

Abstract
Chaperonins assist in the folding of nascent and misfolded proteins, though the mechanism of folding within the lumen of the chaperonin remains poorly understood. The archeal chaperonin from Methanococcus marapaludis, Mm-Cpn, shares the eightfold double barrel structure with other group II chaperonins, including the eukaryotic TRiC/CCT, required for actin and tubulin folding. However, Mm-Cpn is composed of a single species subunit, similar to group I chaperonin GroEL, rather than the eight subunit species needed for TRiC/CCT. Features of the β-sheet fold have been identified as sites of recognition by group II chaperonins. The crystallins, the major components of the vertebrate eye lens, are β-sheet proteins with two homologous Greek key domains. During refolding in vitro a partially folded intermediate is populated, and partitions between productive folding and off-pathway aggregation. We report here that in the presence of physiological concentrations of ATP, Mm-Cpn suppressed the aggregation of HγD-Crys by binding the partially folded intermediate. The complex was sufficiently stable to permit recovery by size exclusion chromatography. In the presence of ATP, Mm-Cpn promoted the refolding of the HγD-Crys intermediates to the native state. The ability of Mm-Cpn to bind and refold a human β-sheet protein suggests that Mm-Cpn may be useful as a simplified model for the substrate recognition mechanism of TRiC/CCT.
Keywords: chaperone, protein folding, crystallin
Introduction
Molecular chaperones are important elements of protein folding pathways and are necessary for the productive folding of a number of key structural and metabolic proteins in the cell. Among these are proteins critical to cell survival, including tubulin and actin in all eukaryotes, and rubisco in plants.1–4 The chaperone superfamily consists of several distinct groups of highly conserved proteins, many of which also function in the heat shock and stress response pathways.5–7 The chaperonins, members of the chaperone superfamily, assist in the folding of nascent proteins in an ATP dependent manner. In the eukaryotic cytosol, chaperonins are often found proximal to the ribosome, and in complex with other chaperones, including Hsc70 and prefoldin (GimC).8–13
All chaperonins share similar structure; two stacked rings of 7–9 subunits each.14 These subunits can be homogeneous as in the E. coli chaperonin GroEL/ES,15 and the M. maripalidus chaperonin Mm-Cpn,16 or heterogeneous, as in the thermosome from T. acidophilum,17 and the eukaryotic chaperonin TRiC/CCT.18 The chaperonin family can be further divided into two subgroups, group I, found in mitochondria, chloroplasts, and the bacterial cytosol, and group II, found in the cytosol of archaea and eukaryotes.18 Group I chaperonins require an additional ring of protein subunits which function as a lid, closing the lumen of the complex once the substrate is encapsulated.19 Group II chaperonins have a built-in lid, and do not require any additional protein subunits to function.14 The underlying mechanism by which these proteins recognize nascent or misfolded polypeptides, and assist them in folding to a native state is of great interest. Models for the mechanism of folding have been proposed,14,20–24 however, the manner in which the chaperonin recognizes and interacts with unfolded substrates is not entirely understood.
The group I chaperonin GroEL/ES is perhaps the best understood of the chaperonins, and is often used as a general model for both groups I and II chaperonin function.18,25 These comparisons, however, are limited by several differences between groups I and II chaperonins. Unlike group II chaperonins, GroEL requires the GroES lid complex for function,19 and there are significant differences in the mechanism of subunit rearrangement upon ATP binding.14,18,26,27 The archaeal chaperonins have less subunit complexity than TRiC/CCT, with one or two discrete subunits, as opposed to the eight individual subunits found in TRiC/CCT.14,25,28 However, in contrast to GroEL/ES, the archaeal chaperonins undergo conformational changes upon ATP binding that are similar to TRiC/CCT and may more accurately model the mechanisms of substrate recognition and binding of the eukaryotic chaperonin TRiC/CCT.26,27 As with all group II chaperonins, the exact nature of the mechanism of Mm-Cpn recognition and refolding of substrates is still unclear. Although the subunit homogeneity of Mm-Cpn may suggest a functional similarity to GroEL/ES, sequence homology and structural mechanisms suggest that Mm-Cpn is more closely related to TRiC/CCT, and thus may serve as a more appropriate model for TRiC/CCT function than GroEL/ES.27
Tubulin and actin, the major substrates of TRiC/CCT, do not refold in the absence of TRiC/CCT.3,29 As a result, the tubulin and actin folding pathways are poorly defined, and very little is known about the conformation of partially folded intermediates. The pathway of β-sheet folding has been particularly difficult to elucidate, despite the availability of high-resolution structures of predominantly β-sheet proteins.30–32 Analysis of the TRiC/CCT interactome indicates that the cellular substrates of TRiC/CCT are enriched in proteins with β-sheet motifs, which are more prone to slow folding or aggregation.33 In addition, it has been shown that TRiC/CCT is required for the folding of WD40 β-propeller proteins,13 as well as the VHL tumor suppressor, whose TRiC/CCT binding sites have been identified as extended β-sheet regions.34 These results suggest that the presence of a β-sheet region is a hallmark of the client proteins of TRiC/CCT, and an understanding of the β-sheet substrate/chaperonin interaction may provide insight into the mechanism of substrate interaction by group II chaperonins.
To characterize the role of the β-sheet region in substrate recognition by group II chaperonins, a conserved β-sheet motif with a well-defined folding pathway is the most appropriate model. The Greek key motif is one of the most common β-sheet folds, seen in prokaryotes, Archaea and eukaryotes, and there are a number of proposed folding pathways,30–32,35 including the pathway beginning with the initial formation of a very long β-hairpin, which then folds into the Greek key motif.36 The folding of the lens γ and β crystallins has been studied in some detail.37–43 The γ crystallins share a similar β-sheet structure containing two Greek key motifs organized into distinct N and C terminal domains.40 A detailed model of the folding pathway for the γ crystallins has been determined, utilizing the unusually efficient quenching of the tryptophan fluorescence when the protein is in the native state.44,45
Cataract, the leading cause of blindness worldwide,46 is caused by the aggregation or precipitation of primarily the β and γ crystallins, the main structural components of lens of the human eye. The γ crystallins contain four buried tryptophans, two in each domain, which exhibit unusual fluorescence properties. In the native state, the tryptophans show very efficient quenching, providing an unusually sensitive reporter of the folding state of the protein.47,48 The N and C terminal domains of HγD-Crys have different stabilities, with the N terminal domain unfolding before the C terminal domain.41,42,44,49,50 This differential stability of the domains results in a three state unfolding pathway, on which HγD-Crys proceeds via a partially folded intermediate.43 This partially folded intermediate consists of a folded C-terminal domain and an unfolded N-terminal domain, and is relatively stable and long-lived.43
We therefore set out to characterize the group II chaperonin interaction with a eukaryotic Greek key substrate, human eye lens crystallin, for which the folding pathway is well defined. In this study, we report the use of HγD-Crys as a substrate for the chaperonin Mm-Cpn, to investigate the ability of the group II chaperonins to prevent aggregation, as well as refold HγD-Crys to a native-like conformation. Aggregation suppression studies, size exclusion chromatography, fluorescence spectroscopy, and SDS gel electrophoresis results suggest that Mm-Cpn prevents HγD-Crys aggregation, and further functions to refold HγD-Crys to a native-like conformation.
Results
Expression and purification of Mm-Cpn
The group II chaperonin Mm-Cpn was purified from E. coli, following the protocol described in Reissman et al.51 Figure 1 shows a transmission electron micrograph of purified chaperonin. The purified Mm-Cpn exhibits morphology consistent with previous reports; a barrel-shaped structure lacking density in the center, and an approximate diameter of 125 Å.51 To be certain that the purified protein was a functional and well-organized complex, we determined the Cryo-EM structure of purified Mm-Cpn in the presence of the ATP-AlFx, which traps ATP-bound molecules in the closed state. Figure 2 shows the Cryo-EM structure of Mm-Cpn in the closed state, at a resolution of 18 Å, demonstrating that the purified protein was capable of adopting a closed conformation.
Figure 1.

Electron micrograph of purified Mm-Cpn at 5 mg/mL, 150k magnification. The micrograph shows the ring-shaped structure characteristic of groups I and II chaperonins, consistent with previously observed chaperonins.52–54
Figure 2.

Cryo-EM reconstruction of purified Mm-Cpn in the closed state. Panel a: representative area of CCD image for Mm-Cpn in the closed state at 60,000× magnification. Panel b: 2D class-averages of Mm-Cpn raw images showing it is in the closed state. Panel c: top, tilted and side view of the 3D density map of Mm-Cpn in the closed state. Final density map is amplitude normalized and low-pass filtered to 18 Å. This structure is consistent with previously observed chaperonin Cryo-EM structures.27,54
Mm-Cpn can prevent HγD crystallin aggregation
Upon dilution from 5.5 M GuHCl into buffer, HγD-Crys folding intermediates partition between aggregation and productive refolding pathways.42 If the dilution from 5.5 M GuHCl is done with low protein concentrations (10–20 μg/mL), the productive refolding pathway is dominant, and unfolded HγD-Crys will refold to a native structure. At higher protein concentrations (50–100 μg/mL) the aggregation pathway is favored, and the partially folded HγD-Crys will aggregate rapidly.42 The formation of the aggregates as a function of time can be monitored by solution turbidity. Figure 3 shows a typical solution turbidity curve for HγD-Crys at 50 μg/mL, alone, or in the presence of a 1:1 molar ratio of Mm-Cpn:HγD-Crys, and 2 mM ATP, at 25 and 37 C. At 37 C, the average t1/2 of HγD-Crys aggregation in the absence of chaperonin was 40 ± 7 seconds, and the average ΔA was 0.22 ± 0.05 OD. The presence of 2 mM ATP did not have an effect on the aggregation of HγD-Crys alone. In the presence of Mm-Cpn, the t1/2 of HγD-Crys aggregation was 436 ± 73 seconds, and the average ΔA was 0.13 ± 0.02 OD. The continuous increase in intensity observed in the presence of Mm-Cpn likely reflects the rapid aggregation of HγD, which, at 37 C, is much faster than the ability of Mm-Cpn to bind and refold the aggregation-prone protein. However, the smaller overall intensity change in the presence of Mm-Cpn suggests that the chaperonin was binding HγD-Crys and suppressing aggregation. At 25 C, the average t1/2 of HγD-Crys aggregation without chaperonin was 142 ± 11 seconds, and the average ΔA was 0.30 ± 0.03 OD. When Mm-Cpn and 2 mM ATP were included, the average t1/2 of aggregation was 508 ± 77 seconds, and the average ΔA was 0.10 ± 0.01 OD. The change in intensity in the presence and absence of Mm-Cpn is greater at 25 C relative to 37 C (ΔΔA = 0.20 at 25 C vs. 0.09 at 37 C). This difference, which is also observed in the calculated aggregation rates, is likely the result of the slower aggregation of HγD-Crys at 25 C relative to 37 C. The slower aggregation kinetics of HγD-Crys at 25 C may allow for better recognition of the unfolded substrate by Mm-Cpn, resulting in more efficient suppression of aggregation.
Figure 3.

Kinetic traces of the aggregation of HγD-Crys in the absence of Mm-Cpn (black) and the presence of a 1:1 molar ratio of Mm-Cpn, and 2 mM ATP (gray), at 37 C and 25 C. In the presence of Mm-Cpn, the aggregation of HγD-Crys is markedly decreased relative to HγD-Crys alone.
Mm-Cpn can refold HγD crystallin in vitro
The ability of the chaperonins to refold damaged or misfolded proteins, as well as assist in the folding of nascent proteins, is one of the distinguishing characteristics of the chaperonin family. Mm-Cpn has been previously shown to have in vitro folding activity,51 however, Human γD Crystallin is not a physiological substrate for Mm-Cpn, and thus there is no implied expectation of folding activity.
Figure 4(a) shows size exclusion chromatograms of Mm-Cpn alone in the absence of ATP (blue), Mm-Cpn in the presence of HγD-Crys and 1 mM ATP (red), native HγD-Crys alone (green) and aggregated HγD-Crys (black). The Mm-Cpn only chromatogram showed a large peak at ∼8 mL, consistent with a protein of 931 kDa, and likely represents Mm-Cpn in an open conformation, as no ATP was present in the buffer, a second, smaller peak was also observed at ∼13 mL, which has been attributed to Mm-Cpn in a partially closed conformation (one ring open, one closed). The native HγD-Crys chromatogram showed a single peak at ∼21 mL, consistent with a protein of 22 kDa.
Figure 4.
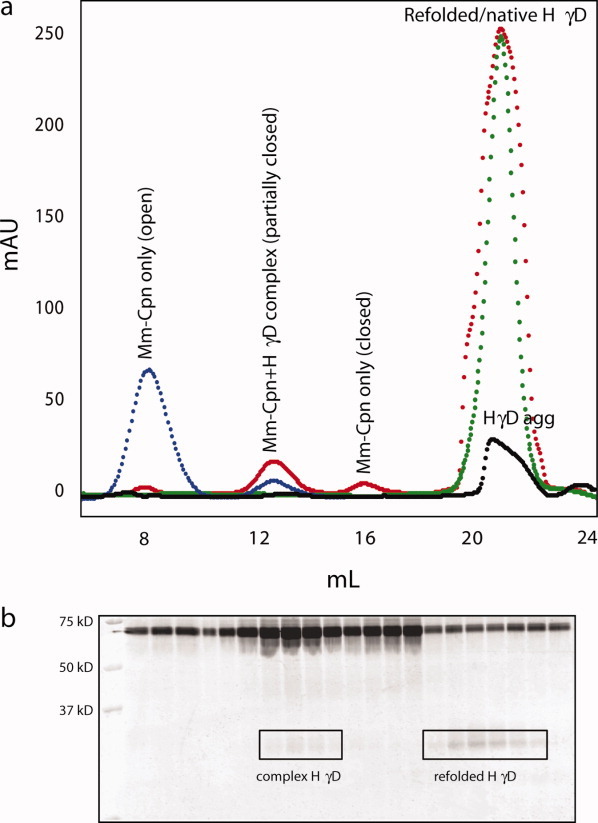
Panel a: Size exclusion chromatograms of Mm-Cpn alone (blue), Mm-Cpn+HγD-Crys and 1 mM ATP (red), native HγD-Crys (green) and HγD-Crys aggregate (black). In the presence of HγD-Crys and ATP, there is a large peak present at ∼18 mL, which is not seen in the Mm-Cpn alone chromatogram. This large peak, which elutes at approximately the same volume as native HγD-Crys, is refolded HγD-Crys. Panel b shows a 14% SDS gel corresponding to the Mm-Cpn+HγD, 1 mM ATP chromatogram. In the fractions corresponding to the Mm-Cpn peak, both a 60 kDa band, the size of the single subunits of Mm-Cpn, and a faint band at ∼20 kDa can be seen. In the fractions corresponding to the refolded HγD-Crys peak, the dominant bands were seen at 20 kDa, the size of HγD-Crys monomer. The bands appearing at 60 kDa in the refolded HγD lanes are likely degraded subunits of Mm-Cpn, which remain associated with the HγD.
In contrast to the chromatograms of Mm-Cpn and HγD-Crys alone, the chromatogram of the Mm-Cpn-HγD-Crys complex contains four distinct peaks. The peak appearing at ∼8 mL has been attributed to Mm-Cpn with both rings in the open conformation, the peak appearing at ∼13 mL contains the Mm-Cpn-HγD-Crys complex [Fig. 4(b)], and may also represent a partially closed conformation (one ring open, one closed). This peak is significantly larger in the Mm-Cpn-HγD-Crys chromatograph, consistent with the formation of Mm-Cpn-HγD-Crys complex. The peak appearing at ∼16 mL has been attributed to Mm-Cpn with both rings in the closed conformation. These data suggest that Mm-Cpn exists in an ensemble of conformational states in the presence of ATP, which may have implications for substrate binding.
The Mm-Cpn-HγD-Crys chromatogram also features a large, intense peak at ∼21 mL. This peak elutes at nearly the exact volume as native HγD-Crys alone (green trace), and thus has been attributed to HγD-Crys that has been refolded to a native or native-like conformation. The chromatogram of the HγD-Crys aggregation reaction in the absence of Mm-Cpn shows a broad, low intensity peak at ∼20 mL, indicating that the large peak seen at 21 mL in the complex chromatogram is not simply aggregated or spontaneously refolded HγD-Crys. The large difference in intensity between the Mm-Cpn and HγD-Crys peaks is due to the presence of tryptophan residues in HγD-Crys, which are absent in Mm-Cpn, resulting in lower absorbance at 280 nm.
When the fractions corresponding to the peaks of the Mm-Cpn-HγD-Crys chromatogram were electrophoresed through a 14% SDS gel [Fig. 4(b)], both the presence of the Mm-Cpn-HγD complex (lanes 7-10), and the presence of the refolded HγD-Crys were confirmed. Lanes 7-10 show the highest concentration of 20 kDa subunit; these lanes correspond to the 13 mL peak, where complexes are expected. Lanes 15-21 contain the fractions obtained from the peak at 21 mL, and the 20 kDa subunit is more prominent, indicating the presence of HγD-Crys. The bands appearing at 60 kDa are most likely single subunits of Mm-Cpn, which may have arisen from degraded native Mm-Cpn, and associated with the refolded substrate.
Fluorescence spectroscopy further confirms the presence of Mm-Cpn refolded HγD-Crys. The fluorescence spectra of denatured and native HγD-Crys are significantly different; the emission maximum of native HγD-Crys is ∼325 nm, while the emission maximum of denatured HγD-Crys is ∼350 nm.42,47,49 Figure 5 shows the fluorescence emission spectra of native (dotted line), denatured (dashed line), and Mm-Cpn-refolded HγD-Crys (solid line). The Mm-Cpn-refolded HγD-Crys spectrum showed an emission maximum at ∼330 nm, consistent with previously observed emission spectra of native HγD-Crys47,49 and indicating the presence of native-like HγD-Crys in the samples. The refolded spectrum showed no significant fluorescence intensity at 350 nm, indicating that there was little to no unfolded HγD-Crys present. The increased intensity at longer wavelengths (>360 nm) in the refolded sample relative to the native HγD sample is likely due to the presence of degraded Mm-Cpn subunits seen in Figure 4(b), which possess some intrinsic fluorescence, despite the absence of Trp residues. These data further confirm that Mm-Cpn is capable of refolding HγD-Crys to a native-like state.
Figure 5.

Fluorescence emission spectra of native HγD-Crys (dotted line), denatured HγD-Crys (dashed line), and Mm-Cpn-refolded HγD-Crys (solid line). Both native HγD-Crys and Mm-Cpn-refolded HγD-Crys show an emission maximum at ∼330 nm, consistent with previous reports.42,49 No maximum is seen in the Mm-Cpn-refolded sample at 350 nm, the emission maximum for denatured HγD-Crys.
Mm-Cpn does not interact with natively folded HγD crystallin
When Mm-Cpn was mixed with native HγD-Crys, in the presence of 1 mM ATP no association was observed (data not shown). The size exclusion chromatograph of the Mm-Cpn/Native HγD-Crys mixture is shown in Figure 6(a). Four distinct peaks are observed in the chromatograph, a small peak at ∼8 mL, corresponding to Mm-Cpn in the open state, a more prominent peak at ∼13 mL, which may correspond to partially closed Mm-Cpn, a small peak at ∼16 mL, corresponding to completely closed Mm-Cpn, and a large peak at ∼21 mL, corresponding to HγD-Crys. When unfolded HγD-Crys is present, the peak appearing at ∼13 mL corresponds with the Mm-Cpn-HγD-Crys complex shown in Figure 4(a), further suggesting that this peak represents a partially closed conformation. SDS gel electrophoresis of the all the peaks indicates that only the peak at 21 mL contains HγD-Crys, while the other peaks contain Mm-Cpn only [Fig. 6(b)], and no complex was detected. These data indicate that Mm-Cpn does not interact with natively folded HγD-Crys, and suggests that a damaged or exposed region of protein is required for recognition of substrate by Mm-Cpn.
Figure 6.

Panel a: Size exclusion chromatogram of Mm-Cpn mixed with natively folded HγD-Crys, in the presence of 1 mM ATP. Peak 1 (8 mL) corresponds to Mm-Cpn with both rings open, peak 2 (13 mL) corresponds to Mm-Cpn in a partially closed conformation, peak 3 (16 mL) corresponds to Mm-Cpn with both rings closed, and peak 4 (21 mL) corresponds to natively folded HγD-Crys. Panel b shows a 14% SDS gel of the four peaks, showing a 60 kDa band in the peaks identified as Mm-Cpn, and a 20 kDa band in the peak identified as HγD-Crys.
In the absence of ATP, Mm-Cpn can bind but not refold HγD crystallin
Mm-Cpn, like all chaperonins, functions to fold nascent proteins or refold misfolded proteins, in an ATP-dependent manner. Previous work has indicated that there is no refolding of denatured protein by Mm-Cpn in the absence of ATP,51 though the degree of chaperonin-substrate interaction in the absence of ATP is not well characterized. Figure 7 shows the solution turbidity curves for HγD-Crys alone, and HγD-Crys in the presence of Mm-Cpn, without ATP. The total change in absorbance for HγD-Crys alone was 0.30 ± 0.02 OD, and the total change in absorbance for HγD-Crys+Mm-Cpn, 0 mM ATP, was 0.20 ± 0.04 OD. This ∼30% decrease in HγD-Crys aggregation in the presence of Mm-Cpn is less than what was observed when ATP was present, and likely represents transient binding of the substrate by the chaperonin. Figure 8 shows the percent aggregation of HγD-Crys in the presence of Mm-Cpn, as a function of ATP concentration.
Figure 7.

Kinetic traces of HγD-Crys aggregation alone (black) and in the presence of Mm-Cpn, without ATP (gray). In the presence of Mm-Cpn, but the absence of ATP, the rate of HγD-Crys aggregation is slowed, but there is no productive refolding of the HγD-Crys to a native-like conformation. This lack of refolded HγD-Crys is consistent with the ATP dependence of group II chaperonins.20
Figure 8.
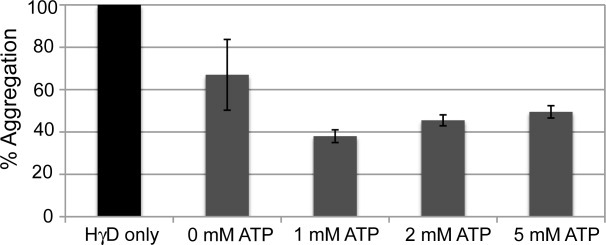
Histogram of HγD-Crys aggregation in the presence and absence of Mm-Cpn, as a function of ATP concentration at 37 C. The largest decrease in % aggregation occurs in the presence of 1 mM ATP, and increasing the ATP concentration does not further suppress aggregation. When no ATP is present, there is some loss of HγD-Crys aggregation, but the high degree of error suggests that this is due to transient binding of HγD-Crys to the open Mm-Cpn, and not the result of productive binding and refolding.
A transient binding effect is further supported by the change in the calculated t1/2 of aggregation in the presence and absence of Mm-Cpn and ATP. As discussed in the previous section, the t1/2 of aggregation of HγD-Crys alone at 37 C was 40 ± 7 seconds, and in the presence of Mm-Cpn and 2 mM ATP, increased to 436 ± 73 seconds. However, in the presence of Mm-Cpn and absence of ATP, the average t1/2 was 200 ± 173 seconds. Figure 9 shows the average t1/2 for HγD-Crys aggregation at various ATP concentrations. The increased t1/2 relative to HγD-Crys alone suggests that HγD-Crys and Mm-Cpn are weakly interacting, consistent with the formation of a transient chaperonin-substrate complex. The large error indicates a significant amount of variation in the calculated t1/2, which is also indicative of weak or nonproductive binding of substrate to the chaperonin. There was no refolded HγD-Crys observed by size exclusion chromatography in the absence of ATP, consistent with previous reports.51
Figure 9.

Histogram of the t½ of HγD-Crys aggregation in the absence of Mm-Cpn (dark gray), and presence of Mm-Cpn (light gray), as a function of ATP concentration. The rates of aggregation are virtually unchanged at 1, 2, and 5 mM ATP, further suggesting that there is no advantage to the presence of an excess of ATP. At 0 mM ATP, there is an increase in the t½ of aggregation, however, the large error again suggests that the interaction is a transient binding of HγD-Crys to the open Mm-Cpn, as opposed to productive refolding.
Dependence on ATP concentration of Mm-Cpn activity with HγD crystallin
Previous studies of group II chaperonins have shown that an excess of ATP (>1 mM) produces negative cooperativity between the two rings, and the rate of ATP hydrolysis is decreased,51 however the influence of this negative cooperativity on substrate binding and refolding is not well understood A similar negative effect was seen on the ability of Mm-Cpn to suppress aggregation of HγD-Crys in the presence of increasing concentrations of ATP. Figure 8 shows the percent aggregation of HγD-Crys, in the presence of chaperonin, relative to HγD in the absence of chaperonin, at various ATP concentrations. As discussed in the previous section, when Mm-Cpn but not ATP was present, there was a minor decrease in the percent aggregation. However, no productive refolding of HγD-Crys was observed. The greatest decrease in percent aggregation was seen at 1 mM ATP, and increasing concentrations of ATP resulted in an increase in percent aggregation relative to HγD-Crys alone. These data suggest that there is an optimal ATP concentration for suppression of aggregation and substrate refolding, and further that the negative cooperativity observed by Reissman et al.51 to affect ATP hydrolysis at high ATP concentrations, may extend to substrate binding and refolding.
Discussion
Mm-Cpn binds and refolds HγD crystallin in vitro
The experiments described above show that Mm-Cpn binds partially folded HγD-Crys, as evidenced by a decrease in both the solution turbidity and the rate of aggregation in the presence of the chaperonin. In addition to simple binding of the unfolded HγD-Crys, Mm-Cpn was also observed to refold HγD-Crys, as indicated by both a decrease in the overall amount of aggregation (Fig. 3, Table I), and by the presence of refolded HγD-Crys detected by size exclusion chromatography (Fig. 4), and fluorescence spectroscopy (Fig. 5). The partially folded HγD-Crys species that is at the junction between productive refolding and aggregation has a native-like C-terminal domain, and an unfolded N-terminal domain, destined to become the Greek key β-sheet native fold.42,43 Mm-Cpn is an archaeal chaperonin, and thus would not a priori be expected to bind and refold human protein substrates. However, the Greek key fold of the crystallin β-sheets is found in archeal proteins, and the ability of Mm-Cpn to refold this substrate may reflect the requirement of an archeal chaperonin to bind and refold a native Greek key protein. Kinetic assays with Mm-Cpn show no gain in suppression of crystallin aggregation at ATP concentrations higher than the physiological concentration of 1 mM, which is also observed in other members of the chaperonin family.55,56 Previous studies of the group II chaperonins have observed varying effects on substrate refolding at high ATP concentrations. In the case of Mm-Cpn, it was observed that at ATP concentrations greater than 1 mM, overall ATP hydrolysis becomes less efficient, as excess ATP forces the chaperonin into a conformation with less than optimal ATPase activity.51 The results presented here are consistent with those observations. However, studies with the eukaryotic chaperonin TRiC/CCT observed an increase in actin refolding rates at ATP concentrations greater than 6 mM.57 These varying results may be reflective of the differences between Mm-Cpn and TRiC/CCT, in particular the role of the various subunits of TRiC/CCT on ATPase activity.
Table I.
t, ΔA, and Percent Aggregation Values for HγD in the Presence and Absence of Mm-Cpn at 37 C and Various ATP Concentrations
| ATP | |||||
|---|---|---|---|---|---|
| 0 mM | 1 mM | 2 mM | 5 mM | ||
| HγD | t½ (s) | 40 ± 16 | 48 ± 1 | 40 ± 6 | 59 ± 5 |
| ΔA | 0.3 ± 0.02 | 0.3 ± 0.01 | 0.25 ± 0.04 | 0.3 ± 0.02 | |
| % aggregation | 100 | 100 | 100 | 100 | |
| Mm-Cpn + HγD | t½ (s) | 200 ± 174 | 398 ± 122 | 436 ± 73 | 402 ± 77 |
| ΔA | 0.2 ± 0.03 | 0.1 ± 0.02 | 0.1 ± 0.02 | 0.17 ± 0.02 | |
| % aggregation | 67 ± 16 | 38 ± 9 | 46 ± 8 | 50 ± 3 | |
The mechanism by which chaperonins refold an encapsulated protein is complex, and remains relatively undefined. A number of models of the mechanism of folding have been developed, based on experimental and computational results. The simplest of these is a passive model in which the newly translated or unfolded protein is encapsulated in the center lumen, and isolated from nonproductive interactions.58 This encapsulation prevents the nascent protein from aggregating with other chains, and allows it to fold to a native state.22,23 A revision of this model suggests that the interior of the chaperonin, rather than providing a dilute environment for the nascent peptide to fold, functions to restrict the conformational space available to the peptide. This restriction of the conformational space reduces the free energy, driving the nascent peptide to adopt the correct fold.23,59,60
Alternatively, a number of models suggest that the chaperonin plays a more active role in the folding of nascent proteins. It has been demonstrated that GroEL/ES is capable of unfolding incorrectly folded substrates and correctly folding them to a native state.61,62 This model has been expanded to an iterative annealing model, in which chaperonins bind proteins trapped in non-native conformations, disrupt the misfolded structure, and release them back into the cytosol.63 This repeated disruption and release gives the nascent protein numerous chances to fold into a native conformation.
The suggestion that the a β-sheet structure may be more easily recognized by group II chaperonins is of particular interest for determining the mechanism of recognition and refolding of HγD-Crys by Mm-Cpn. It has been suggested that the subunit heterogeneity of TRiC/CCT is related to the recognition of specific structural motifs within the substrate proteins.64,65 Kubota et al. identified a pattern of strong recognition by TRiC/CCT of the WD40 proteins, in particular at hydrophobic β-strands, and suggest that this motif may be a key element of chaperonin-substrate recognition for the group II chaperonins.66 In addition, while TRiC/CCT had originally been thought to recognize a very specific set of proteins in the cell, however, more recent data indicates that TRiC/CCT recognizes a large variety of substrates in vivo.33,67 A large number of substrates identified as absolutely requiring TRiC/CCT for folding, including α and β tubulin, G-protein β, VHL tumor suppressor, and cell cycle proteins Cdh1, Cdc20, and Plk1, contain β-sheet motifs, suggesting that the presence of a β-sheet may be a recognition signal for the chaperonin.29,67–69
While the list of client proteins of group II chaperonins continues to expand, the exact site of substrate recognition by the chaperonin remains unclear. Potential recognition sites include exposed hydrophobic surfaces, and stretches of disordered peptide chains. As discussed previously, the partially folded intermediate of HγD-Crys appears to have one folded domain (CTD) and one disordered domain (NTD), leaving one face of the domain interface, which is buried in the native state, exposed to solvent. Mm-Cpn binding to the exposed interface would prevent aggregation, while encapsulation would allow productive folding to occur in isolation. This binding event may be analogous to previously proposed aggregation models, and may represent the mechanism by which Mm-Cpn recognizes and refolds HγD-Crys.
Alternatively, the disordered N-terminal domain may itself provide the recognition motif. Studies have shown that hydrophobic sequences are recognized by chaperones such as Hsp70 and GroEL/ES.11,14,70,71 The interior of HγD-Crys is very hydrophobic- each domain has seven tyrosines, two tryptophans, three phenylalanines, and more than 30 other hydrophobic residues. Most of these residues are buried in the native state, but would be exposed when the protein is in an unfolded conformation, and thus available for Mm-Cpn binding.
Mm-Cpn as a model group II chaperonin
Much of the current understanding of the general mechanism of chaperonins is derived from studies of the structure and function of GroEL/ES. It has been suggested that a number of bacteria have evolved specific variants of GroEL/ES to suit the specific needs of the organism72 but GroEL is only 20–25% identical to the eukaryotic group II chaperonins.73 In addition, the mechanism by which GroEL/ES undergoes the structural change from the open to closed form is dramatically different than the structural change from the open to closed form for group II chaperonins.27 Therefore, there is a limit to the parallels that can be drawn between the mechanism of GroEL/ES function and that of the group II chaperonins.
Archaea have group II chaperonins, which are more closely related to the eukaryotic chaperonin TRiC/CCT, than to the prokaryotic GroEL/ES.74 The archaeal chaperonins represent an intermediate level of complexity between GroEL/ES, and the group II chaperonin TRiC/CCT, as they possess a built-in lid, but have less subunit heterogeneity.75 Archaeal chaperonins share a common architecture with TRiC/CCT, but are generally composed of three or less subunits. The group II chaperonin from T. acidophilum has been has been crystallized, and extensively used as a model group II chaperonin.17,76,77 Group II chaperonin structure and function has also been modeled using chaperonins from other archeael species, including Thermococcus,78 Pyrococcus furiosus, Pyrococcus horikoshii, and Methanococcus jannaschii.79 Chaperonins from these species have provided a greater understanding of the structure and mechanism of group II chaperonins, as well as informed more complicated studies using TRiC/CCT.
The hetero-octomeric nature of TRiC/CCT makes it extremely complex, and it has been suggested that each subunit has a specific role in substrate binding.18 This complexity makes TRiC/CCT a challenging protein to use to understand substrate binding and refolding mechanisms. The homo-oligomeric nature of Mm-Cpn removes some of the inherent complexity of TRiC/CCT, and thus has been used for mechanistic studies of refolding and substrate binding.16,51,74 In addition, the single subunit configuration of Mm-Cpn is conducive to expression in E. coli, and also allows for the creation of mutant chaperonins, which are extremely valuable for exploring the mechanism of protein folding by chaperonins.51 It is therefore clear that Mm-Cpn can serve as an excellent model for the more complex TRiC/CCT, and that much of what is learned from studies using Mm-Cpn can be applied to the general understanding of the group II chaperonins.
Materials and Methods
Purification of Mm-Cpn
The plasmid containing recombinant Mm-Cpn was a generous gift from the laboratory of Judith Frydman (Stanford University). Wild type Mm-Cpn was purified as described in Reissmann et al.,51 with the addition of 1 mM ATP to the elution buffers. The resulting purified protein was evaluated by transmission electron microscopy and cryo-electron microscopy (Fig. 1), and was observed to be consistent with previous reports.
Electron microscopy
Purified Mm-Cpn was visualized using transmission electron microscopy. Carbon grids (Ted Pella) were glow-discharged for 30 seconds. Immediately following glow-discharge, 2 μL of purified protein was spotted onto each grid, and allowed to sit for 2 minutes. Excess sample was blotted off using filter paper, and stained with 1.5% Uranyl-Acetate (Sigma). Micrographs were visualized on a JEOL SX 1200 transmission electron microscope, and digital micrographs taken using an AMT 16000S camera system. Digital images were analyzed using Adobe Photoshop™ (Adobe).
Cryo-electron microscopy
2.3 μM Mm-Cpn was added to buffer solution (50 mM Tris-HCl, 50 mM KCl, 5 mM MgCl2, 5 mM DTT, 1 mM ATP, pH 7.5). Five millimolar Al(NO3)3 and 30 mM NaF were added to form ATP*AlFx nonhydrolyzable analog. Sample was incubated for 30 minutes, then centrifuged at 19,600 g for 20 minutes. Supernatant was spotted on the grid (Quantifoil), then blotted with a Vitrobot (FEI) using a single blot for 7 seconds, immediately followed by freezing in liquid ethane. 7 images were taken on a JEM2010F cryo-electron microscope at 83,100X magnification, on a Gatan 4KX4K CCD Camera. 977 particles were automatically boxed out using e2boxer.py in EMAN2.80 Contrast transfer functions of the images were automatically fitted using fitcf.py,81 and manually adjusted by ctfit in EMAN.82 All particle images were phase-flipped then submitted to the program refine2d.py for 2D analysis.80 The 3D density map was generated using the standard refinement strategy in EMAN. The final resolution of the map was 18 Å according to the Fourier Shell Correlation,83 with a 0.5 cut-off criterion.
Purification of human γD crystallin
Wild type HγD Crystallin was expressed and purified as described by Kosinski-Collins et al.,49 with protocol modifications described in Mills et al.43
Aggregation suppression assay
The aggregation of HγD-Crys in the presence and absence of Mm-Cpn was monitored by solution turbidity at 350 nm, following the procedure described in Acosta-Sampson and King.84 Briefly, 2.3 μM substrate (HγD-Crys) was unfolded overnight at 37 C in unfolding buffer (5.5 M GuHCl, 50 mM Tris-HCl, 5 mM DTT, pH 7.5). The unfolded protein was allowed to equilibrate at the reaction temperature in the spectrophotometer for 5 minutes. Refolding buffer (50 mM Tris-HCl, 1 mM DTT, 50 mM KCl, 5 mM MgCl2, pH 7.5), and the desired concentration of ATP was incubated at the reaction temperature for at least 10 minutes. At high ATP concentrations (>2 mM), MgCl2 concentration was increased to 10 mM in the refolding buffer, to provide a twofold excess of Mg2+ to ATP. To initiate the aggregation process, the unfolded protein was diluted 1:10 with refolding buffer in the presence and absence of Mm-Cpn at a 1:1 molar ratio of chaperonin to substrate. Aggregation kinetics were measured on a temperature-controlled Cary 50 UV/Vis spectrophotometer (Varian Inc.), using the Varian Kinetics program. Solution turbidity was monitored at 350 nm for 1800 seconds, at 25 and 37 C. Kinetic traces were fit to the 2- state rate equation:
where Ya and Yb represent the initial and final absorbances, respectively. Data analysis was done using Kaleidograph version 4.0 (Synergy Software).
Analytical size exclusion chromatography
A Superose 6 size exclusion column (GE Healthcare) was equilibrated with 3 column volumes of MQ-A buffer.51 Samples obtained from aggregation suppression assays were filtered through 0.2 μM filters (Millipore). One milliliter of sample was loaded onto the Superose 6 column at a flow rate of 0.3 mL/min, and 0.5 mL fractions were collected. Fractions corresponding to peaks on the resulting chromatogram were run on a 14% SDS gel, stained with Krypton™ stain (Pierce), and imaged on a Typhoon™ variable mode imager (GE Healthcare).
Fluorescence spectroscopy
Fluorescence spectra of native HγD-Crys, unfolded HγD-Crys, and refolded HγD-Crys were measured on a Hitachi F-4500 spectrometer. An excitation wavelength of 295 nm was used to selectively excite tryptophan residues, and emission spectra were recorded over a wavelength range from 300 to 400 nm, using a 10 mm slit width for both excitation and emission. Refolded HγD-Crys sample was prepared by pooling size exclusion chromatography fractions identified as containing refolded HγD-Crys by SDS gel electrophoresis, and concentrating to 0.5 mL.
Acknowledgments
The authors thank Dr. Ishara Mills-Henry and Shea Jameel for initiating the project. They thank Dr. Ligia Acosta-Sampson, Dr. Jiejin Chen, Dr. Kate Moreau, Oksana Sergeeva, and Ginger Yang for their helpful discussions and other technical assistance. Judith Frydman and the Frydman laboratory, generously provided the Mm-Cpn plasmid and purification protocol, as well as technical assistance.
References
- 1.Yaffe MB, Farr GW, Miklos D, Horwich AL, Sternlicht ML, Sternlicht H. TCP1 complex is a molecular chaperone in tubulin biogenesis. Nature. 1992;358:245–248. doi: 10.1038/358245a0. [DOI] [PubMed] [Google Scholar]
- 2.Sternlicht H, Farr GW, Sternlicht ML, Driscoll JK, Willison K, Yaffe MB. The t-complex polypeptide 1 complex is a chaperone for tubulin and actin in vivo. Proc Natl Acad Sci USA. 1993;90:9422–9426. doi: 10.1073/pnas.90.20.9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunn AY, Melville MW, Frydman J. CellularsSubstrates of the eukaryotic chaperonin TRiC/CCT. J Struct Biol. 2001;135:176–184. doi: 10.1006/jsbi.2001.4380. [DOI] [PubMed] [Google Scholar]
- 4.Grantham J, Brackley K, Willison K. Substantial CCT activity is required for cell cycle progression and cytoskeletal organization in mammalian cells. Exp Cell Res. 2006;312:2309–2324. doi: 10.1016/j.yexcr.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 5.Fink AL. Chaperone-mediated protein folding. Physiol Rev. 1999;79:425–449. doi: 10.1152/physrev.1999.79.2.425. [DOI] [PubMed] [Google Scholar]
- 6.Gibbs SJ, Barren B, Beck KE, Proft J, Zhao X, Noskova T, Braun AP, Artemyev NO, Braun JEA. Hsp40 couples with the CSPalpha chaperone complex upon induction of the heat shock response. PLoS ONE. 2009;4:e4595. doi: 10.1371/journal.pone.0004595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu L, Voloboueva LA, Ouyang Y, Emery JF, Giffard RG. Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J Cereb Blood Flow Metab. 2009;29:365–374. doi: 10.1038/jcbfm.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thulasiraman V, Yang C-F, Frydman J. In vivo newly translated polypeptides are sequestered in a protected folding environment. EMBO J. 1999;18:85–95. doi: 10.1093/emboj/18.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melville MW, McCellan AJ, Meyer AS, Darveau A, Frydman J. Hsp 70 and TRiC/CCT chaperone systems cooperate in vivo to assemble the Von Hippel-Lindeau tumor suppressor complex. Mol Cell Biol. 2003;23:3141–3151. doi: 10.1128/MCB.23.9.3141-3151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Albanese V, Frydman J. Where chaperones and nascent polypeptides meet. Nat Struct Biol. 2002;9:716–718. doi: 10.1038/nsb1002-716. [DOI] [PubMed] [Google Scholar]
- 11.Cuellar J, Martin-Benito J, Scheres SHW, Sousa R, Moro F, Lopes-Vinas E, Gomez-Puertas P, Muga A, Carrascosa JL, Valpuesta JM. The structure of CCT-Hsc70nbd suggests a mechanism for Hsp70 delivery of substrates to the chaperonin. Nat Struct Mol Biol. 2008;15:858–864. doi: 10.1038/nsmb.1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hansen WJ, Cowan NJ, Welch WJ. Prefoldin-nascent chain complexes in the folding of cytoskeletal proteins. J Cell Biol. 1999;145:265–277. doi: 10.1083/jcb.145.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siegers K, Bolter B, Schwarz JP, Bottcher UMK, Guha S, Hartl FU. TRiC/CCT cooperates with different upstream chaperones in the folding of distinct protein classes. EMBO J. 2003;22:5230–5240. doi: 10.1093/emboj/cdg483. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Horwich AL, Fenton WA, Chapman E, Farr GW. Two families of chaperonPhysiology and mechanism. Ann Rev Cell Dev Biol. 2007;23:115–145. doi: 10.1146/annurev.cellbio.23.090506.123555. [DOI] [PubMed] [Google Scholar]
- 15.Hendrix RW, Tsui L. Role of the host in virus assembly: cloning of the Escherichia Coli groE gene and identification of its protein product. Proc Natl Acad Sci USA. 1978;75:136–139. doi: 10.1073/pnas.75.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kusmierczyk AR, Martin J. Nested cooperativity and salt dependence of the ATPase activity of the archaeal chaperonin Mm-Cpn. FEBS. 2003;547:201–204. doi: 10.1016/s0014-5793(03)00722-1. [DOI] [PubMed] [Google Scholar]
- 17.Bigotti MG, Clarke AR. Cooperativity in the thermosome. J Mol Biol. 2005;348:13–26. doi: 10.1016/j.jmb.2005.01.066. [DOI] [PubMed] [Google Scholar]
- 18.Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- 19.Horwich AL, Farr GW, Fenton WA. GroEL-GroES-mediated protein folding. Chem Rev. 2006;106:1917–1930. doi: 10.1021/cr040435v. [DOI] [PubMed] [Google Scholar]
- 20.Spiess C, Meyer AS, Reissmann S, Frydman J. Mechanism of the eukaryotic chaperonprotein folding in the chamber of secrets. Trends Cell Biol. 2004;14:598–604. doi: 10.1016/j.tcb.2004.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thirumaliai D, Lorimer GH. Chaperonin-mediated protein folding. Annu Rev Biophys Biomol Struct. 2001;30:245–269. doi: 10.1146/annurev.biophys.30.1.245. [DOI] [PubMed] [Google Scholar]
- 22.Ellis RJ. Molecular chaperones: inside and outside the Anfinsen cage. Curr Biol. 2001;11:1038–1040. doi: 10.1016/s0960-9822(01)00620-0. [DOI] [PubMed] [Google Scholar]
- 23.Altschuler GM, Willison KR. Development of free-energy-based models for chaperonin containing TCP-1 mediated folding of actin. J R Soc Interface. 2008;5:1391–1408. doi: 10.1098/rsif.2008.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bigotti MG, Clarke AR. Chaperonins: the hunt for the group II mechanism. Arch Biochem Biophys. 2008;474:331–339. doi: 10.1016/j.abb.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 25.Lund PA, Large AT, Kapatai G. The chaperonins: perspectives from the Archaea. Biochem Soc Trans. 2003;31:681–685. doi: 10.1042/bst0310681. [DOI] [PubMed] [Google Scholar]
- 26.Clare DK, Stagg S, Quispe J, Farr GW, Horwich AL, Saibil HR. Multiple states of a nucleotide-bound group 2 chaperonin. Structure. 2008;16:528–534. doi: 10.1016/j.str.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Baker ML, Schroder GF, Douglas NR, Reissmann S, Jakana J, Dougherty M, Fu CJ, Levitt M, Ludtke SJ, Frydman J, Chiu W. Mechanism of folding chamber closure in a group II chaperonin. Nature. 2010;463:379–383. doi: 10.1038/nature08701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rommelaere H, Van Troys M, Gao Y, Melki R, Cowan NJ, Vandekerckhove J, Ampe C. Eukaryotic cytosolic chaperonin contains t-complex polypeptide 1 and seven related subunits. Proc Natl Acad Sci USA. 1993;90:11975–11979. doi: 10.1073/pnas.90.24.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang P, MacRae TH. Molecular chaperones and the cytoskeleton. J Cell Sci. 1997;110:1431–1440. doi: 10.1242/jcs.110.13.1431. [DOI] [PubMed] [Google Scholar]
- 30.Ohki S-Y, Kariya E, Hiraga K, Wakamiya A, Isobe T, Oda K, Kainosho M. NMR structure of Streptomyces killer toxin-like protein SKLP: further evidence for the wide distrubution of single domain bg crystallin superfamily proteins. J Mol Biol. 2001:305–120. doi: 10.1006/jmbi.2000.4244. [DOI] [PubMed] [Google Scholar]
- 31.Wu B, Yee A, Pineda-Lucena A, Semesi A, Ramelot TA, Cort JR, Jung J-W, Edwards A, Lee W, Kennedy M, Arrowsmith CH. Solution structure of ribosomal protein S28E from Methanobacterium thermoautotrophicum. Protein Sci. 2003;12:2831–2837. doi: 10.1110/ps.03358203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Basak A, Bateman O, Slingsby C, Pande A, Asherie N, Ogun O, Benedek GB, Pande J. High-resolution X-ray crystal structures of human gD crystallin (1.25 A) and the R58H mutant (1.15 A) associated with aculeiform cataract. J Mol Biol. 2003;328:1137–1147. doi: 10.1016/s0022-2836(03)00375-9. [DOI] [PubMed] [Google Scholar]
- 33.Yam AY, Xia Y, Lin H-TJ, Burlingame A, Gerstein M, Frydman J. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat Struct Mol Biol. 2008;15:1255–1262. doi: 10.1038/nsmb.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feldman DE, Spiess C, Howard DE, Frydman J. Tumorigenic mutations in VHL disrupt folding in vivo by interfering with chaperonin binding. Mol Cell. 2003;12:1213–1224. doi: 10.1016/s1097-2765(03)00423-4. [DOI] [PubMed] [Google Scholar]
- 35.Salem GM, Hutchinson EG, Orengo CA, Thornton JM. Correlation of observed fold frequency with the occurrence of local structural motifs. J Mol Biol. 1999;287:969–981. doi: 10.1006/jmbi.1999.2642. [DOI] [PubMed] [Google Scholar]
- 36.Richardson JS. The anatomy and taxonomy of protein structure. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 37.Smith MA, Bateman OA, Jaenicke R, Slingsby C. Mutation of interfaces in domain-swapped human bB2-crystallin. Protein Sci. 2007;16:615–625. doi: 10.1110/ps.062659107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takata T, Woodbury LG, Lampi KJ. Deamidation alters interatctions of beta-crystallins in hetero-oligomers. Mol Vision. 2009;15:241–249. [PMC free article] [PubMed] [Google Scholar]
- 39.Wenk M, Herbst R, Hoeger D, Kretschmar M, Lubsen NH, Jaenicke R. Gamma S-crystallin of bovine and human eye lens: solution structure, stability and folding of the intact two domain protein and its separate domains. Biophys Chem. 2000;86:95–108. doi: 10.1016/s0301-4622(00)00161-7. [DOI] [PubMed] [Google Scholar]
- 40.Bloemendal H, de Jong W, Jaenicke R, Lubsen NH, Slingsby C, Tardieu A. Ageing and vision: structure, stability and function of lens crystallins. Prog Biophys Mol Biol. 2004;86:407–485. doi: 10.1016/j.pbiomolbio.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 41.Flaugh SL, Mills IA, King JA. Glutamine deamidation destabilizes human gammaD-crystallin and lowers the kinetic barrier to unfolding. J Biol Chem. 2006;281:30782–30793. doi: 10.1074/jbc.M603882200. [DOI] [PubMed] [Google Scholar]
- 42.Kosinski-Collins MS, King J. In vitro unfolding, refolding and polymerization of human gammaD crystallin, a protein involved in cataract formation. Protein Sci. 2003;12:480–490. doi: 10.1110/ps.0225503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mills IA, Flaugh SL, Kosinski-Collins MS, King JA. Folding and stability of the isolated Greek key domains of the long-lived human lens proteins gammaD-crystallin and gammaS-crystallin. Protein Sci. 2007;16:2427–2444. doi: 10.1110/ps.072970207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flaugh SL, Kosinski-Collins MS, King J. Contributions of hydrophobic domain interface interactions to the folding and stability of human gammaD-crystallin. Protein Sci. 2005;14:569–581. doi: 10.1110/ps.041111405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Das P, King JA, Zhou R. B-strand interactions at the domain interface critical for the stability of human lens gD-crystallin. Protein Sci. 2010;19:131–140. doi: 10.1002/pro.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clark JJ. Principle and practice of ophthalmology. Philadelphia: Saunders College Publishing; 1994. [Google Scholar]
- 47.Chen J, Flaugh SL, Callis PR, King J. Mechanism of the highly efficient quenching of tryptophan fluorescence in human gammaD-Crystallin. Biochemistry. 2006;45:11552–11563. doi: 10.1021/bi060988v. [DOI] [PubMed] [Google Scholar]
- 48.Chen J, Callis PR, King J. Mechanism of the very efficient quenching of tryptophan fluorescence in human gamma D- and gamma S-crystallins: the gamma-crystalin fold may have evolved to protect tryptophan residues from ultraviolet photodamage. Biochemistry. 2009;48:3708–3716. doi: 10.1021/bi802177g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kosinski-Collins MS, Flaugh SL, King J. Probing folding and florescence quenching in human gammaD crystallin Greek key domains using triple tryptophan mutant proteins. Protein Sci. 2004;13:2223–2235. doi: 10.1110/ps.04627004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, Petty S, Trojanowski A, Knee K, Goulet D, Mukerji I, King J. Formation of amyloid fibrils in vitro from partially unfolded intermediates of human gC-crystallin. Invest Ophthalmol Vis Sci. 2010;51:672–678. doi: 10.1167/iovs.09-3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reissmann S, Parnot C, Booth CR, Chiu W, Frydman J. Essential function of the built-in lid in the allosteric regulation of eukaryotic and archaeal chaperonins. Nat Struct Mol Biol. 2007;14:432–440. doi: 10.1038/nsmb1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trent JD, Nimmesgern E, Wall JS, Hartl FU, Horwich AL. A molecular chaperone from a thermophilic archaebacterium is related to the eukaryotic protein t-complex polypeptide-1. Nature. 1991;354:490–493. doi: 10.1038/354490a0. [DOI] [PubMed] [Google Scholar]
- 53.Booth CR, Meyer AS, Cong Y, Topf M, Sali A, Ludtke SJ, Chiu W, Frydman J. Mechanism of lid closure in the eukaryotic chaperonin TRiC/CCT. Nat Struct Mol Biol. 2008;15:746–753. doi: 10.1038/nsmb.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen D-H, Song J-L, Chuang DT, Chiu W, Ludtke SJ. An expanded conformation of single-ring GroEL-GroES complex encapsulates an 86 kDa substrate. Structure. 2006;14:1711–1722. doi: 10.1016/j.str.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 55.Chapman E, Farr GW, Fenton WA, Johnson SM, Horwich AL. Requirement for binding multiple ATPs to convert a GroEL ring to the folding-active state. Proc Natl Acad Sci USA. 2008;105:19205–19210. doi: 10.1073/pnas.0810657105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grason JP, Gresham JS, Lorimer GH. Setting the chaperonin timer: A two-stroke, two-speed, protein machine. Proc Natl Acad Sci USA. 2008;105:17339–17344. doi: 10.1073/pnas.0807418105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCormack EA, Altschuler GM, Dekker C, Filmore H, Willison KR. Yeast phosducin-like protein 2 acts as a stimulatory co-factor for the folding of actin by the chaperonin CCT via a ternary complex. J Mol Biol. 2009;391:192–206. doi: 10.1016/j.jmb.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 58.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 59.Minton AP. Confinement as a determinant of macromolecular structure and reactivity. Biophys J. 1992;63:1090–1100. doi: 10.1016/S0006-3495(92)81663-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horwich AL, Fenton WA. Chaperonin-mediated protein folding: using a central cavity to kinetically assist polypeptide chain folding. Q Rev Biophys. 2009;42:83–116. doi: 10.1017/S0033583509004764. [DOI] [PubMed] [Google Scholar]
- 61.Todd MJ, Viitanen PV, Lorimer GH. Dynamics of the chaperonin ATPase cycle: Implications for facilitated protein folding. Science. 1992;265:659–666. doi: 10.1126/science.7913555. [DOI] [PubMed] [Google Scholar]
- 62.Weissman JS, Kashi Y, Fenton WA, Horwich AL. GroEL-mediated protein folding proceeds by multiple rounds of binding and release of nonnative forms. Cell. 1994;78:693–702. doi: 10.1016/0092-8674(94)90533-9. [DOI] [PubMed] [Google Scholar]
- 63.Todd MJ, Lorimer GH, Thirumalai D. Chaperonin-facilitated protein folding: optimization of rate and yield by an iterative annealing mechanism. Proc Natl Acad Sci USA. 1996;93:4030–4035. doi: 10.1073/pnas.93.9.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hynes GM, Willison KR. Individual subunits of the eukaryotic cytosolic chaperonin mediate interactions with binding sites located on subdomains of beta-actin. J Biol Chem. 2000;275:18985–18994. doi: 10.1074/jbc.M910297199. [DOI] [PubMed] [Google Scholar]
- 65.Pappenberger G, Wilsher JA, Roe SM, Counsell DJ, Willison KR, Pearl LH. Crystal structure of the CCT gamma apical domaimplications for substrate binding to the eukaryotic cytosolic chaperonin. J Mol Biol. 2002;318:1367–1379. doi: 10.1016/s0022-2836(02)00190-0. [DOI] [PubMed] [Google Scholar]
- 66.Kubota S, Kubota H, Nagata K. Cytosolic chaperonin protects folding intermediates of Gb from aggregation by recognizing hydrophobic b-strands. Proc Natl Acad Sci USA. 2006;103:8360–8365. doi: 10.1073/pnas.0600195103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dekker C, Stirling PC, McCormack EA, Filmore H, Paul A, Brost RL, Costanzo M, Boone C, Leroux MR, Willison KR. The interaction network of the chaperonin CCT. EMBO J. 2008;27:1827–1839. doi: 10.1038/emboj.2008.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brackley KI, Grantham J. Activities of the chaperonin containing TCP-1(CCT): implications for cell cycle progression and cytoskeletal organisation. Cell Stress Chaperones. 2009;14:23–31. doi: 10.1007/s12192-008-0057-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Howlett AC, Gray AJ, Hunter JM, Willardson BM. Role of molecular chaperones in G protein b5/regulator of G protein signaling dimer assembly and G protein by dimer specificity. J Biol Chem. 2009;284:16386–16399. doi: 10.1074/jbc.M900800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–580. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 71.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goyal K, Qamra R, Mande SC. Multiple gene duplication and rapid evolution in the groEL gene: functional implications. J Mol Evol. 2006;63:781–787. doi: 10.1007/s00239-006-0037-7. [DOI] [PubMed] [Google Scholar]
- 73.Archibald JM, Blouin C, Doolittle WF. Gene duplication and the evolution of group II chaperonins: implications for structure and function. J Struct Biol. 2001;135:157–169. doi: 10.1006/jsbi.2001.4353. [DOI] [PubMed] [Google Scholar]
- 74.Kusmierczyk AR, Martin J. Nucleotide-dependent protein folding in the type II chaperonin from the mesophilic archaeon Methanococcus maripaludis. Biochem J. 2003;371:669–673. doi: 10.1042/BJ20030230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Archibald JM, Roger AJ. Gene duplication and gene conversion shape the evolution of Archaeal chaperonins. J Mol Biol. 2002;316:1041–1050. doi: 10.1006/jmbi.2002.5409. [DOI] [PubMed] [Google Scholar]
- 76.Ditzel L, Lowe J, Stock D, Stetter KO, Huber H, Huber R, Steinbacher S. Crystal structure of thermosome, the Archaeal chaperonin and homolog of CCT. Cell. 1998;93:125–138. doi: 10.1016/s0092-8674(00)81152-6. [DOI] [PubMed] [Google Scholar]
- 77.Klumpp M, Baumeister W, Essen EO. Structure of the substrate binding domain of the thermosome, an archaeal group II chaperonin. Cell. 1997;91:263–270. doi: 10.1016/s0092-8674(00)80408-0. [DOI] [PubMed] [Google Scholar]
- 78.Kanzaki T, Iizuka R, Takahashi K, Maki K, Masuda R, Sahlan M, Yebenes H, Valpuesta JM, Oka T, Furutani M, Ishii N, Kuwajima K, Yohda M. Sequential action of ATP-dependent subunit conformational change and interaction between helical protrusions in the closure of the built-in lid of group II chaperonins. J Biol Chem. 2008;283:34773–34784. doi: 10.1074/jbc.M805303200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hongo K, Hirai H, Uemura C, Ono S, Tsunemi J, Higurashi T, Mizobata T, Kawata Y. A novel ATP/ADP hydrolysis activity of hyperthermostable group II chaperonin in the presence of cobalt or manganese ion. FEBS Lett. 2006;580:34–40. doi: 10.1016/j.febslet.2005.11.043. [DOI] [PubMed] [Google Scholar]
- 80.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: An extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 81.Yang C, Jiang W, Chen D-H, Adiga U, Ng EG, Chiu W. Estimating contrast transfer function and associated parameters by constrained non-linear optimization. J Microsc. 2009;233:391–403. doi: 10.1111/j.1365-2818.2009.03137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 83.Harauz G, van Heel M. Exact filters for general geometry three dimensional reconstruction. Optik. 1986;73:146–156. [Google Scholar]
- 84.Acosta-Sampson L, King J. Partially folded aggregation intermediates of human gammaD-, gammaC-, and gammaS-crystallin are recognized and bound by human alphaB-crystallin chaperone. J Mol Biol. 2010;401:134–152. doi: 10.1016/j.jmb.2010.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]