 207–239, with a majority of these residues folding into eight β-strands.
Perhaps the most interesting motif appears to be in the fourth domain (Domain 4) of CusB. This region, comprising residues 156–205, forms an all-helical domain. Surprisingly, this domain is folded into an antiparallel, three-helix bundle.36 This structural feature, not found in other known protein structures in the MFP family, highlights the uniqueness of the CusB protein. The helix bundle creates an ∼27 Å long helix-turn-helix-turn-helix secondary structure, making it at least 20 Å shorter than the two-helical hairpin domains of MexA33–35 and AcrA.32 To date, CusB is the only periplasmic protein in the MFP family that possesses this three-helical domain instead of a two-helical hairpin motif.
The crystal structure of CusB represents the first structure of any MFP that is associated with an HME-RND-type transporter. Among the structures of the MFP family, including AcrA32 and MacA,38 the CusB,36 MexA,35 and most recently determined ZneB39 proteins exhibit the most complete three-dimensional structures. Like MexA and ZneB, the structure of CusB revealed that this MFP consists of four major domains, including three β-strand domains and one α-helical domain. However, CusB is folded into a distinct secondary structure when compared with the other available MFP structures. This distinct structural feature may underscore the unique functionality of CusB in the MFP family.
Crystal structures of the CusB-Cu(I) and CusB-Ag(I) complexes
There is strong evidence that members of the MFP family play a functional role in the efflux of substrates. It has been found that the MFP EmrA is able to directly bind different transported drugs.40 Recently, it has also been shown that the CusB41 and ZneB39 proteins directly interact with the metal ions. Thus, it is expected that in addition to their role as adaptors to bridge the inner and outer membrane efflux components, these MFPs may participate in recognizing and extruding their substrates. To elucidate how CusB interacts with the metal ions, we have resolved the crystal structures of the CusB-Cu(I) and CusB-Ag(I) complexes.36 These structures indeed demonstrated that CusB contains multiple binding sites for Cu(I) and Ag(I). Based on the CusB-Cu(I) complex (pdb code: 3H9T), we have found two copper-binding sites (designated as C1 and C2). Site C1 is located at the upper portion of Domain 1 of the protein (Fig. 2). Interestingly, Domain 1 of CusB has also been found to directly interact with the periplasmic domain of CusA (see below). Coordinating with the bound Cu+ ion at this site are the side chains of residues M324, F358, and R368. The binding of Cu+ in site C2 of CusB is located close to the center of the three-helix bundle in Domain 4. This α-helical domain may make a direct contact with the OMC CusC. Cu+ in this location is bound by the side chains of M190, W158, and Q162.
Figure 2.
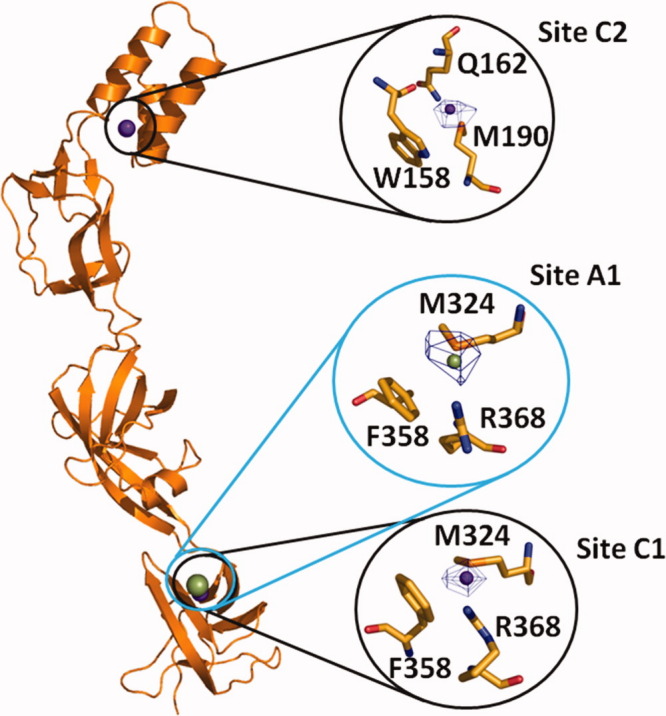
Cu+ and Ag+ binding sites of CusB. Cu+ and Ag+ ions are represented by purple and green spheres, respectively. The overall locations of sites C1, C2, and A1 are circled. Anomalous difference Fourier maps are contoured at 4.6σ, 4.0σ, and 4.2σ for sites C1, C2, and A1, respectively.
For the CusB-Ag(I) complex crystal (pdb code: 3H94), we have found one bound Ag+ in each CusB molecule. This Ag+ binding sites, designated as A1, is observed right next to M324 of molecule A (Fig. 2). It appears that the location of this Ag+-binding site is the same as that of site C1 for Cu+ binding. Thus, the bound Ag+ at site A1 is coordinated with the side chains of M324, F358, and R368.
Conformational flexibility of CusB
Two distinct conformations of CusB were captured in the single crystal, suggesting that this MFP is quite flexible in nature.36 A comparison of the structures of the two molecules of CusB indicates that these two molecules are quite different, presumably representing two transient states of the MFP. However, the two conformations are related by a small hinge motion. Superimposition of these two molecules gives an overall rmsd of 2.6 Å calculated over the Cα atoms. Comparison of these two structures reveals that one of the molecules of CusB adopts a more open conformation, while the other one exhibits a relatively compact form of the structure (Fig. 3). Thus, these two molecules may correspond to the open and closed states of this MFP.
Figure 3.
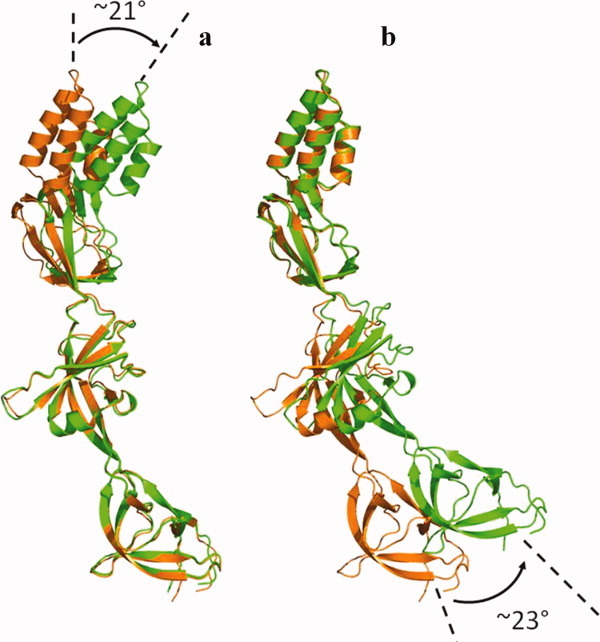
Comparison of the two conformations of CusB observed in the crystal. (a) Superimposition of Domains 1 + 2 of the two CusB protomers, displaying an ∼21° overall shift of the three-helix bundle of Domain 4. (b) Superimposition of Domains 3 + 4 of the two molecules of CusB, displaying an overall shift of the β-strands of Domain 1 by ∼23°.
It appears that Domains 1 + 2 of the two molecules of CusB can easily be superimposed with high structural similarity, giving an overall rmsd of 0.8 Å (168 Cα atoms). Superposition of Domains 3 + 4 alone of these two molecules can also be calculated, showing an overall rmsd of 0.8 Å (118 Cα atoms). Using Domains 1 + 2 as references, the α-helical domains of these two CusB molecules are found to differ by ∼21° overall [Fig. 3(a)]. When Domains 3 + 4 are superimposed, the orientation of the β-barrels of Domain 1 of these two CusB protomers display an overall shift of ∼23° [Fig. 3(b)]. Taken together, these superimpositions suggest that the linker region, which is composed of two loops (residues 116–120 and 240–242) between Domains 2 and 3, forms a flexible hinge in the MFP. This hinge region effectively permits the protein to shift from one conformation to another simply by performing a rigid-body movement at Domains 1 + 2 with respect to Domains 3 + 4.
The fact that two copies of CusB have been found in a single crystal suggests the conformational flexibility of this MFP. Indeed, it has been observed that members of the MFP family, including MexA,35 AcrA,32 and MacA,38 are highly flexible. It has also been demonstrated that binding of metal ions trigger significant conformational changes to the CusB41 and ZneB39 proteins. Thus, these MFPs are also capable of altering their conformations in the presence of their corresponding ligands. Interestingly, it has been reported that the linker region between the membrane proximal and β-barrel domains of MexA forms a flexible hinge to allow this protein to flip from the “unrotated” form to the “rotated” state.35 This flexible linker should be related to the linker region between Domains 1 and 2 in the CusB structure. In the case of AcrA,32 the flexible hinge between the α-helical hairpin and lipoyl domain (corresponds to the linker region between Domains 3 and 4 of CusB) is attributed to the freedom of this protein. As mentioned earlier, our CusB structures indicate that the flexible hinge is located between Domains 2 and 3 of the protein. Thus, it is very likely that all of these linker regions located between different domains of these MFPs potentially can provide additional flexibility for the proteins to perform their biological function.
CusA and CusB interaction
To determine how CusB interacts with the CusA efflux pump and the relative orientation of CusB in the efflux complex, we cross-linked the purified CusA and CusB proteins using the lysine-lysine cross-linker disuccinimidyl suberate. The resulting product was then digested with trypsin and examined using LC-MS/MS. Analysis of the mass spectral data suggests that the lysine residue of the polypeptide IDPTQTQNLGVKTATVTR originating from the N-terminal residues (84–101) of CusB directly interacts with the lysine residue of the peptide SGKHDLADLR (residues 148–157 of CusA), which is supposed to form part of the N-terminal periplasmic loop of the CusA efflux pump.36 When we performed this cross-linking experiment, the structure of CusA was not yet available. We thus generated a structural model of the CusA transporter based on the crystal structure of AcrB and alignment of protein sequences of these two transporters (Fig. 4). The model indicates that the polypeptide SGKHDLADLR (residues 148–157 of CusA) is located directly above the vestibule region of CusA, facing the periplasm. This position should correspond to the PN2 region of AcrB.8 If this is the case, then the C-terminus of CusB should interact with CusA at a position corresponding to the PC1 region of AcrB (Fig. 4). This result is indeed in good agreement with the cross-linking study that the N and C-termini of AcrA directly interact with PN2 and PC1 of the periplasmic domain of AcrB, respectively.35 Thus, together with the crystal structure of CusB and the mass spectrometric data, we suggest that Domain 1, formed by the N and C-terminal ends, of CusB should interact with the periplasmic domain of the CusA transporter.
Figure 4.
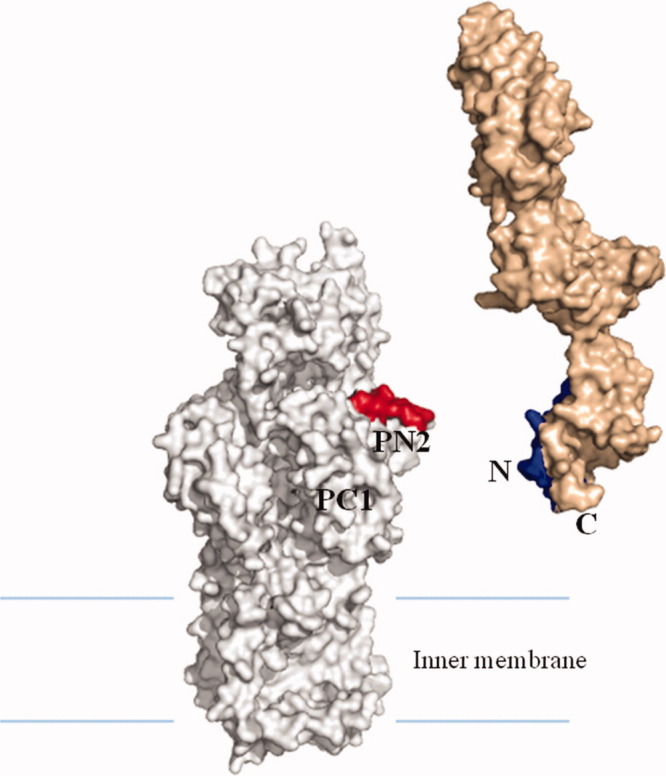
Specific interaction between CusA and CusB. The model of CusA (gray) was created based on protein sequence alignment and the crystal structure of AcrB. Mass spectral data suggest that the periplasmic domain of CusA specifically interacts with the N-terminus of CusB (light brown). Polypeptides α and β were in red and blue, respectively.
Crystal structure of the CusA heavy-metal efflux pump
The crystal structure of the CusA efflux pump37 was resolved to a resolution of 3.52 Å, with 98% of the residues (residues 5–504 and 516–1040) included in the final model (pdb code: 3KO7) (Fig. 5). Overall, the crystal structure of CusA is quite distinct from those of the HAE-RND pumps, AcrB, and MexB. Superimposition of the structure of CusA with the structure of AcrB8 results in a high RMSD of 11.4 Å for 1003 Cα atoms, suggesting highly significant differences between these two transporters. CusA exists as a homotrimer, and each subunit of CusA consists of 12 transmembrane helices (TM1–TM12) and a large periplasmic domain formed by two periplasmic loops between TM1 and TM2, and TM7 and TM8, respectively [Fig. 5(a)]. In the transmembrane region, the relative locations of TM1–TM6 are related to those of TM7–TM12 by pseudo-twofold symmetry. These TM helices are arranged in such a way that TM4 and TM10 form the center of the core and are surrounded by the other TM helices. Differently from AcrB and MexB, four helices, TM4, TM5, TM10, and TM11, extend into the cytoplasm, forming the cytoplasmic domain of the pump. Two other helices, TM2 and TM8, protrude into the periplasm and contribute part of the periplasmic domain. It is important to note that TM2, TM4, and TM5 of the N-terminal half correspond to TM8, TM10, and TM11 of the C-terminal half, respectively, in the pseudo-twofold symmetry.
Figure 5.

Crystal structure of the CusA efflux pump. (a) Ribbon diagram of the CusA homotrimer viewed in the membrane plane. Each subunit of CusA is labeled with a different color. Sub-domains DN, DC, PN2, PC1, and PC2 are labeled on the front protomer (green). The location of PN1 in this protomer is behind PN2, PC1, and PC2 (see text). (b) Top view of the CusA trimer. The six sub-domains are labeled yellow (DN), blue (DC), pink (PN1), orange (PN2), green (PC1), and red (PC2). In the apo-CusA structure, the cleft between PC1 and PC2 is closed.
Like AcrB and MexB, the periplasmic domain of CusA can be divided into six sub-domains, PN1, PN2, PC1, PC2, DN, and DC [Fig. 5(b)]. Sub-domains PN1, PN2, PC1, and PC2 form the pore domain, with PN1 making up the central pore and stabilizing the trimeric organization. Sub-domains DN and DC, however, contribute to form the docking domain, presumed to be interacting with the OMC CusC. The trimeric CusA structure suggests that sub-domains PN2, PC1, and PC2 are located at the outermost core of the periplasmic domain, facing the periplasm. In AcrB, sub-domains PC1 and PC2 form a large external cleft and are presumed to create the entrance for drugs from the periplasm. However, the apo-CusA structure shows that the gap between PC1 and PC2 is completely closed [Fig. 5(b)]. As mentioned above, in vitro cross-linking coupled with mass spectrometry suggested that sub-domain PN2 of CusA should interact with the N-terminus of the MFP CusB. Thus, sub-domains PN2 and PC1 of CusA most likely form the binding surface for CusB.
Perhaps the most interesting secondary structural feature appears in the cleft region of the periplasmic domain. Residues located on the left side of the wall (Fig. 6), formed by one α-helix (residues 690–706) and three β-sheets (residues 681–687, 711–716, and 821–827), appear to tilt into the cleft to close the opening. Surprisingly, residues 665–675, located at the bottom of the cleft, form an α-helix. This structural feature, not found in AcrB and MexB, likely governs the specificity of the CusA pump. The α-helix orients horizontally and roughly divides the transmembrane and periplasmic domains into two compartments. Located just above this horizontal helix, we find three proximal methionine residues, M573, M623, and M672, presumably creating a three-methionine specific binding site for Cu(I) and Ag(I) ions.42,43 Notably M672 is also one of the residues within the horizontal helix. Site-directed mutagenesis had suggested that these three methionine residues are essential for mediating copper resistance.26 Indeed, a strong peak at the copper edge was observed at the center of these three methionines in our CusA-Cu(I) crystal derivative, indicating the binding of a Cu(I) ion in a three-sulfur binding site.
Figure 6.

Comparison of the apo and metal bound structures of CusA. (a) Superposition of a monomer of apo-CusA (red) onto a monomer of Cu(I) bound-CusA (green). The bound Cu(I) is pink. The arrow represents a major swing of the PC2 sub-domain initiated by Cu(I) binding. (b) Conformational changes of the periplasmic domain of CusA. The conformation of each sub-domain of CusA before (left) and after (right) Cu(I) binding. The periplasmic cleft formed between PC1 and PC2 is opened after Cu(I) binding. The bound coppers in the CusA-Cu(I) structure are blue. M573, M623, and M672 forming a metal binding site at the periplasmic cleft are shown in stick form (yellow). (c) The changes in conformation of the horizontal helix and TM8, are shown in a superimposition of the structures of apo (red) and Cu(I)-bound (green) CusA. The bound Cu(I) is shown as a pink sphere. Anomalous map of the bound Cu(I), contoured at 8σ, is in blue. M573, M623, and M672 are shown as sticks. (d) The Ag(I) binding site. The bound Ag(I) is shown as a gray sphere. Anomalous map of the bound Ag(I), contoured at 10σ, is in red.
Crystal structures of the CusA-Cu(I) and CusA-Ag(I) complexes
Intriguingly, the overall structure of the CusA-Cu(I) complex (pdb code: 3KSS)37 is quite distinct from that of apo-CusA (Fig. 6). Superimposition of these two structures gives an overall RMSD of 3.9 Å (for 1006 Cα atoms). We used the copper edge wavelength (1.3779 Å) to collect the x-ray diffraction data of the CusA-Cu(I) crystal, thus allowing us to identify the location of the bound Cu(I). As mentioned above, the bound Cu(I) ion was found to coordinate residues M573, M623, and M672. These three methionines specifically form a typical three-methionine coordination site for binding Cu(I)/Ag(I).42,43 Binding of Cu(I) initiates significant conformational changes in the periplasmic as well as transmembrane domains of CusA. Perhaps, the most noticeable difference between the apo and ion-bound structures appears in the PC2 region [Fig. 6(a)]. Cu(I) binding leads to a 30° swing of the entire PC2 sub-domain. This motion shifts PC2 away from the PC1 sub-domain. The hinge for this rotational movement appears to be at the junction between sub-domains PC2 and DC with residues G721 and P810 forming the hinge. As a consequence, the gap between PC1 and PC2 appears to open up after binding this metal ion [Fig. 6(b)]. This gap presumably creates an entrance for metal ions from the periplasmic space. The horizontal helix, residues 665–675, located inside the cleft also makes a substantial movement. The C-terminal end of this helix is found to tilt upward by 21° in the Cu(I)-bound structure with respect to the apo form [Fig. 6(c)]. This tilting motion allows M672 to move closer to M573 and M623 to complete the three-methionine coordination site. Coupled with this movement, TM8 also shifts in position to a more vertical orientation while retaining its α-helical structure. Overall, the N-terminal end of TM8 is found to shift away from the core by 10 Å after Cu(I) binding.
For the CusA-Ag(I) complex (pdb code:3KSO),37 an anomalous difference Fourier peak was found at the center of residues M573, M623, and M672, indicating that the bound Ag+ ion is coordinated by these three methionines [Fig. 6(d)]. The overall conformational changes triggered by Cu(I) and by Ag(I) binding are nearly identical. Superimposition of the CusA-Cu(I) and CusA-Ag(I) structures gives an overall RMSD of 1.0 Å (for 1021 Cαs). Based on the crystal structures, the horizontal helix in the cleft directly interacts with the N-terminal end of TM8. The movement of TM8 may relate directly to transmembrane signaling and could initiate the translocation of a proton across the membrane. Indeed, in the AcrB pump, there is evidence that proton translocation is coupled to the conformational change in TM8.13,15 The change in conformation takes place in TM8 by reeling in some random coil residues to extend the α helix.13 In addition, when individual residues of the proton-relay network was changed to alanine, disrupting the hydrogen bonds in the system, it was reported that TM8 of AcrB becomes longer and extends into the periplasmic domain.15 In contrast to AcrB, the helical structure of the N-terminus of TM8 in CusA is retained after metal binding. Instead, the N-terminal end of TM8 moves outward, together with the movement of the PC2 sub-domain. According to the crystal structures of the apo and bound-CusA, we agree with the prior speculation that the structure of apo-AcrB is actually the detergent-bound form.29
Intriguingly, it appears that the binding of Cu(I) or Ag(I) also triggers significant conformational changes in the other transmembrane helices of the pump. In addition to the movement of TM8, all other transmembrane helices except TM2 shift horizontally by as much as 4 Å, mimicking the motion of TM8. Further, TM1, TM3, and TM6 readjust in ∼3 Å upward shift with respect to the inner membrane surface. The net result is that all three of these transmembrane helices move toward the periplasm by one turn. Mutagenesis studies of the transmembrane domain of CusA indicated the conserved charged residue D405 of TM4 to be essential for transporter function.26 Based on the crystal structure, this acidic residue interacts with E939 and K984 (Fig. 7). It is possible that these three charged residues participate in forming the proton-relay network in the transmembrane region of the pump.
Figure 7.
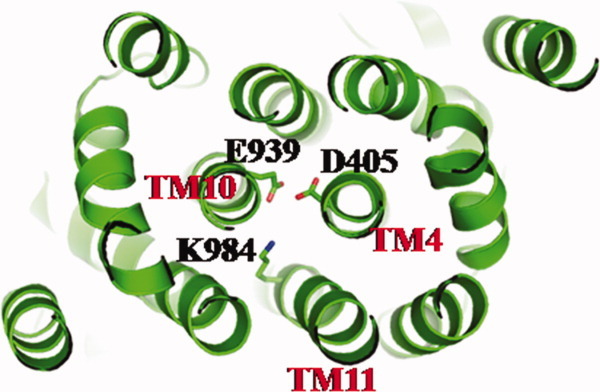
Ion pairs in the transmembrane domain. Residues D405 of TM4, E939 of TM10, and K984 of TM11 that form ion pairs, which may play an important role in proton translocation, are in green sticks.
Coupled with the above conformational changes, the sub-domain PN1 at the periplasmic domain was also found to undergo substantial movement. Overall, PN1 also shifts upward by 3 Å. This moves the central pore helix upward by one turn upon metal binding. It should be noted that the binding of metal ions does not significantly affect the conformation of the sub-domains DN, DC, PN2, or the transmembrane helix TM2. PN2 has been found to interact with the N-terminus of the CusB MFP. Thus, we expect that PN2 and TM2 could move in the presence of CusB, to accommodate the binding of this MFP. If TM8 were responsible for signaling the binding of metal ions, it would suggest that TM2 is important for detecting the presence of the membrane fusion component CusB.
The methionine-residue ion relay network
The full-length CusA includes 34 methionine residues with 18 of them located in the transmembrane domain. Of the 18 methionines, six are paired up to form three distinct methionine pairs. These methionine pairs are M410 of TM4 and M501 of TM6; M403 of TM4 and M486 of TM6; and M391 of TM4 and M1009 of TM12 (Fig. 8). It is known that copper tolerance proteins, such as CusF,27,28 CueR,44 and Atx1,45 frequently utilize two-methionine or two-cysteine binding pockets to carry their Cu(I)/Ag(I) cargos. Thus, these methionine pairs could potentially form binding sites for Ag(I) and Cu(I) in the transmembrane region of the pump. If this is the case, then CusA could transport these metal ions from the cytoplasm along these methionine pairs. In the periplasmic domain of CusA, we have found another methionine pair (M271–M755) in addition to the three-methionine metal binding site (Fig. 8). In view of the crystal structures, these five methionine pairs/clusters are deemed to be important for binding and transport of metal ions.
Figure 8.
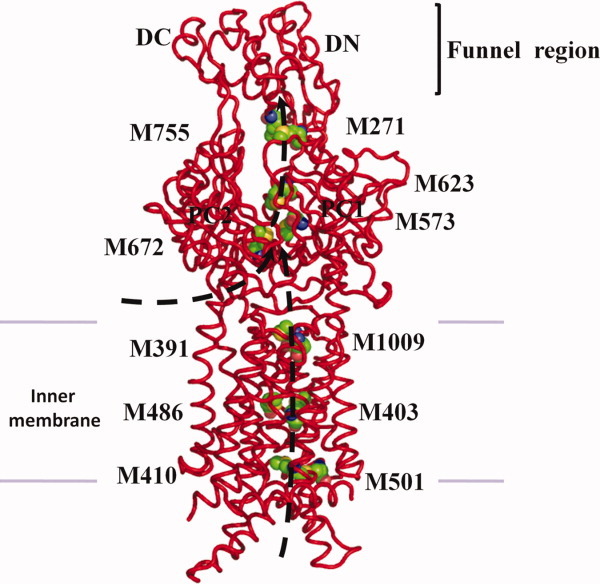
Proposed metal transport pathway of the CusA efflux pump. The five methionine pairs/clusters of the apo-CusA transporter form a pathway for metal export. These five methionines are shown as spheres (C, green; O, red; N, blue; S, orange). The pair M271–M755 is located at the bottom of the periplasmic funnel where the metal ion could then be released for final extrusion. The paths for metal transport through the periplasmic cleft and transmembrane region are illustrated with black curves.
We then determined whether the CusA protomer forms a channel by using the program CAVER (http://loschmidt.chemi.muni.cz/caver) and found that each protomer of CusA forms a channel spanning the entire transmembrane region up to the bottom of the periplasmic funnel (Fig. 9). Intriguingly, the channel includes four methionine pairs, three (M410–M501, M403–M486, M391–M1009) from the transmembrane region and one (M271–M755) from the periplasmic domain, as well as the three-methionine binding site formed by M573, M623, and M672 (Fig. 8). Taken together, these five methionine pairs/clusters are likely to form a relay network facilitating metal ion transport. Remarkably, this channel spans almost the entire length of each protomer, from the transmembrane domain through to the bottom of the periplasmic funnel region, and is likely to represent a real path for transporting the metal ion from both the cytoplasm and periplasm to the periplasmic funnel for extrusion.
Figure 9.
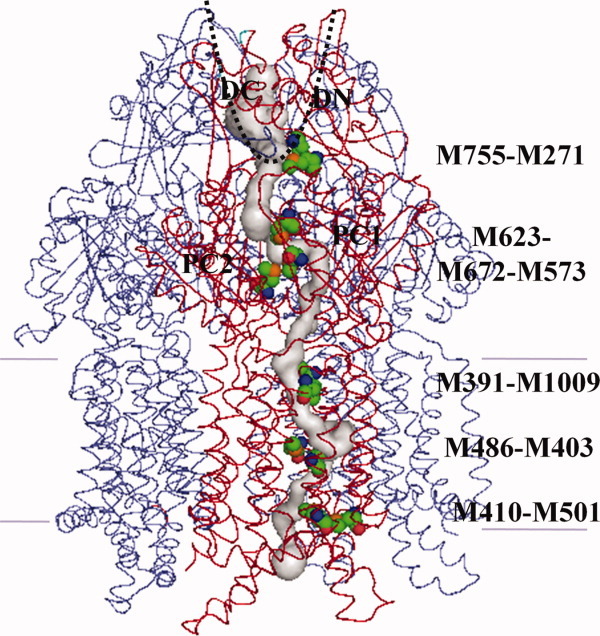
Channel in the CusA pump. The channel formed by the front protomer of apo-CusA (red) leading through the transmembrane and periplasmic domains is in gray color. This channel was calculated using the position of the sulfur atom of residue M672 as a starting point. The 11 methionines forming the relay network are in spheres (green, carbon; red, oxygen; blue, nitrogen; orange, sulfur). Two other CusA protomers behind the front protomer are shown as blue wires.
In vivo metal susceptibility assay
We made an E. coli knocked-out strain BL21(DE3)ΔcueOΔcusA that lacks both the cueO and cusA genes. We mutated M573, M623, and M672, which are members of the binding site triad inside the cleft of the periplasmic domain, into isoleucines (M573I, M623I, and M672I). We then expressed these three mutant transporters in BL21(DE3)ΔcueOΔcusA and tested their ability to confer copper and silver resistance in vivo. We found that the CusA mutants, M573I, M623I, and M672I are unable to relieve the copper or silver sensitivity of strain BL21(DE3)ΔcueOΔcusA, thus agreeing well with the work of Franke et al.26 in which M573, M623, and M672 were shown to be essential for the function of CusA.
We mutated M410 into an isoleucine (M410I) to disrupt the pair formed by M410 and M501 at the bottom of the transmembrane and also replaced M486 and M391, which pair with M403 and M1009, with isoleucines (M486I and M391I). Expression of these three mutants in BL21(DE3)ΔcueOΔcusA showed a significant decrease in the level of copper and silver tolerances when compared with cells expressing wild-type CusA. In addition, we introduced M755I in the periplasmic domain. Again, when expressed in BL21(DE3)ΔcueOΔcusA, this mutant transporter (M755I) showed a decrease in copper/silver tolerance in comparison with cells harboring the wild-type transporter. Together, these results strongly support the hypothesis that these methionine pairs/clusters engage in the methionine-residue ion relay channel.
Based on the crystal structure of CusA, it is expected that the charged residues D405, E939, and K984 are important for the proton-relay network of the pump, and these in turn were replaced with alanines (D405A, E939A, and K984A) to disrupt the hydrogen-bonded network. Indeed, cells expressing this mutant were unable to tolerate copper and silver, demonstrating that these three charged residues are essential for the transporter's functioning.
In vitro metal transport assay
To investigate whether CusA can transport metal ions from the cytoplasm, we reconstituted the purified CusA protein into liposomes containing the fluorescent indicator Phen Green SK in the intravesicular space. The intravesicular and extravesicular pHs of these proteoliposomes were 6.6 and 7.0, respectively. We then determined whether these proteoliposomes can capture metal ions from the extravesicular medium with a stopped-flow transport assay. (The charged metal ions are unlikely to diffuse spontaneously across membrane bilayers.) When Ag+ ions were added into the extravesicular medium, we detected the quenching of the fluorescence signal, suggesting the uptake of Ag+ into the intravesicular space [Fig. 10(a)]. The uptake into proteoliposomes is presumably due to the CusA active transport activity.
Figure 10.
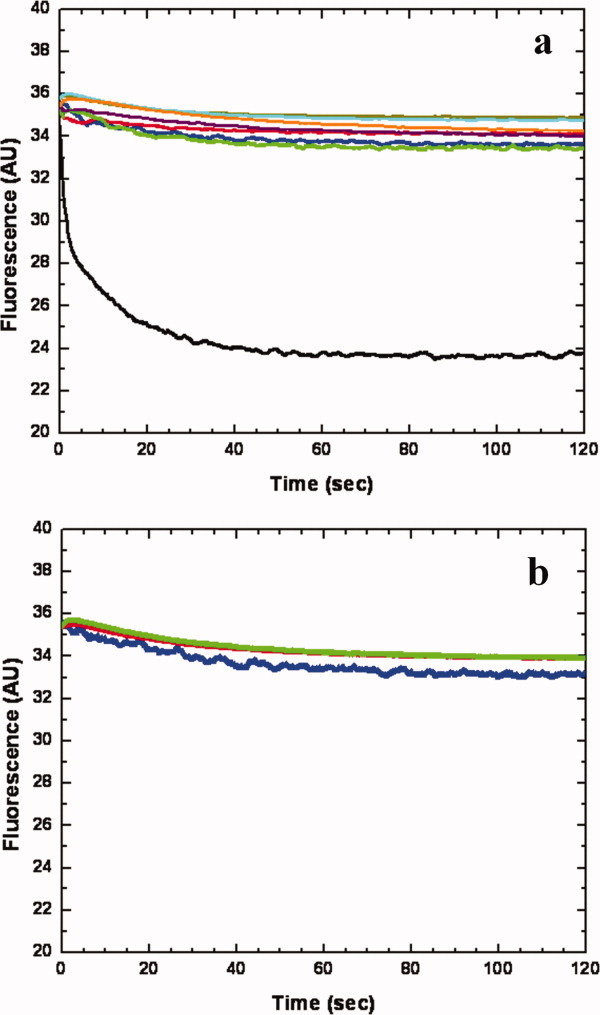
Stopped-flow transport assay of reconstituted CusA with extravesicular Ag+ ion. (a) Mutants of the methionine-residue relay network. The decrease in fluorescence signal of PGSK mediated by proteoliposomes of wild-type CusA indicates the transport of Ag+ across the membrane. The stopped-flow traces are the cumulative average of four successive recordings (wild-type CusA, black curve; M486I, green curve; M391I, brown curve; M573I, red curve; M623I, purple curve; M672I, orange curve; M755I, cyan curve; control liposome, blue curve). (b) Mutants of the proton-relay network. The traces are the cumulative average of four successive recordings (D405A, blue curve; E939A, red curve; K984A, green curve).
In addition to the above experiment, we investigated the methionine residues that were shown to be important for copper and silver tolerances with the above transport assay. We expressed, purified and reconstituted the CusA mutants, M573I, M623I, and M672I, into liposomes encapsulated with the same fluorescence indicator. As expected, these mutant transporters do not take up Ag+ from the extravesicular medium of the proteoliposomes [Fig. 10(a)]. It is clear that even single point mutations M573I, M623I, or M672I abolish the process of metal transport across the membrane. Thus, it is expected that this three-methionine site is requisite for both metal binding and export.
We then purified the mutants, M391I, M486I, and M755I, and reconstituted them into liposomes. In these three cases, the fluorescence signals did not attenuate as indicated by the stopped-flow assay [Fig. 10(a)]. When compared with the result from the wild-type CusA, this suggests that the M391I, M486I, and M755I mutant transporters are unable to actively transport Ag+ across the membrane. Our collective experiments provide direct compelling evidence that CusA is capable of taking up Ag+ from the cytoplasm.
The crystal structure of CusA suggests that the charged residues D405, E939, and K984 in the transmembrane domain may be important for proton translocation. When reconstituted into liposomes, mutant transporters (D405A, E939A, and K984A) do not take up Ag+ into the intravesicular space [Fig. 10(b)], thus confirming the importance of D405, E939, and K984 for the function of the pump.
Proposed mechanism for metal-ion export
It has been demonstrated that the divalent cation efflux pump CzcA is capable of catalyzing the transport of divalent cations, such as Zn2+, from the cytosol.46 It has also been observed that the AcrD multidrug efflux pump can capture aminoglycosides from both the periplasm and cytoplasm.18 Thus, we hypothesize that CusA can pick up metal ions from both the cytoplasm and periplasm. We propose that this transporter utilizes methionine pairs/clusters to export Cu(I) and Ag(I). The periplasmic cleft of CusA presumably remains closed when there is no Cu(I)/Ag(I). In the presence of Cu(I) or Ag(I) ions, the periplasmic cleft opens. Metal ions could enter the three-methionine binding site inside the cleft directly through the periplasmic cleft or via the methionine pairs within the transmembrane region (Fig. 8). Thus, transport of a metal ion within the membrane is likely to involve a stepwise process that shuttles the metal ion from one methionine pair to another along the pathway. The locations of these methionine pairs/clusters allow us to depict a direct pathway for metal transport and extrusion. The metal ion bound at the three-methionine binding site of the periplasmic cleft could then be released to the nearest methionine pair, formed by M271 and M755, following which the metal ion could then be released into the central funnel to reach the CusC channel for final extrusion. Interestingly, the proposed metal export pathway in the periplasmic domain, based on the location of the methionine pairs/clusters, is similar to the proposed drug export pathway in AcrB.13 Nonetheless, our mechanism for export of metal ions intermediated by binding to sequential methionine pairs/clusters is fully consistent with the body of evidence in all of our structural and mutagenesis studies.
Concluding Remarks
It is clear from the published literature that the RND efflux pumps have significant functions in conferring resistance to clinically relevant antimicrobials.47 Thus, it is very important to understand how these tripartite pumps work and assemble. To date, a crystallographic model of these tripartite efflux complexes has not yet been reported. One direct approach to obtain the complete picture of these efflux complexes is to elucidate the structures of individual components as well as the assembled complexes of these efflux systems. This approach should be able to allow us to rational design inhibitors that prohibit different components of the pumps to assemble, thus disallowing these pumps to function. In the case of the Cus efflux system, the availability of the co-crystal structures of the adaptor-transporter CusBA, the channel-adaptor CusCB, and the complete tripartite CusCBA efflux complexes are essential to achieve this task although co-crystallization of different components of these RND efflux systems have been proven to be extremely difficult.
Acknowledgments
This work is supported by NIH Grants GM 074027 (E.W.Y.) and GM 086431 (E.W.Y.). This paper is dedicated to the memory of Prof. Victor S. Lin, a colleague and a friend.
References
- 1.Silver S. Bacterial silver resistance: molecular biology and uses and misuses of silver compounds. FEMS Microbiol Rev. 2003;27:341–353. doi: 10.1016/S0168-6445(03)00047-0. [DOI] [PubMed] [Google Scholar]
- 2.Percival SL, Bowler PG, Russell D. Bacterial resistance to silver in wound care. J Hosp Infect. 2005;60:1–7. doi: 10.1016/j.jhin.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 3.Ercal N, Gurer-Orhan H, Aykin-Burns N. Toxic metals and oxidative stress part I: mechanisms involved in metal induced oxidative damage. Curr. Topics Med Chem. 2001;1:529–539. doi: 10.2174/1568026013394831. [DOI] [PubMed] [Google Scholar]
- 4.Tseng TT, Gratwick KS, Kollman J, Park D, Nies DH, Goffeau A, Saier MH., Jr The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development protein. J Mol Microbiol Biotechnol. 1999;1:107–125. [PubMed] [Google Scholar]
- 5.Nies DH. Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol Rev. 2003;27:313–339. doi: 10.1016/S0168-6445(03)00048-2. [DOI] [PubMed] [Google Scholar]
- 6.Zgurskaya H, Nikaido H. Bypassing the periplasm: reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proc Natl Acad Sci USA. 1999;96:7190–7195. doi: 10.1073/pnas.96.13.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishino K, Yamaguchi A. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J Bacteriol. 2001;183:5803–5812. doi: 10.1128/JB.183.20.5803-5812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- 9.Yu EW, McDermott G, Zgruskaya HI, Nikaido H, Koshland DE., Jr Structural basis of multiple drug binding capacity of the AcrB multidrug efflux pump. Science. 2003;300:976–980. doi: 10.1126/science.1083137. [DOI] [PubMed] [Google Scholar]
- 10.Yu EW, Aires JR, McDermott G, Nikaido H. A periplasmic drug-binding site of the AcrB multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J Bacteriol. 2005;187:6804–6815. doi: 10.1128/JB.187.19.6804-6815.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature. 2006;443:173–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- 12.Seeger MA, Schiefner A, Eicher T, Verrey F, Dietrichs K, Pos KM. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science. 2006;313:1295–1298. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- 13.Sennhauser G, Amstutz P, Briand C, Storchengegger O, Grütter MG. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 2007;5:e7. doi: 10.1371/journal.pbio.0050007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das D, Xu QS, Lee JY, Ankoudinova I, Huang C, Lou Y, Degiovanni A, Kim R, Kim SH. Crystal structure of the multidrug efflux transporter AcrB at 3.1 Å resolution reveals the N-terminal region with conserved amino acids. J Struct Biol. 2007;158:494–502. doi: 10.1016/j.jsb.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su C-C, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, Yu EW. Conformation of the AcrB multidrug efflux pump in mutants of the putative proton relay pathway. J Bacteriol. 2006;188:7290–7296. doi: 10.1128/JB.00684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Törnroth-Horsefield S, Gourdon P, Horsefield R, Brive L, Yamamoto N, Mori H, Snijder A, Neutze R. Crystal structure of AcrB in complex with a single transmembrane subunit reveals another twist. Structure. 2007;15:1663–1673. doi: 10.1016/j.str.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg EY, Ma D, Nikaido H. AcrD of Escherichia coli is an aminoglycoside efflux pump. J Bacteriol. 2000;182:1754–1756. doi: 10.1128/jb.182.6.1754-1756.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aires JR, Nikaido H. Aminoglycosides are captured from both periplasm and cytoplasm by the AcrD multidrug efflux transporter of Escherichia coli. J Bacteriol. 2005;187:1923–1929. doi: 10.1128/JB.187.6.1923-1929.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst E. Molecular cloning of acrA and acrE genes of Escherichia coli. J Bacteriol. 1993;175:6299–6313. doi: 10.1128/jb.175.19.6299-6313.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lau SY, Zgurskaya HI. Cell division defects in Escherichia coli deficient in the multidrug efflux transporter AcrEF-TolC. J Bacteriol. 2005;187:7815–7825. doi: 10.1128/JB.187.22.7815-7825.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baranova N, Nikaido H. The BaeSR two-component regulatory system activates transcription of yegMNOB (mdtABCD) transporter gene cluster in Escherichia coli and increases its resistance to novobiocin and deoxycholate. J Bacteriol. 2002;184:4168–4176. doi: 10.1128/JB.184.15.4168-4176.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagakubo S, Nishino K, Hirata T, Yamaguchi A. The putative response regulator BaeR stimulates multidrug resistance of Escherichia coli via a novel multidrug exporter system, MdtABC. J Bacteriol. 2002;184:4161–4167. doi: 10.1128/JB.184.15.4161-4167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bohnert JA, Schuster S, Fähnrich E, Trittler R, Kern WV. Altered spectrum of multidrug resistance associated with a single point mutation in the Escherichia coli RND-type MDR efflux pump YhiV (MdtF) J Antimicrob Chemother. 2007;6:1216–1222. doi: 10.1093/jac/dkl426. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi A, Hirakawa H, Hirata T, Nishino K, Yamaguchi A. Growth phase-dependent expression of drug exporters in Escherichia coli and its contribution to drug tolerance. J Bacteriol. 2001;16:5693–5703. doi: 10.1128/JB.00217-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franke S, Grass G, Nies DH. The product of the ybdE gene of the Escherichia coli chromosome is involved in detoxification of silver ions. Microbiol. 2001;147:965–972. doi: 10.1099/00221287-147-4-965. [DOI] [PubMed] [Google Scholar]
- 26.Franke S, Grass G, Rensing C, Nies DH. Molecular analysis of the copper-transporting efflux system CusCFBA of Escherichia coli. J Bacteriol. 2003;185:3804–3812. doi: 10.1128/JB.185.13.3804-3812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue Y, Davis AV, Balakrishnan G, Stasser JP, Staehlin BM, Focia P, Spiro TG, Penner-Hahn JE, O'Halloran TV. Cu(I) recognition via cation-π and methionine interactions in CusF. Nature Chem Biol. 2008;4:107–109. doi: 10.1038/nchembio.2007.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loftin IR, Franke S, Blackburn NJ, McEvoy MM. Unusual Cu(I)/Ag(I) coordination of Escherichia coli CusF as revealed by atomic resolution crystallography and x-ray absorption spectroscopy. Protein Sci. 2007;16:2287–2293. doi: 10.1110/ps.073021307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sennhauser G, Bukowska MA, Briand C, Grütter MG. Crystal structure of the multidrug exporter MexB from Pseudomonas aeruginosa. J Mol Biol. 2009;389:134–145. doi: 10.1016/j.jmb.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405:914–919. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- 31.Akama H, Kanemaki M, Yoshimura M, Tsukihara T, Kashiwag T, Yoneyama H, Narita S, Nakagawa A, Nakae T. Crystal structure of the drug discharge outer membrane protein, OprM, of Pseudomonas aeruginosa. J Biol Chem. 2004;279:52816–52819. doi: 10.1074/jbc.C400445200. [DOI] [PubMed] [Google Scholar]
- 32.Mikolosko J, Bobyk K, Zgurskaya HI, Ghosh P. Conformational flexibility in the multidrug efflux system protein AcrA. Structure. 2006;14:577–587. doi: 10.1016/j.str.2005.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Higgins MK, Bokma E, Koronakis E, Hughes C, Koronakis V. Structure of the periplasmic component of a bacterial drug efflux pump. Proc Natl Acad Sci USA. 2004;101:9994–9999. doi: 10.1073/pnas.0400375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akama H, Matsuura T, Kashiwag S, Yoneyama H, Narita S, Tsukihara T, Nakagawa A, Nakae T. Crystal structure of the membrane fusion protein, MexA, of the multidrug transporter in Pseudomonas aeruginosa. J Biol Chem. 2004;279:25939–25942. doi: 10.1074/jbc.C400164200. [DOI] [PubMed] [Google Scholar]
- 35.Symmons M, Bokma E, Koronakis E, Hughes C, Koronakis V. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc Natl Acad Sci USA. 2009;106:7173–7178. doi: 10.1073/pnas.0900693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su C-C, Yang F, Long F, Reyon D, Routh MD, Kuo DW, Mokhtari AK, Van Ornam JD, Rabe KL, Hoy JA, Lee YJ, Rajashankar KR, Yu EW. Crystal structure of the membrane fusion protein CusB from Escherichia coli. J Mol Biol. 2009;393:342–355. doi: 10.1016/j.jmb.2009.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Long F, Su C-C, Zimmermann MT, Boyken SE, Rajashankar KR, Jernigan RL, Yu EW. Crystal structures of the CusA efflux pump suggest methionine-mediated metal transport. Nature. 2010;467:484–488. doi: 10.1038/nature09395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yum S, Xu Y, Piao S, Sim S-H, Kim H-M, Jo W-S, Kim K-J, Kweon H-S, Jeong M-H, Lee K, Ha N-C. Crystal structure of the periplasmic component of a tripartite macrolide-specific efflux pump. J Mol Biol. 2009;387:1286–1297. doi: 10.1016/j.jmb.2009.02.048. [DOI] [PubMed] [Google Scholar]
- 39.De Angelis F, Lee JK, O'Connell JD, III, Miercke LJW, Verschueren KH, Srinivasan V, Bauvois C, Govaerts C, Robbins RA, Ruysschaert J-M, Stroud RM, Vandenbussche G. Metal-induced conformational changes in ZneB suggest an active role of membrane fusion proteins in efflux resistance systems. Proc Natl Acad Sci USA. 2010;107:11038–11043. doi: 10.1073/pnas.1003908107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borges-Walmsley MI, Beauchamp J, Kelly SM, Jumel K, Candlish D, Harding SE, Price NC, Walmsley AR. Identification of oligomerization and drug-binding domains of the membrane fusion protein EmrA. J Biol Chem. 2003;278:12903–12912. doi: 10.1074/jbc.M209457200. [DOI] [PubMed] [Google Scholar]
- 41.Bagai I, Liu W, Rensing C, Blackburn NJ, McEvoy MM. Substrate-linked conformational change in the periplasmic component of a Cu(I)/Ag(I) efflux system. J Biol Chem. 2007;282:35695–35702. doi: 10.1074/jbc.M703937200. [DOI] [PubMed] [Google Scholar]
- 42.Zhou H, Thiele DJ. Identification of a novel high affinity copper transport complex in the fission yeast Schizosaccharomyces pombe. J Biol Chem. 2001;276:20529–20535. doi: 10.1074/jbc.M102004200. [DOI] [PubMed] [Google Scholar]
- 43.Jiang J, Nadas IA, Kim MA, Franz KJ. A Mets motif peptide found in copper transport proteins selectively binds Cu(I) with methionine-only coordination. Inorg Chem. 2005;44:9787–9794. doi: 10.1021/ic051180m. [DOI] [PubMed] [Google Scholar]
- 44.Changela A, Chen K, Xue Y, Holschen J, Outten CE, O'Halloran TV, Mondragón A. Molecular basis of metal-ion selectivity and zeptomolar sensitivity by CueR. Science. 2003;301:1383–1387. doi: 10.1126/science.1085950. [DOI] [PubMed] [Google Scholar]
- 45.Arnesano F, Banci L, Bertini I, Huffman DL, O'Halloran TV. Solution structure of the Cu(I) and apo-forms of the yeast metallochaperone, Atx1. Biochemistry. 2001;40:1528–1539. doi: 10.1021/bi0014711. [DOI] [PubMed] [Google Scholar]
- 46.Goldberg M, Pribyl T, Juhuke S, Nies DH. Energetics and topology of CzcA, a cation/proton antiporter of the resistance-nodulation-cell division protein family. J Biol Chem. 1999;274:26065–26070. doi: 10.1074/jbc.274.37.26065. [DOI] [PubMed] [Google Scholar]
- 47.Piddock LJV. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin Microbiol Rev. 2006;19:382–402. doi: 10.1128/CMR.19.2.382-402.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]