

Abstract
Proteins that switch conformations in response to a signaling event (e.g., ligand binding or chemical modification) present a unique solution to the design of reagent-free biosensors as well as molecules whose biological functions are regulated in useful ways. The principal roadblock in the path to develop such molecules is that the majority of natural proteins do not change conformation upon binding their cognate ligands or becoming chemically modified. Herein, we review recent protein engineering efforts to introduce switching properties into binding proteins. By co-opting natural allosteric coupling, joining proteins in creative ways and formulating altogether new switching mechanisms, researchers are learning how to coax conformational changes from proteins that previously had none. These studies are providing some answers to the challenging question: how can one convert a lock-and-key binding protein into a molecular switch?
Keywords: folding, unfolding, intrinsic disorder, allostery, sensor, circular permutation, alternate frame folding
Introduction
A biomolecular switch is a protein or nucleic acid that can change between two or more distinct conformations in response to a stimulus. These molecules are useful because they can transduce a variety of biological signals into an equally diverse assortment of functional responses. Examples of input signals include ligand binding, light absorption, covalent modification, and changes in pH or temperature. To generate a functional output, each conformation is typically associated with a different biological activity. For instance, one form may be catalytically active, whereas the other is not, or they may fluoresce at different wavelengths. Indeed, the development of molecular switches has been largely driven by two applications—biosensing and regulating protein function—and this review discusses protein switching mechanisms with these purposes in mind.
The biosensor field in particular has seen tremendous growth over the past decade, fueled by the desire to detect disease markers, pathogens, environmental toxins, and chemical/biological threats to security. A biosensor is a detection device consisting of a biological recognition element and a transducer. Proteins possess three properties that make them ideal recognition modules. First, they typically contact their substrates via a large, three-dimensional surface. This extensive interface enables binding to be both tight and highly specific. Second, proteins can be made to bind many targets of interest. Nature already provides us with an abundance of pre-existing binding proteins, as well as a means to generate new proteins (in the form of antibodies) that bind previously unrecognized ligands. Importantly, for small protein scaffolds, the latter process can be carried out in vitro by directed evolution methods.1 The basic problem, however, is that most proteins do not change conformation upon binding their cognate ligands (or absorbing light, changing pH/temperature, etc.). We refer to this type of binding as lock-and-key [Fig. 1(A)]. The foremost challenge facing the researcher who wishes to build a functional switch is therefore to transduce the binding event or other stimulus into a measurable output signal.
Figure 1.
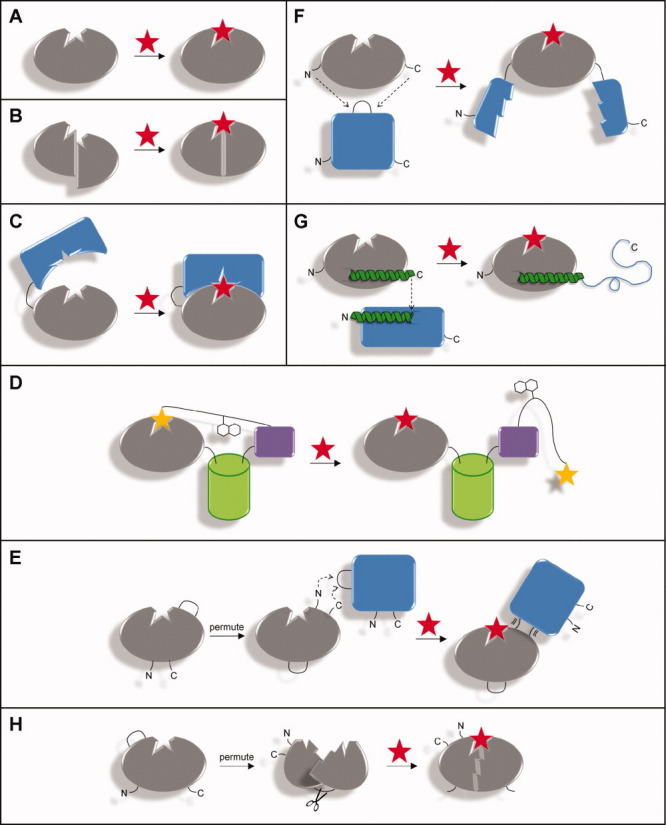
Approaches for converting a binding protein into a switch. The binding protein is shown in gray and the ligand as a red star. (A) Lock-and-key binding of an unmodified protein, in which the free and bound conformations are identical. (B) Altering specificity of a protein with a pre-existing conformational change. (C) Affinity clamp technology. An enhancer domain (blue) is fused to the binding protein via a short linker (black line). The open and closed forms are shown at left and right, respectively. (D) SNAP-tag methodology. The binding protein is joined to an FP (green) and SNAP (purple). A synthetic molecule, consisting of a chemical analog of the ligand (yellow star), a fluorescent acceptor, and O6-benzylguanine (covalently bound to SNAP) links the binding protein to SNAP. Displacement of the ligand analog causes the protein to switch from the closed (left) to the open (right) form. (E) Protein-in-protein insertion with circular permutation. In this example, the binding protein is first permuted so that its new termini are positioned close to the binding site. It is then inserted into a surface loop of the reporter protein (blue), to which binding-induced conformational changes are propagated through the nascent linkage. (F) Mutually exclusive folding by domain insertion. Ligand binding causes the binding protein to fold, which mechanically unfolds the reporter protein (blue). (G) Overlapping sequences. The C-terminus of the binding protein and the N-terminus of the reporter protein (blue) contain an α-helix of similar amino acid sequence. The C-terminus of the binding protein is linked to the N-terminus of the reporter protein such that they share a portion of that α-helix. Ligand binding enables the former to “steal” the helix from the latter. (H) Forced circular permutation. Circularly permuting the protein with a short linker forces its original termini together, unfolding it or distorting the binding site. Chemical or enzymatic cleavage eliminates the conformational strain and restores binding.
The third quality that proteins possess addresses this need and is of primary interest to this review. Proteins are unique in nature in that they offer multiple built-in mechanisms for reversible switching. Binding can induce an allosteric conversion between two or more native structures, as in the well-known (but rare) examples of hemoglobin and calmodulin. The folding reaction is an even more dramatic conformational change that is ubiquitous to all proteins. Because three-dimensional binding interfaces typically exist only when the protein is folded, binding and folding are naturally coupled processes. An example of this phenomenon occurs in nature in the form of intrinsically disordered proteins (IDPs). It has become clear that a significant percentage of proteins in the cell (perhaps as high as 55%)2 contain regions of intrinsic disorder, which may become structured upon binding.3,4
There are two general approaches for transducing a protein's binding event into a detectable signal. The first is to immobilize it on an optical, electrochemical, or piezoelectric device. A binding-dependent conformational change is not necessary and in most cases none occurs. Rather, binding is recorded by the difference in optical activity, mass, index of refraction, or charge between the free receptor and the receptor–ligand complex. The recognition molecules can usually be used with minimal modification, although it can be thorny to affix them to the surface of the transducer without diminishing their biological function. For a more detailed discussion of these technologies, the reader is referred to recent articles on the subject.5–9 The second strategy is to use a single protein as both the recognition and transduction element. Combining these functionalities reduces the need for complex and expensive detection equipment as well as potential problems associated with surface adsorption. Moreover, it opens the door for the creation of hybrid proteins in which biological function is coupled to molecular recognition in new and creative ways. The hurdle with this approach is that much of the engineering burden is transferred to the design of the biomolecule.
Switchable proteins offer a solution to this challenge. Researchers are learning how to build switches by co-opting existing allosteric mechanisms (reviewed recently in Ref.10–15). Less well understood is how one can introduce switching properties into proteins that previously had none. This issue is crucial to the further development and widespread availability of biological sensors. Herein, we focus on one specific question: how can one convert an ordinary binding protein—even one that exhibits lock-and-key binding—into a molecular switch?
Altering Specificity of Proteins that Exhibit Existing Conformational Change
Perhaps the most well known demonstration of how to turn a binding protein into a biosensor is the conversion of calmodulin into cameleon, a fluorescent calcium sensor.16 The general idea behind cameleon and related sensors is to attach environment or distance-sensitive chromophores at positions that respond to a conformational change between free and bound states. This method is reliable when the conformational change is large, as shown schematically in Fig. 1(B). In the case of cameleon, the change is extreme: calcium binding causes the calmodulin domain to wrap around the M13 peptide, which is fused to its C-terminus. The N- and C-termini of the molecule approach each other as a result. A donor fluorescent protein (FP; e.g., cyan FP) and acceptor FP (e.g., yellow FP) attached each terminus report on this event by an increase in Förster resonance energy transfer (FRET). Cameleon is therefore ratiometric as well as genetically encoded, which are the two highly desirable characteristics of a biological sensor. A similar approach was used to create sensors for phosphorylated peptides.17–20
Calmodulin is an example of a protein whose allosteric conformational change is obvious and dramatic. This type of highly tailored switching mechanism is not easily transferred to other proteins. What is needed is a protein that changes conformation upon binding its cognate ligand, but whose binding pocket can be modified to accommodate alternate substrates [Fig. 1(B)]. This is not the same as turning an arbitrary binding protein into a switch, but it does offer the potential for converting an existing switch to one that responds to an arbitrary ligand. Hellinga and coworkers created a family of biosensors based on the bacterial periplasmic binding protein (PBP) scaffold.21,22 PBPs contact their substrates (mostly amino acids and small sugars) in a cleft between two domains, which close by ∼30° upon binding. They reported using computational methods to redesign the binding sites of various PBPs to recognize TNT, l-lactate, serotonin, and pinacolyl methyl phosphonic acid (the hydrolytic product of the nerve agent soman).23,24 These studies have been challenged, however, by the finding that several of the re-engineered PBPs do not appear to bind to their targets.25 At the present time, design of binding sites seems to be best tackled by directed evolution techniques, although computational methods are improving rapidly.26
Affinity Clamps
One recent approach in which directed evolution figures prominently is the affinity clamp methodology of Koide.27–29 An existing binding domain (capture domain) is joined via a short linker to a small antibody-like protein [enhancer domain; Fig. 1(C)]. The newly-formed domain interface is then optimized by randomizing the antigen binding loops of the surrogate antibody and panning the library using phage display. When the capture domain was PDZ, which binds a short peptide from a p120-related catenin with low-micromolar affinity, and the enhancer was fibronectin type III domain, the resulting clamp bound the peptide substrate with low nanomolar affinity and high specificity. Importantly, binding can be detected by attaching FPs to the N- and C-termini, in much the same manner as cameleon. Affinity clamp technology is significant because capture domains can be mixed and matched with different enhancer domains to maximize chances of success.
SNAP-Tag
Johnsson and coworkers have devised a semisynthetic method for eliciting a similar closed-to-open conformational change from a generic binding protein.30 The genetically encoded portion of their sensor is comprised of three proteins joined end-to-end: SNAP-tag (a small protein based on O6-alkylguanine-DNA alkyltransferase), an FP, and the binding protein of interest [Fig. 1(D)]. The synthetic component is an artificial molecule consisting of O6-benzylguanine on one end, the ligand to the binding protein on the other end, and a fluorescent acceptor group in the middle. The synthetic molecule acts as a tether; it binds covalently to the SNAP-tag at one end and noncovalently to the binding protein at the other. This arrangement defines the closed state wherein the FP and the fluorescent acceptor group are close in space. Binding of the natural ligand displaces the synthetic molecule from the binding protein, which causes the protein to assume the open conformation and the donor–acceptor distance to increase. The readout is a ratiometric change in FRET signal. The SNAP-tag technique is powerful because it is well-suited for sensing and imaging in vivo as long as the artificial tether is readily synthesizable and cell permeable. For metabolites and other small ligands this is likely to be the case.
Naturally Occuring Fold Switches
Although proteins generally adopt a single structure that corresponds to the global minimum of free energy, some can sample alternate native conformations whose energy minima are only slightly higher. If one fold binds a ligand with higher affinity than the others, then such a protein can exhibit switching behavior. A striking example is provided by proteins that can switch between unrelated folds.31 Lymphotactin adopts the canonical α/β chemokine fold at low temperature and in the presence of NaCl.32 At higher temperature and lower salt concentration, however, it takes on a completely different all-β structure.33 The switch is driven by binding, as the all-β form is dimeric. The mitotic arrest deficiency two protein (Mad2) interconverts between two functionally distinct conformations (O-Mad2 and C-Mad2).34 Binding of C-Mad2 (as part of the C-Mad2/Mad1 complex) to a partially unfolded form of O-Mad2 is thought to catalyze the conversion of the latter to C-Mad2.34 Still other proteins can be induced to switch conformations by changing a relatively small number of amino acids, as demonstrated by the classic experiments of Sauer with Cro and Arc repressors,35,36 Regan with protein G,37 and more recently, Cordes with Cro homologs.38
The above examples illustrate the extent to which proteins are capable of rearranging their structures. However, it is still largely out of reach to engineer this type of fold-switching mechanism into a generic binding protein. One notable exception is the design of metal-dependent switches.13 Here, efforts are aided by the relative simplicity of metal binding sites. Zinc, for example, is coordinated by four atoms (typically supplied by side chains of His and/or Cys) arranged in tetrahedral geometry. Several groups have created proteins that switch to zinc finger39,40 or helix-loop-helix41 conformations from different folded structures, in response to zinc binding.
Internal Protein-in-Protein Fusions
When one wishes to fuse two proteins in such a way as to minimize their potential interaction, one typically links the C-terminus of the first to the N-terminus of the second using a long, structureless peptide tether. Common examples include attaching glutathione S-transferase or an FP to one of the ends of a target protein to facilitate its purification or to visualize its cellular localization, respectively. In such applications, the goal is for each protein to be oblivious to the fact that it is covalently bonded to the other. Conversely, joining two proteins in a more intimate fashion—at an internal position and by using short linkers—increases the likelihood that changes in one domain will be communicated to the second domain; for example, ligand binding to a receptor domain effecting a structural or functional change in a reporter domain [Fig. 1(E)].11,12 This type of communication requires the presence of a binding-induced conformational change. However, the change can be less dramatic than in proteins such as cameleon because even a subtle structural change can alter optical or enzymatic activity of the attached protein,42,43 sometimes in a manner that was not forseen.44
An effective strategy for optimizing interdomain communication is to insert the reporter into the receptor at a location near the receptor's ligand binding site, or to insert the receptor into the reporter at a location near the reporter's active site. It is desirable to place the “guest” protein into a surface loop of the “host” protein (rather than into an α-helix or β-strand) to avoid perturbing the structure of the host. Linker length is likewise a critical factor: a very long linker decouples interdomain communication, whereas a very short linker can compress the guest and stretch the host. This effect, which occurs when the N-to-C distance of the guest exceeds the distance between the ends of the loop in the host, can itself be exploited as a switching mechanism (see Mutually Exclusive Folding). A more gentle means of insertion is to first circularly permute the guest. Circular permutation creates a new surface loop connecting the original termini, and new termini at a chosen site elsewhere in the protein [usually at a former surface loop; Fig. 1(E)]. Thus, the termini of a permuted protein are always close in space. Not only does permutation help ensure that the host–guest loop distances are compatible, it also adds a valuable combinatorial aspect to the process. All told, one can experiment with the following parameters to optimize coupling: permutation site in the guest, linker used for permutation, insertion site in the host, and linkers used to join the proteins.
Not surprisingly, many of the most successful implementations of protein-in-protein switches have leveraged the power of genetic screens or selections to optimize the design. FPs, and to a lesser extent luminescent proteins, have figured prominently in this regard due to their compatibility with cell sorting and other rapid screening methods. Tsien and coworkers made a key advance when they discovered that green FP (GFP) can tolerate insertions of foreign sequences in the loop around Tyr145, and that GFP can be permuted in numerous locations, while remaining fluorescent.45 They inserted calmodulin and a zinc finger domain from zif268 at position 145 and observed that metal binding changed the GFP chromophore environment such that fluorescence was enhanced. Reversing the host and the guest, Nagai et al. inserted circularly permuted GFP in between calmodulin and the M13 peptide to create a family of calcium sensors called pericams.43 The spectral properties of GFP were altered by calcium binding but the structural basis for these changes remains unclear. To illustrate this point, one variant became brighter upon calcium binding, another became dimmer, and a third exhibited a ratiometric response in which the emission wavelength shifted. They were able to elicit these different behaviors by mutating a few amino acids near the chromophore and by changing the length and sequence of the linker used to permute GFP. Fan et al. inserted the cAMP-binding domain B from protein kinase A into a circular permutant of firefly luciferase.46 By experimenting with different permutation sites as well as linkers used to join the N- and C-termini, they generated a sensor that increased its luminescence by 19-fold in the presence of cAMP.
Enzymes have also drawn attention as reporter domains, because they allow binding to be transduced into a functional output. Ostermeier and coworkers screened large libraries of mutants (∼106) in which β-lactamase was randomly permuted and inserted into random or specific positions in maltose binding protein.47–49 By monitoring how well cells grew in the presence of ampicillin and maltose they were able to identify several variants whose enzymatic activity was inhibited or enhanced by sugar binding by as much as 600-fold. Edwards et al. took a related approach to create a cytochrome b562/β-lactamase fusion in which tolerance to antibiotic was dependent on heme in the growth medium.50
Mutually Exclusive Folding
When constructing protein-in-protein fusions, one can take the opposite tack and deliberately make the N-to-C distance of the guest much larger than the end-to-end distance of the loop in the host. This condition results in a structural tug-of-war in which the folding free energy of one protein is used to mechanically unfold the other [Fig. 1(F)]. This effect was demonstrated by inserting ubiquitin (38 Å N-to-C distance)51–53 or the GCN4 DNA binding domain (75 Å N-to-C distance)54 into a surface loop of barnase (10 Å end-to-end loop distance). When the tethering linkers are sufficiently short, then only one protein can be folded at any given time. Peng and Li were able to directly observe this tug-of-war in another chimera (the 27th Ig domain of titin inserted into the GB1-L5 protein) using fluorescence and atomic force microscopy.55
Thermodynamic and structural coupling between domains provides a pathway for transducing binding to conformational change. For example, the presence of a cognate DNA sequence causes GCN4 to fold and barnase to unfold and lose its ribonuclease activity.54 Barnase is thus converted to a DNA-dependent molecular switch. With regard to generality of the design, the extent of coupling is determined chiefly by ratio of the guest and host distances mentioned above. Very long linkers decouple the domains such that they fold independently. Mutually exclusive folding appears to require a minimum guest:host distance of ∼2:1, where the length of any linkers (in an extended conformation) used to join the proteins is added to the host distance.53 One potential caveat to this design is that some host proteins, upon unfolding by the guest, may refold with a second, identically unfolded host molecule by a three-dimensional domain-swap mechanism. In this case, the switch is from a monomeric protein to a dimeric (or oligomeric) protein. Ubiquitin-unfolded and GCN4-unfolded barnase may form domain-swapped dimers,53,54 but Ig-unfolded GB1-L5 apparently does not.55
Overlapping Sequences
If two proteins contain a similar stretch of amino acids, then it may be possible to join them such that they compete for that common sequence. Ligand binding can then drive one protein to fold and the other to unfold in a manner akin to that of mutually exclusive folding [Fig. 1(G)]. Sallee et al. interrogated the protein data bank for pairs of ligand-binding domains whose N- and C-termini partially overlap.56 They constructed 25 switches consisting of overlaps of two domains, one domain and one peptide, or two peptides, and tested them for mutually exclusive binding of the two ligands. Peptide–peptide constructs were the most successful switches and domain–domain fusions were the least successful. This trend is likely explained by the fact that domains are folded and their stabilities can be quite disparate, so one conformation of the switch can dominate regardless of whether the ligand is present or not. “Tuning” the stabilities of the two domains so that their populations change in the presence of ligand is critical to all strategies that link binding to folding; we will return to that concept in Optimizing Switch Response.
Competing for a shared sequence need not engender mutually exclusive folding behavior to effect switching. At the C-terminus of the photoactive LOV2 domain of Avena sativa phototropin-1 is a 20-residue α-helix that dissociates and unfolds on exposure to light. Strickland et al. joined LOV2 to the N-terminus of Eschericia coli Trp repressor (TrpR) to generate the LovTAP chimera.57 They truncated the first 11 amino acids of the 21-residue N-terminal helix of TrpR to make the fusion, thus effectively linking the two proteins via a shared helix. Both proteins remain folded because the C-terminal helix is only weakly associated with the core of LOV2. Nevertheless, the LOV2 C-terminal helix is able to “steal” a few residues from the N-terminal helix of TrpR, reducing its DNA binding affinity. Upon receipt of a photon by the LOV2 domain, the TrpR domain reclaims those residues and DNA binding is restored. Woolley and coworkers created another photoswitchable DNA binding protein by fusing photoactive yellow protein (PYP) to the C-terminus of GCN4.58,59 They replaced the N-terminal 25 amino acids of PYP with those of the GCN4 coiled-coil domain, aligned in such a way as to maximize the sequence similarity between the two proteins. Similar to LovTAP, absorption of a photon causes partial unfolding of PYP and activation of DNA binding.
The sequence overlap approach described above is limited to pairs of proteins that share common sequences at their ends. Several related strategies, however, use overlap within a single protein to achieve switching. Matthews and coworkers duplicated in tandem the 10-residue helix B of T4 lysozyme.60,61 The C-terminus of helix B is stabilized by a loop that includes Arg63. Mutation of Arg63 to Ala destabilized the C-terminal copy of helix B, causing it to partially unfold and form an extended, flexible loop. Addition of guanidinium stabilized the C-terminal copy of helix B by binding to the loop in place of the Arg63 side chain. As a consequence, the C-terminal copy of helix B became fully helical and the N-terminal copy partially unfolded and looped out into solution. The net result was that the helix translocated by ∼20 Å. The extent to which this design can be applied to other proteins depends on the ability to identify or engineer binding sites at locations flanking the duplicated amino acid sequence. Another duplication strategy, termed as alternate frame folding (AFF), addresses this limitation and is discussed below.
Irreversible Switches
When cells require a signaling event to produce the strongest, longest lasting response possible, they turn to protein switches that are triggered irreversibly. Examples include serpins and the zymogen class of enzymes (e.g., caspases and blood clotting enzymes). In both cases, the signal is site-specific cleavage of the polypeptide chain, which turns on the catalytic and inhibitory activities of the respective proteins. Taking this cue from nature, researchers have developed proteins, which function as artificial zymogens or as sensors of specific protease activity. Ribonucleases have been the target of several studies because of their toxicity to human cells and certain pathogens. If their activity can be suppressed in healthy cells and unleashed only in diseased cells, then RNases offer the potential to treat diseases such as cancer and viral infections. Raines and coworkers circularly permuted pancreatic RNase A and bridged its original termini with peptides that contained cleavage sequences for HIV-1 protease, NS3 protease from hepatitis C, and plasmepsin II from Plasmodium falciparium.62–64 The linkers reduced catalytic efficiency of the enzyme by partially occluding the active site. Proteolytic cleavage increased kcat/KM by ∼100-fold. In a technically similar but mechanistically different approach, Butler et al. circularly permuted barnase using very short linkers to force the original termini together and thereby introduce conformational strain into the molecule [Fig. 1(H)].65 Cleaving the linker with a chemical reagent relieved the strain and increased catalytic efficiency.
Although the above strategies can be successful for some proteins, they rely on specific properties of the target enzyme and are therefore not general. The linker used to circularly permute an enzyme does not typically obscure the active site, nor is cleaving the linker guaranteed to remove the obstruction. Similarly, N- to C-terminal distances in many proteins are too short to induce significant conformational strain when linked. Even in the case where this distance was compressed from 27 Å to that of a single amino acid, the protein structure was able to absorb the strain and enzymatic activity remained high.65 Mitrea et al. recently introduced a switching mechanism that addresses the issue of generality.66 We defer discussion of this study to the AFF section below.
Binding-Induced Folding
Intrinsically disordered proteins
In our view, binding-induced folding is the most generally applicable strategy for converting a binding protein into a switch. The reasons are as follows. (i) Proteins constantly cycle through all possible conformations—from native to partially folded to unfolded—according to their Boltzmann distributions. Thus, even a lock-and-key protein is already a reversible switch, though it spends nearly all of its time in one state. (ii) It is straightforward to convert a folded protein into a pseudo-IDP by manipulating the relative populations of native and unfolded forms. The latter state can be made dominant by decreasing stability of the former by point mutation, truncation, insertion, and so forth. Indeed, we have already seen examples in mutually exclusive folding and overlapping sequences. The native conformation can then be restored by ligand binding, which, as mentioned, is thermodynamically linked to folding. In principle, any protein is capable of such a reversible transformation. (iii) The folding reaction is the most dramatic conformational change that a protein undergoes. So many of its properties change (structure, dynamics, and charge distribution to name a few) that it is likely that a means to detect binding can be devised.67 As a simple example, one can attach donor and acceptor fluorophores at the N- and C-termini. As the ends of globular proteins are frequently proximal68,69 (or can be made so by circular permutation) and are expected to be more distant (on average) in the unfolded state, binding can potentially be monitored by increase in FRET. Parenthetically, the opposite is true for short peptide “beacons”.67,70–72 These unstructured peptides are designed to bind their targets in a rigid, extended conformation. Binding can be detected by the increase in distance between ends of the peptide as monitored by loss of quenching between fluorescent donor and quencher groups.
Kohn and Plaxco elegantly demonstrated principles of binding-induced folding by progressively deleting amino acids from the C-terminus of the SH3 domain from Fyn tyrosine kinase until it unfolded.73 Binding the cognate phosphorylated peptide induced SH3 to fold, which was detected by fluorescence of the intrinsic Trp residue or that of a BODIPY group previously attached to a strategically placed Cys side chain. Impressively, their sensor worked even when tested in the complex, contaminant-ridden environment of blood serum. Others have taken advantage of intrinsically disordered regions in p5374 and BRCA175 that fold upon binding to create fluorescent sensors for their respective ligands.
The principal challenge with the binding-induced folding paradigm is that destabilized and unfolded proteins tend to aggregate or otherwise misfold. Degradation is also a concern, particularly in cellular applications. Naturally occurring IDPs appear to have evolved the ability to remain soluble when unfolded, likely due (at least in part) to their unusually high content of charged groups and low percentage of hydrophobic residues (2). Destabilization of native proteins, on the other hand, is strongly implicated as the root cause of numerous aggregation-related diseases (reviewed for example in76). Truncated, unfolded SH3 (53 residues) is well-behaved and soluble at >100 μM concentration and was not degraded when expressed in E. coli.73 The extent to which these attributes pertain to large or multidomain proteins remains to be tested.
Alternate frame folding
AFF combines several of the themes discussed earlier, including circular permutation, partial sequence overlap, fold switching, and binding-induced folding. The unique feature of AFF is that it couples binding-induced folding to unfolding of another portion of the molecule.66,77,78 These two reactions are balanced so that no net folding or unfolding occurs; rather, the structure is remodeled. The conformational change is from one native structure to another. The AFF mechanism does not require a reporter protein to be fused to the binding protein. In fact, no foreign sequence is added to the binding protein with the exception of a flexible peptide linker. AFF is designed to convert even a lock-and-key binding protein into a molecular switch.
Figure 2 illustrates the basic steps of this conversion. First, a segment from one of the termini of the protein is duplicated and appended to the opposite end. The position and length of this segment is dictated by two considerations. First, it must contain at least one amino acid which, when mutated, abolishes ligand binding. Beyond that criterion, its length is determined by the preference for the newly generated termini of a circularly permuted protein to reside at a former surface loop. For example, if a critical binding residue is close to the C-terminus, one would choose to duplicate the sequence from the C-terminus to the first surface loop on the other side of the binding residue. This minimizes the length of the twinned segment, which may help discourage aggregation and degradation. Other loop sites can be tested if one needs to optimize stability, solubility, signal response (vide infra), or other properties of the molecule.
Figure 2.

Schematic of the alternate frame folding design. A lock-and-key binding protein of 100 amino acids is shown. The duplicate segment is shown in blue and green. The small stars in each duplicate segment denote copies of the same ligand-binding residue; the black star indicates that it has been mutated to a nonbinding amino acid. The linker used to join the original termini in the circularly permuted fold (N′) is shown as the black bar in between the green and gray segments. Fluorophores (diamond and inverted triangle) are placed at the amino terminus (position 80′) and in the surface loop chosen for the permutation site (position 80) in order to report on the binding-induced N to N′ conformational change.
The additional sequence information gives the folding protein a choice. It can use the normal set of amino acids to generate the WT conformation (N), in which case the duplicate segment extends from the amino terminus as a tail (Fig. 2). It can also fold in an alternate frame, using the second set of amino acids, to yield the circularly permuted structure (N′). Here, the extra residues form a carboxy-terminal tail. The molecule cannot adopt N and N′ conformations simultaneously because each competes for the nonduplicated sequence. Accordingly, the protein is expected to interconvert between the two forms with the equilibrium distribution determined by their relative thermodynamic stabilities. The stabilities are tuned (e.g., by point mutation) such that one form is ∼10-fold more populated than the other in the absence of ligand, and the binding mutation is introduced into the more stable fold. Binding triggers the switch to the alternate fold, in which the remodeled active site presents the correct residue for contact with the ligand. This conformational change is readily harnessed to an optical or functional readout. If one places fluorophores at the positions indicated in Figure 2 then they will always be proximal in N′ because, although distant in sequence, they adopt the equivalent of sequential positions in the WT structure. They are expected to be farther apart in N, depending to some degree on whether the “orphaned” amino-terminal peptide is structured or disordered.
Unlike its larger cousin calmodulin, calbindin D9k does not undergo an appreciable conformational change upon calcium binding.79 Nevertheless, Stratton et al. converted it to a calcium-driven molecular switch (calbindin-AFF) using the AFF methodology.77,78 Calbindin proved to be a propitious choice because N and N′ are approximately equal in stability. This property allowed them to show that the switch could be driven in either direction by transferring a Ca2+-binding mutation from the binding site in N to the binding site in N′. The sign of the fluorescence change (and its inversion upon reversal of the switch), as measured by both pyrene excimer fluorescence and BODIPY self quenching, was consistent with the proposed structural model. Their data suggested that the orphaned peptides were mostly disordered but with some residual structure present.
Mitrea et al. used a similar modification to convert barnase into a functional switch.66 Instead of ligand binding, the input signal was proteolytic cleavage. A carboxy-terminal segment of barnase was duplicated as above, and an HIV-1 protease recognition sequence was inserted into the loop that served as the permutation site. The engineered enzyme, barnase-AFF, was primarily in conformation N, and this fold was rendered catalytically inert by mutating the general acid His102 to Ala. Cleavage by the protease released the carboxy-terminal duplicated peptide, which contained the H102A mutation. Barnase-AFF refolded to the catalytically competent N′ conformation and RNase activity increased by 130-fold.
What are the advantages and disadvantages of AFF compared with traditional binding-induced folding switches? As part of the molecule is always unfolded, AFF is subject to the same concerns of aggregation and degradation expressed earlier. One important distinction, however, is that AFF bypasses populating the globally unfolded state, which can be particularly problematic. The length of the unfolded segment is adjustable. It can be a relatively small portion of the molecule. For example, 30% and 7.5% of the residues in calbindin-AFF and barnase-AFF are involved in their respective folding/unfolding reactions. The additional requirements of AFF are that the protein must tolerate circular permutation and not contain covalent linkages (e.g., disulfide bridges) that would physically prevent the fold shift.
Optimizing switch response
The key properties of any switch are affinity, gain (signal change between bound and free forms), and response time. For folding-based switches, these properties translate to the thermodynamics and kinetics of protein folding. Fortunately, decades of work in this area have laid out a path for tailoring the characteristics of the switch to suit a particular application.78,80 In therapeutic implementations where a toxic protein is involved, it is imperative to rigidly enforce the off-state of the switch. Cytotoxic RNase activity of barnase-AFF was suppressed to nearly undetectable levels by introducing point mutations that selectively destabilized the N′ conformation.66 For biosensor applications, the best balance of robust signal change and high affinity is generally when 50–90% of the protein is unfolded in the absence of ligand. This condition allows for the majority of molecules to fold upon binding. Because binding energy is used to drive the unfavorable folding reaction, the apparent dissociation constant (Kd) is weaker than the intrinsic Kd of the native protein by a factor of (1 + Kf)/Kf, where Kf is the equilibrium constant for folding (assuming two-state unfolding). Thus, apparent affinity is reduced by only 2–10-fold in the example above. In other cases, it may be desirable to reduce affinity further so that Kd matches the concentration of analyte. It is straightforward to achieve the desired Kd by tuning the stabilities of native and unfolded forms (or the N and N′ conformations in the AFF mechanism) using point mutations, truncations, or both. This method of affinity tuning can be preferable to directly modifying the binding site, because the latter process can alter specificity as well.14 Destabilizing mutations can be introduced at locations distant from the binding pocket where they are unlikely to affect specificity.
Response time can be a critical parameter of a sensor, particularly when one needs to monitor rapidly changing levels of analyte in real time. Luckily, small proteins tend to fold quickly. The truncated SH3 sensor responded with a time constant of 10 ms.73 This figure is consistent with the time constant reported for folding of full-length SH3. Calbindin-AFF took longer to respond (several seconds), apparently because an unfolding step is at least partially rate-limiting, and unfolding rates can be slow in native conditions. Nevertheless, the switching rate was significantly faster than the rate of global unfolding, indicating that the entire molecule did not need to denature in order to switch folds. It may be possible to adjust response times by modulating folding and unfolding rates.78
Conclusions
Protein conformational change is a natural and powerful means for coupling an input event to an output signal. Many of the most successful existing designs fuse the binding protein to a reporter domain to propagate a conformational change through the covalent linkage, or to engender an open-to-closed domain movement upon binding. A more general approach may be to link binding to folding (and possibly to partial unfolding) within a single protein molecule. With this strategy, even a lock-and-key protein may be converted into a functional switch.
Acknowledgments
The authors thank Jeung-Hoi Ha, Diana Mitrea, and Huimei Zheng for comments and discussions.
References
- 1.Dougherty MJ, Arnold FH. Directed evolution: new parts and optimized function. Curr Opin Biotechnol. 2009;20:486–491. doi: 10.1016/j.copbio.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci USA. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lubin AA, Plaxco KW. Folding-based electrochemical biosensors: the case for responsive nucleic acid architectures. Acc Chem Res. 2010;43:496–505. doi: 10.1021/ar900165x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White RJ, Plaxco KW. Exploiting binding-induced changes in probe flexibility for the optimization of electrochemical biosensors. Anal Chem. 2010;82:73–76. doi: 10.1021/ac902595f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prieto-Simin B, Campas M, Marty JL. Biomolecule immobilization in biosensor development: tailored strategies based on affinity interactions. Protein Pept Lett. 2008;15:757–763. doi: 10.2174/092986608785203791. [DOI] [PubMed] [Google Scholar]
- 8.North SH, Lock EH, Taitt CR, Walton SG. Critical aspects of biointerface design and their impact on biosensor development. Anal Bioanal Chem. 2010;397:925–933. doi: 10.1007/s00216-010-3637-4. [DOI] [PubMed] [Google Scholar]
- 9.Luong JH, Male KB, Glennon JD. Biosensor technology: technology push versus market pull. Biotechnol Adv. 2008;26:492–500. doi: 10.1016/j.biotechadv.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 10.Ambroggio XI, Kuhlman B. Design of protein conformational switches. Curr Opin Struct Biol. 2006;16:525–530. doi: 10.1016/j.sbi.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 11.Ostermeier M. Designing switchable enzymes. Curr Opin Struct Biol. 2009;19:442–448. doi: 10.1016/j.sbi.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ostermeier M. Engineering allosteric protein switches by domain insertion. Protein Eng Des Sel. 2005;18:359–364. doi: 10.1093/protein/gzi048. [DOI] [PubMed] [Google Scholar]
- 13.Wright CM, Heins RA, Ostermeier M. As easy as flipping a switch? Curr Opin Chem Biol. 2007;11:342–346. doi: 10.1016/j.cbpa.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 14.Vallee-Belisle A, Plaxco KW. Structure-switching biosensors: inspired by Nature. Curr Opin Struct Biol. 2010;20:518–526. doi: 10.1016/j.sbi.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koide S. Generation of new protein functions by nonhomologous combinations and rearrangements of domains and modules. Curr Opin Biotech. 2009;20:398–404. doi: 10.1016/j.copbio.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Ma Y, Taylor SS, Tsien RY. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc Natl Acad Sci USA. 2001;98:14997–15002. doi: 10.1073/pnas.211566798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ting AY, Kain KH, Klemke RL, Tsien RY. Genetically encoded fluorescent reporters of protein tyrosine kinase activities in living cells. Proc Natl Acad Sci USA. 2001;98:15003–15008. doi: 10.1073/pnas.211564598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fosbrink M, Aye-Han NN, Cheong R, Levchenko A, Zhang J. Visualization of JNK activity dynamics with a genetically encoded fluorescent biosensor. Proc Natl Acad Sci USA. 2010;107:5459–5464. doi: 10.1073/pnas.0909671107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey CD, Ehrhardt AG, Cellurale C, Zhong H, Yasuda R, Davis RJ, Svoboda K. A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci USA. 2008;105:19264–19269. doi: 10.1073/pnas.0804598105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dwyer MA, Hellinga HW. Periplasmic binding proteins: a versatile superfamily for protein engineering. Curr Opin Struct Biol. 2004;14:495–504. doi: 10.1016/j.sbi.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 22.De Lorimer RM, Smith JJ, Dwyer MA, Looger LL, Sali KM, Paavola CD, Rizk SS, Sadigov S, Conrad DW, Loew L, Hellinga HW. Construction of a fluorescent biosensor family. Protein Sci. 2002;11:2655–2675. doi: 10.1110/ps.021860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allert M, Rizk SS, Looger LL, Hellinga HW. Computational design of receptors for an organophosphate surrogate of the nerve agent soman. Proc Natl Acad Sci USA. 2004;101:7907–7912. doi: 10.1073/pnas.0401309101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Looger LL, Dwyer MA, Smith JJ, Hellinga HW. Computational design of receptor and sensor proteins with novel functions. Nature. 2003;423:185–190. doi: 10.1038/nature01556. [DOI] [PubMed] [Google Scholar]
- 25.Schreier B, Stumpp C, Wiesner S, Hocker B. Computational design of ligand binding is not a solved problem. Proc Natl Acad Sci USA. 2009;106:18491–18496. doi: 10.1073/pnas.0907950106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, St Clair JL, Gallaher JL, Hilvert D, Gelb MH, Stoddard BL, Houk KN, Michael FE, Baker D. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science. 2010;329:309–313. doi: 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang J, Koide S. Rational conversion of affinity reagents into label-free sensors for peptide motifs by designed allostery. ACS Chem Biol. 2010;5:273–277. doi: 10.1021/cb900284c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang J, Koide A, Makabe K, Koide S. Design of protein function leaps by directed domain interface evolution. Proc Natl Acad Sci USA. 2008;105:6578–6583. doi: 10.1073/pnas.0801097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang J, Makabe K, Biancalana M, Koide A, Koide S. Structural basis for exquisite specificity of affinity clamps, synthetic binding proteins generated through directed domain-interface evolution. J Mol Biol. 2009;392:1221–1231. doi: 10.1016/j.jmb.2009.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brun MA, Tan KT, Nakata E, Hinner MJ, Johnsson K. Semisynthetic fluorescent sensor proteins based on self-labeling protein tags. J Am Chem Soc. 2009;131:5873–5884. doi: 10.1021/ja900149e. [DOI] [PubMed] [Google Scholar]
- 31.Bryan PN, Orban J. Proteins that switch folds. Curr Opin Struct Biol. 2010;20:482–488. doi: 10.1016/j.sbi.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuloglu ES, McCaslin DR, Kitabwalla M, Pauza CD, Markley JL, Volkman BF. Monomeric solution structure of the prototypical 'C' chemokine lymphotactin. Biochemistry. 2001;40:12486–12496. doi: 10.1021/bi011106p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tuinstra RL, Peterson FC, Kutlesa S, Elgin ES, Kron MA, Volkman BF. Interconversion between two unrelated protein folds in the lymphotactin native state. Proc Natl Acad Sci USA. 2008;105:5057–5062. doi: 10.1073/pnas.0709518105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo X, Tang Z, Xia G, Wassmann K, Matsumoto T, Rizo J, Yu H. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol. 2004;11:338–345. doi: 10.1038/nsmb748. [DOI] [PubMed] [Google Scholar]
- 35.Anderson TA, Cordes MH, Sauer RT. Sequence determinants of a conformational switch in a protein structure. Proc Natl Acad Sci USA. 2005;102:18344–18349. doi: 10.1073/pnas.0509349102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mossing MC, Sauer RT. Stable, monomeric variants of lambda Cro obtained by insertion of a designed beta-hairpin sequence. Science. 1990;250:1712–1715. doi: 10.1126/science.2148648. [DOI] [PubMed] [Google Scholar]
- 37.Dalal S, Regan L. Understanding the sequence determinants of conformational switching using protein design. Protein Sci. 2000;9:1651–1659. doi: 10.1110/ps.9.9.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roessler CG, Hall BM, Anderson WJ, Ingram WM, Roberts SA, Montfort WR, Cordes MH. Transitive homology-guided structural studies lead to discovery of Cro proteins with 40% sequence identity but different folds. Proc Natl Acad Sci USA. 2008;105:2343–2348. doi: 10.1073/pnas.0711589105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ambroggio XI, Kuhlman B. Computational design of a single amino acid sequence that can switch between two distinct protein folds. J Am Chem Soc. 2006;128:1154–1161. doi: 10.1021/ja054718w. [DOI] [PubMed] [Google Scholar]
- 40.Hori Y, Sugiura Y. Conversion of antennapedia homeodomain to zinc finger-like domaZn(II)-induced change in protein conformation and DNA binding. J Am Chem Soc. 2002;124:9362–9363. doi: 10.1021/ja026577t. [DOI] [PubMed] [Google Scholar]
- 41.Cerasoli E, Sharpe BK, Woolfson DN. ZiCo: a peptide designed to switch folded state upon binding zinc. J Am Chem Soc. 2005;127:15008–15009. doi: 10.1021/ja0543604. [DOI] [PubMed] [Google Scholar]
- 42.Wright CM, Majumdar A, Tolman JR, Ostermeier M. NMR characterization of an engineered domain fusion between maltose binding protein and TEM1 beta-lactamase provides insight into its structure and allosteric mechanism. Proteins. 2010;78:1423–1430. doi: 10.1002/prot.22657. [DOI] [PubMed] [Google Scholar]
- 43.Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+ Proc Natl Acad Sci USA. 2001;98:3197–3202. doi: 10.1073/pnas.051636098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang J, Kim JR, Boock JT, Mansell TJ, Ostermeier M. Ligand binding and allostery can emerge simultaneously. Protein Sci. 2007;16:929–937. doi: 10.1110/ps.062706007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baird GS, Zacharias DA, Tsien RY. Circular permutation and receptor insertion within green fluorescent protein. Proc Natl Acad Sci USA. 1999;96:11241–11246. doi: 10.1073/pnas.96.20.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fan F, Binkowski BF, Butler BL, Stecha PF, Lewis MK, Wood KV. Novel genetically encoded biosensors using firefly luciferase. ACS Chem Biol. 2008;3:346–351. doi: 10.1021/cb8000414. [DOI] [PubMed] [Google Scholar]
- 47.Guntas G, Mitchell SF, Ostermeier M. A molecular switch created by in vitro recombination of nonhomologous genes. Chem Biol. 2004;11:1483–1487. doi: 10.1016/j.chembiol.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 48.Guntas G, Ostermeier M. Creation of an allosteric enzyme by domain insertion. J Mol Biol. 2004;336:263–273. doi: 10.1016/j.jmb.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 49.Guntas G, Mansell TJ, Kim JR, Ostermeier M. Directed evolution of protein switches and their application to the creation of ligand-binding proteins. Proc Natl Acad Sci USA. 2005;102:11224–11229. doi: 10.1073/pnas.0502673102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edwards WR, Busse K, Allemann RK, Jones DD. Linking the functions of unrelated proteins using a novel directed evolution domain insertion method. Nucleic Acids Res. 2008;36:e78. doi: 10.1093/nar/gkn363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Radley TL, Markowska AI, Bettinger BT, Ha J-H, Loh SN. Allosteric switching by mutually exclusive folding of protein domains. J Mol Biol. 2003;332:529–536. doi: 10.1016/s0022-2836(03)00925-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cutler T, Loh SN. Thermodynamic analysis of an antagonistic folding-unfolding equilibrium between two protein domains. J Mol Biol. 2007;371:308–316. doi: 10.1016/j.jmb.2007.05.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cutler T, Mills BM, Lubin DJ, Chong LT, Loh SN. Effect of interdomain linker length on an antagonistic folding-unfolding equilibrium between two protein domains. J Mol Biol. 2009;386:854–868. doi: 10.1016/j.jmb.2008.10.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ha J-H, Butler JS, Mitrea DM, Loh SN. Modular enzyme design: regulation by mutually exclusive protein folding. J Mol Biol. 2006;357:1058–1062. doi: 10.1016/j.jmb.2006.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peng Q, Li H. Direct observation of tug-of-war during the folding of a mutually exclusive protein. J Am Chem Soc. 2009;131:13347–13354. doi: 10.1021/ja903480j. [DOI] [PubMed] [Google Scholar]
- 56.Sallee NA, Yeh BJ, Lim WA. Engineering modular protein interaction switches by sequence overlap. J Am Chem Soc. 2007;129:4606–4611. doi: 10.1021/ja0672728. [DOI] [PubMed] [Google Scholar]
- 57.Strickland D, Moffat K, Sosnick TR. Light-activated DNA binding in a designed allosteric protein. Proc Natl Acad Sci USA. 2008;105:10709–10714. doi: 10.1073/pnas.0709610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morgan SA, Woolley GA. A photoswitchable DNA-binding protein based on a truncated GCN4-photoactive yellow protein chimera. Photochem Photobiol Sci. 2010 doi: 10.1039/c0pp00214c. [DOI] [PubMed] [Google Scholar]
- 59.Morgan SA, Al-Abdul-Wahid S, Woolley GA. Structure-based design of a photocontrolled DNA binding protein. J Mol Biol. 2010;399:94–112. doi: 10.1016/j.jmb.2010.03.053. [DOI] [PubMed] [Google Scholar]
- 60.Yousef MS, Baase WA, Matthews BW. Use of sequence duplication to engineer a ligand-triggered, long distance molecular switch in T4 lysozyme. Proc Natl Acad Sci USA. 2004;101:11583–11586. doi: 10.1073/pnas.0404482101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sagermann M, Baase WA, Matthews BW. Structural characterization of an engineered tandem repeat contrasts the importance of context and sequence in protein folding. Proc Natl Acad Sci USA. 1999;96:6078–6083. doi: 10.1073/pnas.96.11.6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Plainkum P, Fuchs SM, Wiyakrutta S, Raines RT. Creation of a zymogen. Nat Struct Biol. 2003;10:115–119. doi: 10.1038/nsb884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Turcotte RF, Raines RT. Design and characterization of an HIV-specific ribonuclease zymogen. AIDS Res Human Retro. 2008;24:1357–1363. doi: 10.1089/aid.2008.0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnson RJ, Lin SR, Raines RT. A ribonuclease zymogen activated by the NS3 protease of the hepatitis C virus. FEBS J. 2006;273:5457–5465. doi: 10.1111/j.1742-4658.2006.05536.x. [DOI] [PubMed] [Google Scholar]
- 65.Butler JS, Mitrea DM, Mitrousis G, Cingolani G, Loh SN. Structural and thermodynamic analysis of a conformationally-strained circular permutant of barnase. Biochemistry. 2009;48:3497–3507. doi: 10.1021/bi900039e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mitrea DM, Parsons L, Loh SN. Engineering an artificial zymogen by alternate frame protein folding. Proc Natl Acad Sci USA. 2010;107:2824–2829. doi: 10.1073/pnas.0907668107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oh KJ, Cash KJ, Plaxco KW. Beyond molecular beacons: optical sensors based on the binding-induced folding of proteins and polypeptides. Chemistry. 2009;15:2244–2251. doi: 10.1002/chem.200701748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thornton JM, Sibanda BL. Amino and carboxy-terminal regions in globular proteins. J Mol Biol. 1983;167:443–460. doi: 10.1016/s0022-2836(83)80344-1. [DOI] [PubMed] [Google Scholar]
- 69.Krishna MM, Englander SW. The N-terminal to C-terminal motif in protein folding and function. Proc Natl Acad Sci USA. 2005;102:1053–1058. doi: 10.1073/pnas.0409114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oh KJ, Cash KJ, Hugenberg V, Plaxco KW. Peptide beacons: a new design for polypeptide-based optical biosensors. Bioconjug Chem. 2007;18:607–609. doi: 10.1021/bc060319u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oh KJ, Cash KJ, Lubin AA, Plaxco KW. Chimeric peptide beacons: a direct polypeptide analog of DNA molecular beacons. Chem Commun (Camb) 2007:4869–4871. doi: 10.1039/b709776j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oh KJ, Cash KJ, Plaxco KW. Excimer-based peptide beacons: a convenient experimental approach for monitoring polypeptide-protein and polypeptide-oligonucleotide interactions. J Am Chem Soc. 2006;128:14018–14019. doi: 10.1021/ja0651310. [DOI] [PubMed] [Google Scholar]
- 73.Kohn JE, Plaxco KW. Engineering a signal transduction mechanism for protein-based biosensors. Proc Natl Acad Sci USA. 2005;102:10841–10845. doi: 10.1073/pnas.0503055102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Geddie ML, O'Loughlin TL, Woods KK, Matsumura I. Rational design of p53, an intrinsically unstructured protein, for the fabrication of novel molecular sensors. J Biol Chem. 2005;280:35641–35646. doi: 10.1074/jbc.M508149200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cissell KA, Shrestha S, Purdie J, Kroodsma D, Deo SK. Molecular biosensing system based on intrinsically disordered proteins. Anal Bioanal Chem. 2008;391:1721–1729. doi: 10.1007/s00216-007-1819-5. [DOI] [PubMed] [Google Scholar]
- 76.Luheshi LM, Dobson CM. Bridging the gap: from protein misfolding to protein misfolding diseases. FEBS Lett. 2009;583:2581–2586. doi: 10.1016/j.febslet.2009.06.030. [DOI] [PubMed] [Google Scholar]
- 77.Stratton MM, Mitrea DM, Loh SN. A Ca2+-sensing molecular switch based on alternate frame protein folding ACS. Chem Biol. 2008;3:723–732. doi: 10.1021/cb800177f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stratton MM, Loh SN. On the mechanism of protein fold-switching by a molecular sensor. Proteins. 2010;78:3260–3269. doi: 10.1002/prot.22833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Skelton NJ, Kordel J, Chazin WJ. Determination of the solution structure of apo calbindin D9k by NMR spectroscopy. J Mol Biol. 1995;249:441–462. doi: 10.1006/jmbi.1995.0308. [DOI] [PubMed] [Google Scholar]
- 80.Vallee-Belisle A, Ricci F, Plaxco KW. Thermodynamic basis for the optimization of binding-induced biomolecular switches and structure-switching biosensors. Proc Natl Acad Sci USA. 2009;106:13802–13807. doi: 10.1073/pnas.0904005106. [DOI] [PMC free article] [PubMed] [Google Scholar]