

Abstract
The type III secretion system (T3SS) is a protein injection nanomachinery required for virulence by many human pathogenic bacteria including Salmonella and Shigella. An essential component of the T3SS is the tip protein and the Salmonella SipD and the Shigella IpaD tip proteins interact with bile salts, which serve as environmental sensors for these enteric pathogens. SipD and IpaD have long central coiled coils and their N-terminal regions form α-helical hairpins and a short helix α3 that pack against the coiled coil. Using AutoDock, others have predicted that the bile salt deoxycholate binds IpaD in a cleft formed by the α-helical hairpin and its long central coiled coil. NMR chemical shift mapping, however, indicated that the SipD residues most affected by bile salts are located in a disordered region near helix α3. Thus, how bile salts interact with SipD and IpaD is unclear. Here, we report the crystal structures of SipD in complex with the bile salts deoxycholate and chenodeoxycholate. Bile salts bind SipD in a region different from what was predicted for IpaD. In SipD, bile salts bind part of helix α3 and the C-terminus of the long central coiled coil, towards the C-terminus of the protein. We discuss the biological implication of the differences in how bile salts interact with SipD and IpaD.
Keywords: gram-negative bacteria, type III secretion system, tip protein, SipD, bile salts
Introduction
Many Gram-negative bacteria such as Salmonella, Shigella, and Burkholderia species, which are leading agents of infectious diseases and mortality worldwide, utilize the type III secretion system (T3SS) to inject bacterial proteins directly into their host cells to initiate infections. The structural component of the T3SS is a needle apparatus, which is assembled from over 20 different proteins,1 and consists of a base, an external needle and a tip complex (Supporting Information Fig. S1). The tip complex is assembled by the tip proteins, which are bound directly on top of the needle, and by the translocon proteins, which are membrane-spanning proteins. The T3SS tip proteins are SipD2 in the Salmonella typhimurium pathogenicity island 1 (SPI-1), IpaD3 in Shigella flexneri and BipD in Burkholderia pseudomallei. The crystal structures of IpaD4 and BipD4,5 show common features such as a long central coiled coil that imparts an overall oblong shape to these proteins, and an N-terminal α-helical hairpin. The α-helical hairpin motif is also common to PrgI,6,7 MxiH,8 and BsaL,9 which are the T3SS needle proteins of Salmonella typhimurium, Shigella flexneri, and Burkholderia pseudomallei, respectively.
Shigella and Salmonella are enteric pathogens and their invasiveness to human cells is affected by bile salts.10–14 The intestines are enriched in bile salts such as deoxycholate and chenodeoxycholate and Shigella11,12 and Salmonella15 utilize bile salts as environmental sensors. Bile salts affect the activity of the Shigella11,12 and Salmonella15 T3SS, however, bile salts affect the invasiveness of Shigella and Salmonella in an opposite manner. Bile salts activate the Shigella T3SS and increase the invasiveness of Shigella to epithelial cells,11,12 whereas bile salts repress the T3SS and decrease invasiveness in Salmonella.13,14 The opposing responses of Shigella and Salmonella T3SSs to bile salts are poorly understood. To date, the only T3SS proteins that have been shown to interact directly with bile salts are the Shigella IpaD11 and the Salmonella SipD tip proteins.11,16 Further, IpaD12 and SipD2 are present on the bacterial surface before host cell contact. Thus, IpaD and SipD are exposed to the environment before bacterial invasion and could function as sensors for environmental molecules.
How IpaD and SipD interact with bile salts is not well understood. Stensrud et al.11 used computer docking simulation to predict that an IpaD pocket formed between the long central coiled-coil and the N-terminal α-helical hairpin is the binding site for deoxycholate.11 However, using NMR chemical shift mapping, we showed previously that SipD bound to deoxycholate, chenodeoxycholate and taurodeoxycholate in a different manner from what was predicted for the IpaD-deoxycholate interaction.16 Thus, it is not clear why IpaD and SipD, which are expected to share a high degree of structural similarity, would interact with bile salts differently. Here we report the 1.9 Å resolution crystal structures of SipD in complex with deoxycholate and chenodeoxycholate and show that bile salts bind to SipD in a different manner as predicted for IpaD.
Results
SipD crystallization
The N-terminal 30-39 residues were disordered in the BipD5 and IpaD4 crystals, therefore, the corresponding N-terminal 38 residues of SipD were truncated to form the SipD construct (residues 39-343) used in this study. The lone native cysteine residue (C244), which contributed in a slight dimerization of SipD during purification, was mutated into a serine residue. Both wild-type (WT) and C244S mutant SipD afforded high level of expression in E. coli as fusion proteins with GB1 (the 56-residue B1 immunoglobulin domain of Streptococcal protein G). After purification by Ni2+-affinity chromatography and digestion with the tobacco etch virus protease, purified SipD (WT and C244S forms) formed crystals under crystallization conditions. The SipD-deoxycholate complex was obtained by cocrystallization whereas the SipD-chenodeoxycholate complex was obtained by soaking apo SipD crystals in chenodeoxycholate. Other bile salts such as taurodeoxycholate and cholate hydrate failed to crystallize with SipD. The four crystals reported here—the two apo forms (WT and C244S) and the two bound forms (with deoxycholate and chenodeoxycholate) yielded high quality X-ray diffraction data (Table I). The SipD crystals crystallized with C2 space group and contained two molecules (A and B) in the asymmetric unit and were refined to 1.7 Å for apo SipD WT, 1.9 Å for apo SipD C244S; and 1.9 Å for the SipD-deoxycholate and SipD-chenodeoxycholate complexes (Table I). For the SipD-deoxycholate complex, the buried surface area between molecules A and B was 520 Å2 as determined by PISA.17
Table I.
Crystallographic Statistics for the SipD Crystals
| apo SipD | apo SipD C244S | SipD C244S –deoxycholate | SipD-chenodeoxycholate | |
|---|---|---|---|---|
| PDB ID | 3NZZ | 3O00 | 3O01 | 3O02 |
| Data collection | ||||
| Unit-cell parameters (Å,°) | a = 203.06 | a = 203.35 | a = 201.93 | a = 202.22 |
| b = 52.25 | b = 52.15 | b = 52.33 | b = 52.37 | |
| c = 57.52 | c = 57.56 | c = 57.31 | c = 57.32 | |
| β = 90.05 | β = 90.45 | β = 90.3 | β = 90.20 | |
| Space group | C2 (No. 5) | C2 (No. 5) | C2 (No. 5) | C2 (No. 5) |
| Resolution rangea | 50.0–1.65 (1.71–1.65) | 50.0–1.85 (1.92–1.85) | 50.0–1.9 (1.97–1.90) | 50.0–1.9 (1.97 – 1.90) |
| Wavelength (Å) | 1.0000 | 1.000 | 1.000 | 1.000 |
| Temperature (K) | 100 | 100 | 100 | 100 |
| Observed reflections | 256,708 | 195,676 | 170,803 | 169,943 |
| Unique reflections | 69,282 | 50,066 | 46,924 | 46,661 |
| <I/σ(I)>a | 39.2 (1.9) | 36.1 (3.5) | 20.3 (1.9) | 27.9 (3.7) |
| Completeness | 95.5 (86.1) | 97.0 (81.6) | 98.5 (88.1) | 97.9 (83.5) |
| Redundancya | 3.7 (3.0) | 3.9 (3.3) | 3.6 (3.1) | 3.69 (2.8) |
| Rmerge (%)ab | 4.4 (49.6) | 5.3 (32.4) | 5.3 (38.1) | 5.3 (23.0) |
| Refinement | ||||
| Resolution (Å) | 25.35 – 1.65 | 32.07–1.85 | 27.98–1.90 | 24.97–1.90 |
| Reflections (working/test) | 62,042/3, 296 | 46,099/2,477 | 42,527/2,249 | 43,463 / 2,308 |
| Rfactor/Rfree (%)c | 19.7/23.7 | 19.5 / 24.6 | 19.6 / 25.8 | 18.5 / 23.6 |
| No. of atoms (protein (A:B)/Ni2+/ligand/water) | 2,190:2,018/1/348 | 2,186:2,019/1/297 | 2,184:2,053/1/28/252 | 2,177:2,032/1/28/330 |
| Model quality | ||||
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.014 | 0.015 | 0.017 | 0.018 |
| Bond angles (°) | 1.478 | 1.461 | 1.556 | 1.614 |
| Average B factor (Å2) | ||||
| All atoms | 29.6 | 29.7 | 32.0 | 28.2 |
| Protein (chain A/B) | 29.0 / 29.2 | 28.6 / 30.3 | 31.5 / 32.3 | 27.6 / 27.9 |
| Ni2+ | 22.0 | 21.3 | 26.6 | 21.8 |
| Deoxycholate or chenodeoxycholate | – | – | 28.6 | 26.2 |
| Water | 35.4 | 34.0 | 34.6 | 33.5 |
| Coordinate error based on maximum likelihood (Å) | 0.25 | 0.26 | 0.26 | 0.26 |
| Ramachandran plot | ||||
| Most favored (%) | 99.1 | 98.9 | 98.9 | 98.9 |
| Additionally allowed (%) | 0.9 | 1.1 | 1.1 | 1.1 |
Values in parentheses are for the highest resolution shell.
Rmerge = ΣhklΣi|Ii(hkl) − <I(hkl)>|/ΣhklΣi|Ii(hkl), where Ii(hkl) is the intensity measured for the ith reflection and <Ii(hkl)> is the average intensity of all reflections with indices hkl.
Rfactor = Σhkl||Fobs (hkl)| − |Fcalc (hkl)||/Σhkl|Fobs (hkl)|; Rfree is calculated in an identical manner using 5% of randomly selected reflections that were not included in the refinement.
Overall structure of SipD
The crystal structures of SipD in the apo and bound forms are nearly identical (Fig. 1 and Supporting Information Fig. S2). For the SipD-deoxycholate crystal (Fig. 1), the final model includes residues 39-342 for molecule A and 46-336 for molecule B. However, residues 110-132 in molecule A and residues 92-94 and 118-134 in molecule B can not be fit to the electron density due to disorder. SipD is a highly α-helical protein, with 62% of residues in 8 α-helices (α1-α8), 6% residues in 5 short beta strands (β1-β5), and the rest in loops and disordered regions. A prominent structural feature of T3SS tip proteins is a long central coiled coil.4,5,18 In SipD, this long coiled coil is formed by helix α4 and helix α8, which defines a central axis and imparts an overall oblong shape to SipD (Fig. 1). Among the four SipD crystals, there is a slight variability in the length of helix α4 – it is shortest (spanning residues 133-177) in molecule A of apo SipD (WT and C244S), and it is longest (spanning residues 136-177) in molecule B of the SipD-chenodeoxycholate complex. There is also variability regarding the length of helix α8, which is the longest helix in SipD and defines its entire length. Helix α8 can be as long as 55 residues (in molecule A of apo and bound SipD) or as short as 49 residues (in molecule B of apo SipD).
Figure 1.
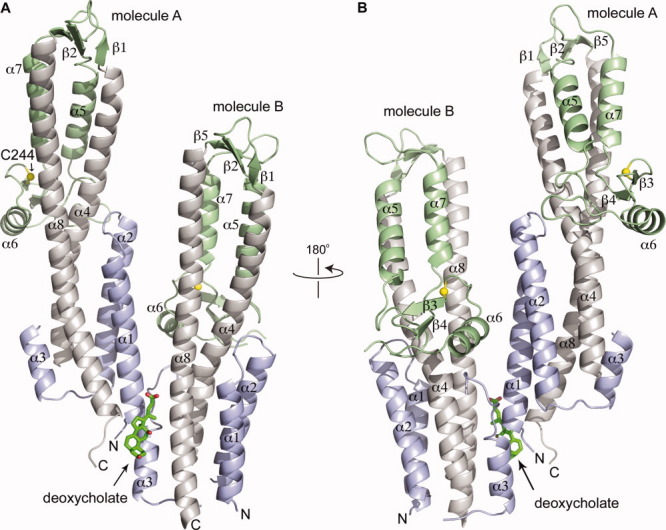
Crystal structure of the SipD-deoxycholate complex. Deoxycholate (shown as a stick model) binds at the interface of molecules A and B of the asymmetric unit. SipD is colored as follows: the coiled coil (helix α4 and α8), gray; the N-terminal region (helix α1-α3), blue; and the mixed α/β domain, green. The crystal structures of SipD-chenodeoxycholate complex and apo SipD (WT and C244S) are similar to the SipD-deoxycholate crystal and are shown in the Supporting Information.
The long central coiled coil as depicted in Figure 1 orients an arbitrary “top” and “bottom” of the molecule. On top of the coiled coil sits three short antiparallel β-strands (β1, β2, and β5) (Fig. 1), which in turn is followed by a small mixed α/β domain formed by three α-helices (α5, α6, and α7) and two β strands (β3 and β4). The mixed α/β domain extends into the middle part of the coiled coil (Fig. 1). Finally, at the bottom of the coiled coil, the SipD N-terminal region (residues 39-132) forms 3 helices (α1, α2, and α3). Helix α1 and α2 form an α-helical hairpin that packs on one face of the coiled coil and on the opposite face packs helix α3 and a long 23-residue loop (residue 110-132). The α-helical hairpin of molecule A is in close contact with molecule B (Fig. 1), which likely contributes in the variability of the lengths of the α-helical hairpins of molecules A and B. The α-helical hairpin is longer in molecule A compared to molecule B.
Binding site of bile salts in SipD
Crystals of the SipD-deoxycholate and SipD-chenodeoxycholate complexes show a large amount of positive difference density (Fo-Fc) that are consistent with deoxycholate [Fig. 2(A)] and chenodeoxycholate [Fig. 2(B)]. The deoxycholate and chenodeoxycholate molecules are located between the ends of a noncrystallographic SipD dimer but are also in close proximity to another molecule A related by a crystallographic (011) translation (Supporting Information Fig. S3). Deoxycholate binds in a hydrophobic pocket [Fig. 2(A)] with a surface area of about 520 Å2 and formed by residues on the C-terminus (K338 and F340) and helix α1 (R41 and I45) of molecule A, and residues of helix α3 towards the loop 110-134 (N104 and A108) and helix α8 (N321, L322 and L318) of molecule B. The carboxylate of deoxycholate is in hydrogen bonding distance with the side chains of K338 and N321 whereas the rest of deoxycholate is surrounded by hydrophobic residues [Fig. 2(A)]. Two residues (S221 and G222) from another molecule A related by a crystallographic (011) translation are also in close proximity to deoxycholate. For the SipD-chenodeoxycholate complex, the same residues that are in close proximity to deoxycholate [Fig. 2(A)] are also involved in binding chenodeoxycholate [Fig. 2(B)]. Binding of deoxycholate and chenodeoxycholate to SipD is mainly through hydrophobic contacts and stabilized by hydrogen bonds. The apo and bound forms of SipD differ by only 0.34 Å Cα backbone rmsd, however, the side chains of F340 and N104 reorient to accommodate the bile salts in the binding pocket [Fig. 2(C)]. By comparison, our previous NMR results showed the largest chemical shift perturbations of SipD residues near (S96) or on loop 110-134 (S114, L116, F117, and E133) upon binding to bile salts.16
Figure 2.
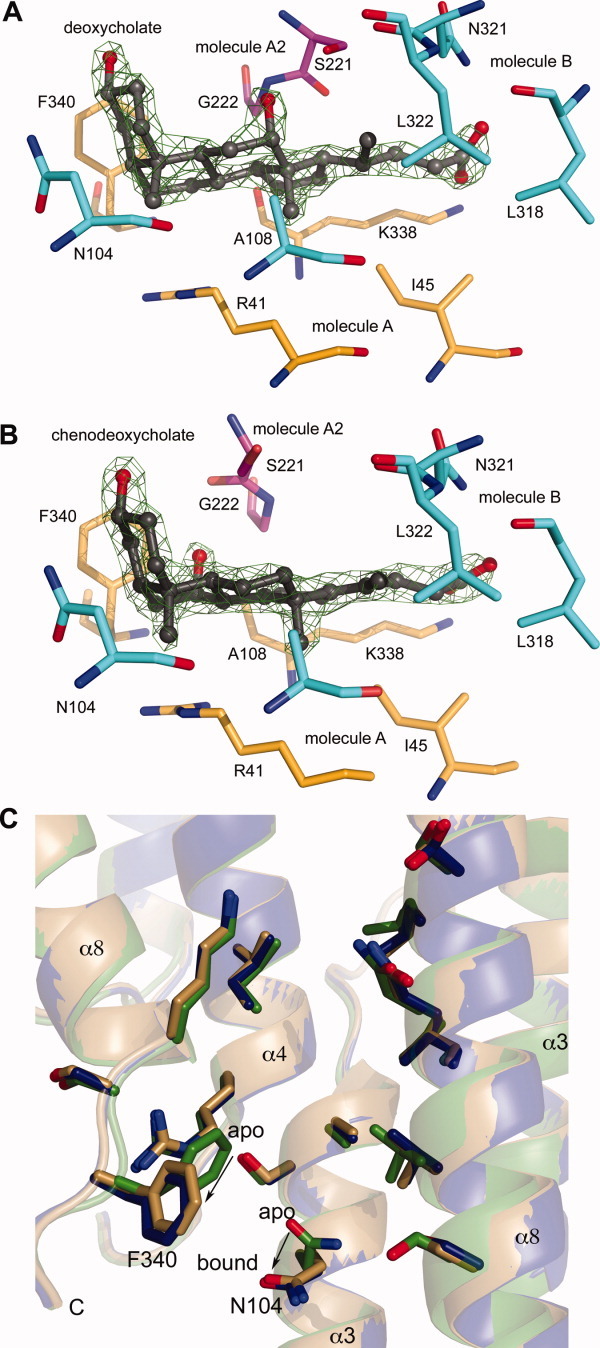
Fo-Fc omit maps contoured at 3σ of (A) deoxycholate and (B) chenodeoxycholate in complex with SipD shown with residues within 5 Å of bile salts (color scheme: molecule A, yellow; molecule B, cyan; and another molecule A related by a crystallographic (011) translation, pink). (C) Conformational changes in SipD upon binding deoxycholate and chenodeoxycholate (color scheme: apo SipD, green; SipD-deoxycholate complex, blue; and SipD-chenodeoxycholate complex, gold). Bile salts have been removed for clarity. Upon binding bile salts, the side chain of F340 shifts by 5.8 Å and that of N104 by 2.1 Å.
Effect of SipD mutations on bacterial invasiveness
To determine which regions of SipD might be important for function, we used the Salmonella invasion assay to assess the effect of SipD mutations on the ability of Salmonella to invade cultured human epithelial cells. We made point mutations and deletions (shown in Supporting Information Fig. S4) in the loop region between helix α3 and α4 (Δ116-124), the β2 beta strand (Δ186-190 and Δ187-188), and the extreme C-terminal five residues (CΔ5) based on the following rationale: previously, we reported that residues in the 23-residue loop between helix α3 and α4 loop and the β2 beta strand showed significant chemical shift perturbation upon titration with bile salts16 and in the crystal structures reported here, the C-terminus of SipD formed an ordered loop that is in close contact with the bile salts [Fig. 1 and Supporting Information Fig. S2(A)]. Results of Salmonella invasion assay with respect to mutations in SipD are shown in Figure 3(A). Overall, point mutations (F117A and Q124A) did not affect bacterial invasiveness, although some point mutations (L116A, V187A, and K188A) decreased invasiveness. A 9-residue deletion (Δ116-124) in the 23-residue loop (spanning residues 110-134) did not affect bacterial invasiveness, thus the long loop between helix α3 and α4, which had no electron density in the crystal structure, was not important in the function of SipD. The most drastic effect on the invasiveness occurred from short deletions in the β2 strand (Δ186-187 and Δ187-188) and the extreme C-terminus (CΔ5), which were noninvasive. The noninvasiveness of the SipD CΔ5 mutant was similar to the results of the Shigella IpaD CΔ5 mutation reported previously.3
Figure 3.
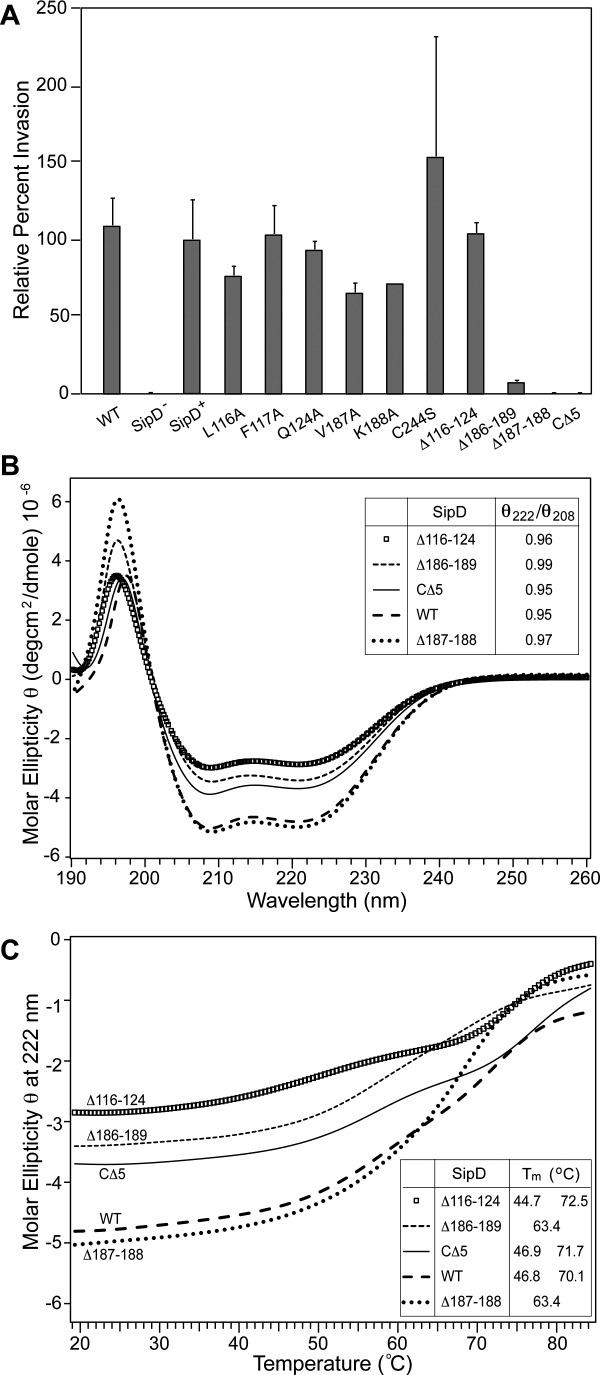
(A) Results of Salmonella invasion assay (legends: WT, wild-type strain; sipD−, sipD null strain; SipD+, wild-type sipD introduced by the plasmid pRK2-sipD into the sipD− strain; L116A, F117A, Q124A, K188A, and C244S are SipD point mutations in pRK2-sipD; Δ116-124, Δ186-189, Δ187-188, and CΔ5 are SipD deletions in pRK2-sipD). (B) CD spectra of SipD and deletion mutants (insert: ratio of molar ellipticity at 222 and 208 nm). (C) CD thermal denaturation monitored by the molar ellipticity at 222 nm of SipD and various deletion mutants (insert: estimated transition temperatures, Tm).
CD and NMR of SipD deletion mutants
We used CD spectroscopy to assess the folding of the SipD deletion mutants (Δ116-124, Δ186-189, Δ187-188, and CΔ5) that were noninvasive. The CD plots for WT and mutant SipD [Fig. 3(B)], showing minima at 208 and 222 indicated folded and highly α-helical proteins. Further, the ratio of molar ellipticity at 222 and 208 nm of nearly 1.0 [insert, Fig. 3(B)] indicated extensive interhelical contacts.19–21 However, the Δ186-189 and Δ187-188 deletions showed different melting behavior compared to WT SipD [Fig. 3(C)]. This suggested conformational changes in the folding of the Δ186-189 and Δ187-188 deletions, which showed only one transition temperature of 63.4° [Fig. 3(C)] compared to two transition temperatures for WT SipD.
NMR spectroscopy indicated that the CΔ5, Δ186-189, and Δ187-188 deletion mutants were folded (Fig. 4). Because the four SipD tryptophan residues (W135, W177, W234, and W290) were located in different regions of SipD (Supporting Information Fig. S4) and their side chain NMR resonances were easily identified (boxed in Fig. 4), we used the tryptophan side chain resonances to assess the overall changes in the conformation of SipD for the CΔ5, Δ186-189, and Δ187-188 deletions. Among the deletions, the Δ186-189 deletion showed the most drastic change in the tryptophan side chain resonances (Fig. 4). The W177 side chain resonance of Δ186-189 [Fig. 4(C)] changed significantly compared to WT SipD [Fig. 4(A)]. W177 was located at an end of helix α4 and faced helix α5, which immediately followed the β2 strand [Supporting Information Fig. S4(B)]. The β2 strand was deleted in Δ186-189, thus changing the local environment of the W177 side chain with respect to helix α5. Nevertheless, the three other tryptophan residues (W135, W234, and W290) showed similar side chain resonances as WT indicating there were no drastic conformation changes in other regions of SipD surrounding W135, W234, and W290. W135 (in helix α4) was buried in a pocket formed by the central coiled coil (helix α4/α8) and the N-terminal helices (α1, α2, and α3), W234 (in helix α6) faced the middle portion of the central coiled coil, and W290 (in helix α8) faced helix α7 of the mixed α/β domain. Based on the tryptophan side chain resonances (Fig. 4), the Δ186-189 deletion resulted in a more significant change in the conformation of SipD compared to the CΔ5 and Δ187-188 deletions.
Figure 4.

Two-dimensional 1H-15N TROSY spectra of (A) SipD (residues 39-343) and the (B) CΔ5, (C) Δ186-189 and the (D) Δ187-188 deletion mutants (boxed: tryptophan side chain peaks). Peak assignments for SipD were reported previously.16 The noise peak (asterisk) in the tryptophan side chain region in (C) is absent in the 2D 1H-15N HSQC spectrum.
Discussion
Previous results have established that bile salts activate the Shigella T3SS11,12 and repress the Salmonella T3SS,14 and that bile salts bind directly to the T3SS tip proteins IpaD11 and SipD.11,16 Here, we report the crystal structure of SipD in complex with the bile salts deoxycholate and chenodeoxycholate. The main significance of this work is that the crystal structures reported here are currently the only available atomic structures of a T3SS tip protein in complex with bile salts. SipD and IpaD have backbone Cα rmsd of 1.4 Å, however, the binding site of bile salts in SipD is different from what was predicted previously for IpaD. Using AutoDock, Stensrud et al.11 predicted that bile salts bind at the interface formed by the N-terminal α-helical hairpin (equivalent to SipD helix α1/α2, Fig. 1) and the coiled coil motif (equivalent to SipD helix α4/α8). For the SipD-bile salt complexes [Fig. 1 and Supporting Information Fig. S2(A)], crystallography shows that bile salts bind at the interface of two SipD molecules, suggesting that SipD oligomerization is important in bile salt binding.
SipD is expected to oligomerize at the tip of the Salmonella needle based on the following observations: the Yersinia pestis22 and Shigella flexneri8,23 tip proteins form complexes on top of their respective needles.22 Further, in Shigella, there are 5.6 needle subunits per turn of the needle,8 thus, it is estimated that the Shigella tip protein forms a pentameric complex on top of the needle.4,23 In solution, the T3SS tip proteins of Shigella flexneri,4 Pseudomonas aeruginosa,24 and Yersinia pestis24 oligomerize into tetramers or pentamers under certain conditions. Recently, Galkin et al.25 showed there are 6.3 needle subunits per turn in the Salmonella needle, thus, it is expected that 6 SipD molecules may form a complex on top of the Salmonella needle. We propose that SipD complex formation on top of the needle could provide the binding pocket for bile salts. The oligomerization of SipD with respect to binding bile salts could explain the different results of crystallography and NMR spectroscopy. NMR previously indicated that the loop 110-134 showed the largest chemical shift perturbations upon bile salt interaction, whereas in the crystal structures presented here, the extreme C-terminus of SipD appears to be the major site involved in bile salt binding. Under NMR conditions, SipD is monomeric and bile salts mainly affect residues that are near or on loop 110-134.16 Upon oligomerization, bile salts bind at the interface of two SipD molecules formed partly by the CΔ5 residues.
What could explain the difference in the bile salts binding sites in IpaD and SipD? It is currently unknown how a T3SS tip protein docks on top of an assembled needle. Based on the structural similarity of the tip protein α-helical hairpin with the needle protein α-helical hairpin, others have hypothesized that the tip protein central coiled coil is the primary binding site for the needle monomers.8,23 We have recently shown by NMR paramagnetic relaxation enhancement (PRE) that a region in the SipD coiled coil facing the N-terminal α-helical hairpin is the binding site for the Salmonella needle protein PrgI (Rathinavelan et al., submitted for publication). The PRE data regarding SipD-PrgI interaction together with the recent atomic model of the Salmonella needle25 constrains the possible orientation of SipD on top of the needle, allowing us to model how SipD docks on top of the needle. In this model, the C-terminal region of helix α8 and the C-terminus of SipD are expected to be facing towards the needle channel. Thus, the bile salts binding site in SipD is expected to be facing towards the needle channel. We hypothesize that bile salts, by binding in a region of SipD that is expected to form the wall of the needle channel, could be in a position to interfere with the passage of other proteins through the needle, which could lead to the inactivation of the Salmonella T3SS in the presence of bile salts.14 On the other hand, in Shigella, bile salts are proposed to bind between the IpaD coiled coil motif and the α-helical hairpin, which in the assembled needle-tip complex is expected to be positioned away from the needle channel. This binding results in the activation of the Shigella T3SS by promoting the assembly of the translocon.12 Thus, the difference in the binding of bile salts in IpaD and SipD probably accounts for the observed differences in the behavior of the Shigella and Salmonella T3SSs with respect to bile salts. It is also possible that the difference in the proposed bile salts binding sites in SipD and IpaD are due to the difference in the techniques used (crystallography vs. AutoDock).
Comparison with other tip proteins
Together with the Salmonella SipD structure presented here, there are currently crystal structures of five T3SS tip proteins (Fig. 5): Yersinia LcrV,18 Shigella IpaD,4 Burkholderia BipD,4,5 and a SipD homolog from a soil bacterium, Chromobacterium violaceum,26 a Gram-negative soil bacterium. Of the five structures, IpaD, BipD and SipD are structurally more similar to each other than LcrV in that their N-terminal regions form α-helical hairpins (colored blue in Fig. 5) that pack at one end of the central coiled coil, whereas LcrV lacks this domain. Instead, LcrV has a protein chaperone, LcrG (95 residues) that is expected to be highly helical and interact with LcrV.27–30 IpaD, BipD and SipD have no known chaperones, hence, the α-helical hairpins were suggested to function as self-chaperones for IpaD and BipD.4
Figure 5.
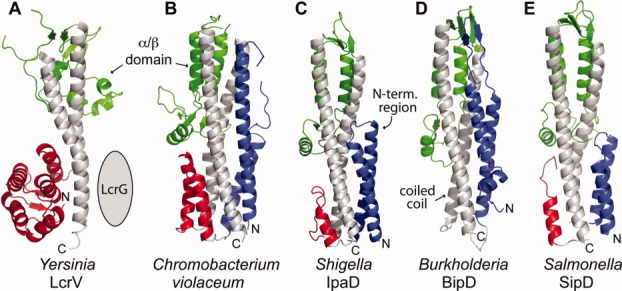
Comparison of five current crystal structures of T3SS tip proteins: (A) Yersinia LcrV,18 (B) a SipD homolog from Chromobacterium violaceum, (C) Shigella IpaD,4 (D) Burkholderia BipD,4,5 and (E) Salmonella SipD. The central coiled coil (gray) and mixed α/β domain (green) are common structural features of T3SS tip proteins. The N-terminal region in (A) forms a globular domain of α-helices and β-strands (red), whereas in (B–E), the N-terminal region forms α-helical hairpins (blue) followed by short α-helices (red) in (B, D, and E).
Another importance of the SipD crystal structure reported here is its usefulness in modeling how SipD is assembled on top of the Salmonella needle and how the translocon is assembled on top of the SipD complex (Supporting Information Fig. S1). Among the T3SS tip proteins (Fig. 5), IpaD and SipD are the closest structural homologs with an overall Cα RMSD of 1.4 Å; thus the IpaD crystal structure might be used to obtain a homology model of the SipD structure. However, there are significant differences in how the N-terminal regions (comprising helix α1-α3) of IpaD and SipD pack against their respective central coiled coils. The α-helical hairpins of IpaD and SipD pack differently by about 18° from each other with respect to their corresponding central coiled coils [Fig. 6(A)], and the packing of helix α3 to the central coiled coil in IpaD and SipD differs by an angle of 26° [Fig. 6(B)]. Because the region of the coiled coil facing the α-helical hairpin on one side and helix α3 on the other side is hypothesized to be important in how the tip protein docks on top of the assembled needle, the significant differences in packing angles may suggest differences in how IpaD and SipD might dock on their respective needles. Additionally, the difference in the number of needle monomers per turn in the Salmonella25 and the Shigella8 needles suggests that the packing of the Salmonella and Shigella tip proteins would be different as well. Thus, using an experimentally derived high-resolution structure of SipD to model the SipD-needle complex would lead to a more accurate result rather than using a homology modeled structure of SipD.
Figure 6.
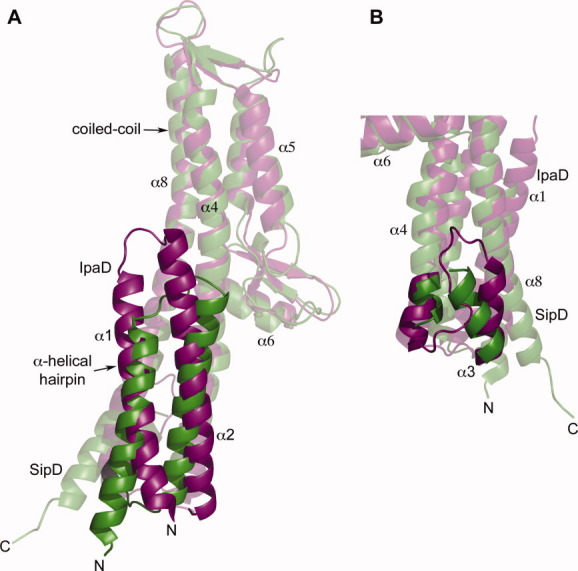
The major differences in the crystal structures of IpaD (purple) and SipD (green) are in the packing of the N-terminal α-helical hairpin with the coiled coil. (A) The SipD α-helical hairpin packs on the central coiled coil at a different angle of 18° compared to the α-helical hairpin of IpaD, and (B) the SipD helix α3 pack at a different angle of 26° compared to helix α3 of IpaD (also, helix α4 of IpaD is longer by one turn).
In summary, we present high-resolution crystal structures of SipD in the apo form and in complex with bile salts. Currently, these are the only available atomic resolution crystal structures of a T3SS tip protein in complex with bile salts. These structures are important in identifying how bile salts interact with a T3SS tip protein and are needed in modeling how SipD is assembled at the tip of the needle.
Materials and Methods
Protein expression and purification
SipD (residues 39-343) with WT or C244S point mutation was overexpressed in Escherichia coli using the pDZ1-SipD plasmid as a fusion protein with the Streptococcal GB1 domain and a hexahistidine tag and purified by nickel affinity chromatography as described.16 Following cleavage of the fusion protein with the tobacco etch virus protease,16 purified SipD was dialyzed against 1 L of buffer (20 mM Tris, 196 mM NaCl, pH 8.0), concentrated to ∼20 mg/mL (∼0.6 mM) and stored at 4°C before crystallization.
CD spectroscopy
Samples for circular dichroism spectroscopy contained 0.15 mM protein in buffer (20 mM Tris, 196 mM NaCl, pH 8.0) and CD spectra were acquired in triplicate using a JASCO J-815 Spectropolarimeter. Wavelength scans were collected at 20°C at a scan rate of 50 nm/min and thermal denaturation curves were monitored at 222 nm at a temperature ramp rate of 15°C/h.
Preparation of Salmonella sipD knockout strain
A Salmonella typhimurium sipD nonpolar knockout strain was constructed using the lambda Red recombinase method.31 WT Salmonella typhimurium SL1344 strain was obtained from Dr. Bradley Jones (University of Iowa) and the plasmids pKD46 and pKD331 were obtained from the E. coli Genetic Stock Center (Yale University). Briefly, WT Salmonella typhimurium SL1344 strain was electroporated with pKD46,31 which contained the genes needed for lambda Red-mediated recombination, and grown at 30°C. The chloramphenicol resistance gene in pKD3 was PCR-amplified using primers that contained complementary sequences to the chloramphenicol resistance gene and the sipD flanking regions and the PCR product was electroporated into the SL1344/pKD46 strain. Transformants were grown at 37°C to expel the temperature-sensitive pKD46 plasmid and selected against chloramphenicol resistance. PCR and DNA sequencing confirmed the sipD nonpolar null mutation. To rescue the sipD null mutation, full-length sipD was subcloned into the NdeI/SalI site of pRK2.32 Mutations in sipD were introduced by PCR in pRK2-sipD for invasion assay and in pDZ1-SipD for protein expression. Mutations were confirmed by DNA sequencing.
Salmonella invasion assay
The effect of sipD mutations on the ability of S. typhimurium to invade a cultured human epithelial cell line (Henle 407) was assayed as follows. Henle 407 cells (American Type Culture Collection CCL-6) were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% newborn calf serum at 37°C in 5% CO2 in 24-well plates before confluence (which typically occurred within 24 hours). The plasmid pRK2-sipD with either WT or mutant sipD was electroporated into the S. typhimurium sipD− strain and single colonies were inoculated into LB media supplemented with 25 mg/L trimethoprim, 50 mg/L ampicillin, 50 mg/L kanamycin and grown in standing cultures overnight. A 10 mL LB culture with 1 mM IPTG was inoculated with 1 mL of overnight bacterial growth, and incubated at 37°C on standing for 2.5-3 hours (until A600 ∼0.4). Approximately 15-30 uL of bacterial suspension was added with 300 uL DMEM into the Henle 407 cells and incubated at 37°C for 60 min to allow invasion before the suspension was removed by aspiration. The Henle 407 cells were incubated with fresh DMEM with 100 mg/L of gentamycin for 1.5 hours, aspirated, rinsed with DMEM, and lysed with 1% Triton X-100 to free the entrapped bacteria. The number of bacterial colonies, which correlated with invasiveness, was estimated by serial dilution and plating. The Salmonella invasion assay was done in triplicate for each sipD construct.
Crystallization of apo SipD
WT and C244S SipD were concentrated to 20 mg/mL in buffer (20 mM Tris pH, 196 mM NaCl pH 8.0) and screened for crystallization in Compact Jr. (Emerald Biosystems) sitting drop plates using 0.75 uL of protein and crystallization solution (Hampton Research) equilibrated against 100 uL of the latter. Crystals were obtained in ∼24 hours at 20°C. The WT SipD crystallized in the Hampton Research Index screen condition 69 (25% (w/v) PEG 3350, 100 mM Tris pH 8.5, 200 mM (NH4)2SO4) and SipD C244S crystallized in Index screen condition 45 (25% (w/v) PEG 3350, 100 mM Tris pH 8.5). Single crystals were sequentially transferred to solutions containing 25% (w/v) PEG 3350, 100 mM Tris pH 8.5 and 10% glycerol and frozen in the same solution containing 15–20% glycerol for X-ray data collection.
Crystallization of SipD-deoxycholate/chenodeoxycholate complexes
A SipD C244S-deoxycholate solution was prepared by mixing 1 μL 400 mM sodium deoxycholate (Amresco) with 100 μL of 0.3 mM SipD C244S. For crystallization, hanging drops were prepared by mixing 1.5 μL of SipD C244S-deoxycholate solution with 1.5 uL crystallization screening solution on a cover slip and placed over 600 μL of reservoir volume and stored at 25°C. The SipD C244S-deoxycholate cocrystals grew in about 12 days in crystallization solution containing 0.2 M magnesium formate dihydrate and 20% (w/v) PEG 3350. The amount of PEG was optimized and diffraction-quality crystals were obtained with 0.2 M magnesium formate dihydrate and 15% PEG 3350 in 8 days. To crystallize the SipD-chenodeoxycholate complex, crystals of WT SipD were soaked for 4 hours in 4 mM chenodeoxycholate (Sigma) dissolved in crystallization solution. Crystals suitable for X-ray diffraction were soaked in 80% crystallization solution and 20% glycerol and flash frozen in liquid nitrogen.
Crystal structure determination
X-ray diffraction data were collected to 1.9 Å at the Advanced Photon Source (APS) beamline 17BM (IMCA-CAT) using an ADSC Q210 CCD detector. Intensities were integrated and scaled using the HKL2000 package.33 For the SipD-deoxycholate cocrystal, indexing indicated a C-centered orthorhombic lattice or a C-centered monoclinic lattice with the β angle ∼90° for the latter. The Laue class was checked using POINTLESS34 which yielded the highest score for 2/m indicating that C2 was indeed the correct space group. A solvent content of 46.2% (Vm = 2.3 Å3/Da) was calculated for two molecules in the asymmetric unit. Additionally, the self rotation function calculated with POLARRFN35 using data between 15 and 3.5 Å resolution and an integration radius of 20 Å yielded a peak on the κ = 180° section at ω = 49.1°, φ = 180° indicating the presence of a noncrystallographic 2-fold axis. Structure solution was carried out by molecular replacement with PHASER36 and the pathogenicity island 1 effector protein from Chromobacterium violaceum (PDB ID: 2P7N)26 served as the search model. The amino acid sequence of 2P7N was 50% identical to SipD and a homology model for molecular replacement was created using CHAINSAW.35 Rotation and translation searches for two molecules in the asymmetric unit yielded a clear solution and initial refinement converged at R = 42%, Rfree = 46%. The structure was improved by employing automated model building with ARP/WARP37 and successive rounds of manual model building and refinement with COOT38 and REFMAC,39 respectively. During successive rounds of refinement, a large difference density peak was observed at the N-terminus of molecule A of the SipD-deoxycholate crystal. Subsequently, residues G36, H37 and M38, which were cloning artifacts, were fit to the residual electron density. Following refinement with these residues included, a large peak of difference density remained that appeared to be coordinated in a square planar arrangement to G36 and H37 of molecule A and H40 from another molecule A related by a crystallographic 2-fold rotation. A Ni2+ ion was ultimately assigned at this site, which was likely obtained during purification by Ni2+-affinity chromatography. Molecular replacement by MOLREP40 was used to solve the structures of apo SipD, apo SipD C244S mutant and the SipD-chenodeoxycholate complex. Only the protein portion of the refined structure of SipD-deoxycholate crystal was used as search model to solve the crystal structures of apo SipD and SipD-chenodeoxycholate complex, whereas the refined structure of apo SipD was used as search model to determine the crystal structure of the apo SipD C244S mutant. Structures were validated using MOLPROBITY41 and analyzed using PYMOL.42 Helix crossing angles were calculated using MOLMOL43 and buried surface areas were calculated using PISA.17
Structure Coordinates
Coordinates have been deposited at the Protein Data Bank with accession codes: 3NZZ, 3O00, 3O01, and 3O02.
Acknowledgments
The authors thank Dr. Audrey Lamb, Dr. Qianyi Luo, and Andrew Ouellette for assistance in crystallography and to Asokan Anbanandam for NMR spectroscopy. They thank the Industrial Macromolecular Crystallography Association, the Hauptman-Woodward Medical Research Institute and the U. S. Department of Energy Office of Basic Energy Sciences (Contract No. W-31-109-Eng-38) for the use of the IMCA-CAT beamline 17-BM at the Advanced Photon Source.
References
- 1.Cornelis GR. The type III secretion injectisome. Nat Rev Microbiol. 2006;4:811–825. doi: 10.1038/nrmicro1526. [DOI] [PubMed] [Google Scholar]
- 2.Lara-Tejero M, Galan JE. Salmonella enterica serovar typhimurium pathogenicity island 1-encoded type III secretion system translocases mediate intimate attachment to nonphagocytic cells. Infect Immun. 2009;77:2635–2642. doi: 10.1128/IAI.00077-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Espina M, Olive AJ, Kenjale R, Moore DS, Ausar SF, Kaminski RW, Oaks EV, Middaugh CR, Picking WD, Picking WL. IpaD localizes to the tip of the type III secretion system needle of Shigella flexneri. Infect Immun. 2006;74:4391–4400. doi: 10.1128/IAI.00440-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson S, Roversi P, Espina M, Olive A, Deane JE, Birket S, Field T, Picking WD, Blocker AJ, Galyov EE, Picking WL, Lea SM. Self-chaperoning of the type III secretion system needle tip proteins IpaD and BipD. J Biol Chem. 2007;282:4035–4044. doi: 10.1074/jbc.M607945200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erskine PT, Knight MJ, Ruaux A, Mikolajek H, Sang NW, Withers J, Gill R, Wood SP, Wood M, Fox GC, Cooper JB. High resolution structure of BipD: an invasion protein associated with the type III secretion system of Burkholderia pseudomallei. J Mol Biol. 2006;363:125–136. doi: 10.1016/j.jmb.2006.07.069. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Ouellette AN, Egan CE, Rathinavelan T, Im W, De Guzman RN. Differences in the electrostatic surfaces of the type III secretion needle proteins PrgI, BsaL, and MxiH. J Mol Biol. 2007;371:1304–1314. doi: 10.1016/j.jmb.2007.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poyraz O, Schmidt H, Seidel K, Delissen F, Ader C, Tenenboim H, Goosmann C, Laube B, Thunemann AF, Zychlinsky A, Baldus M, Lange A, Griesinger C, Kolbe M. Protein refolding is required for assembly of the type three secretion needle. Nat Struct Mol Biol. 2010;17:788–792. doi: 10.1038/nsmb.1822. [DOI] [PubMed] [Google Scholar]
- 8.Deane JE, Roversi P, Cordes FS, Johnson S, Kenjale R, Daniell S, Booy F, Picking WD, Picking WL, Blocker AJ, Lea SM. Molecular model of a type III secretion system needle: Implications for host-cell sensing. Proc Natl Acad Sci USA. 2006;103:12529–12533. doi: 10.1073/pnas.0602689103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L, Wang Y, Picking WL, Picking WD, De Guzman RN. Solution structure of monomeric BsaL, the type III secretion needle protein of Burkholderia pseudomallei. J Mol Biol. 2006;359:322–330. doi: 10.1016/j.jmb.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 10.Pope LM, Reed KE, Payne SM. Increased protein secretion and adherence to HeLa cells by Shigella spp. following growth in the presence of bile salts. Infect Immun. 1995;63:3642–3648. doi: 10.1128/iai.63.9.3642-3648.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stensrud KF, Adam PR, La Mar CD, Olive AJ, Lushington GH, Sudharsan R, Shelton NL, Givens RS, Picking WL, Picking WD. Deoxycholate interacts with IpaD of Shigella flexneri in inducing the recruitment of IpaB to the type III secretion apparatus needle tip. J Biol Chem. 2008;283:18646–18654. doi: 10.1074/jbc.M802799200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olive AJ, Kenjale R, Espina M, Moore DS, Picking WL, Picking WD. Bile salts stimulate recruitment of IpaB to the Shigella flexneri surface, where it colocalizes with IpaD at the tip of the type III secretion needle. Infect Immun. 2007;75:2626–2629. doi: 10.1128/IAI.01599-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prouty AM, Brodsky IE, Manos J, Belas R, Falkow S, Gunn JS. Transcriptional regulation of Salmonella enterica serovar Typhimurium genes by bile. FEMS Immunol Med Microbiol. 2004;41:177–185. doi: 10.1016/j.femsim.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Prouty AM, Gunn JS. Salmonella enterica serovar typhimurium invasion is repressed in the presence of bile. Infect Immun. 2000;68:6763–6769. doi: 10.1128/iai.68.12.6763-6769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gunn JS. Mechanisms of bacterial resistance and response to bile. Microbes Infect. 2000;2:907–913. doi: 10.1016/s1286-4579(00)00392-0. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Nordhues BA, Zhong D, De Guzman RN. NMR Characterization of the interaction of the Salmonella type III secretion system protein SipD and bile salts. Biochemistry. 2010;49:4220–4226. doi: 10.1021/bi100335u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 18.Derewenda U, Mateja A, Devedjiev Y, Routzahn KM, Evdokimov AG, Derewenda ZS, Waugh DS. The structure of Yersinia pestis V-antigen, an essential virulence factor and mediator of immunity against plague. Structure. 2004;12:301–306. doi: 10.1016/j.str.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 19.Zhou NE, Zhu BY, Kay CM, Hodges RS. The two-stranded alpha-helical coiled-coil is an ideal model for studying protein stability and subunit interactions. Biopolymers. 1992;32:419–426. doi: 10.1002/bip.360320419. [DOI] [PubMed] [Google Scholar]
- 20.Kiss RS, Kay CM, Ryan RO. Amphipathic alpha-helix bundle organization of lipid-free chicken apolipoprotein A-I. Biochemistry. 1999;38:4327–4334. doi: 10.1021/bi982597p. [DOI] [PubMed] [Google Scholar]
- 21.Choy N, Raussens V, Narayanaswami V. Inter-molecular coiled-coil formation in human apolipoprotein E C-terminal domain. J Mol Biol. 2003;334:527–539. doi: 10.1016/j.jmb.2003.09.059. [DOI] [PubMed] [Google Scholar]
- 22.Mueller CA, Broz P, Muller SA, Ringler P, Erne-Brand F, Sorg I, Kuhn M, Engel A, Cornelis GR. The V-antigen of Yersinia forms a distinct structure at the tip of injectisome needles. Science. 2005;310:674–676. doi: 10.1126/science.1118476. [DOI] [PubMed] [Google Scholar]
- 23.Blocker AJ, Deane JE, Veenendaal AK, Roversi P, Hodgkinson JL, Johnson S, Lea SM. What's the point of the type III secretion system needle? Proc Natl Acad Sci USA. 2008;105:6507–6513. doi: 10.1073/pnas.0708344105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gebus C, Faudry E, Bohn YS, Elsen S, Attree I. Oligomerization of PcrV and LcrV, protective antigens of Pseudomonasaeruginosa and Yersinia pestis. J Biol Chem. 2008;283:23940–23949. doi: 10.1074/jbc.M803146200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galkin VE, Schmied WH, Schraidt O, Marlovits TC, Egelman EH. The structure of the Salmonella typhimurium type III secretion system needle shows divergence from the flagellar system. J Mol Biol. 2010;396:1392–1397. doi: 10.1016/j.jmb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benach J, Abashidze M, Seetharaman J, Zhao L, Janjua H, Cunningham K, Ma LC, Xiao R, Liu J, Baran MC, et al. Crystal structure of the Pathogenicity island 1 effector protein from Chromobacterium violaceum. Northeast Structural Genomics Consortium (NESGC) target CvR69. (to be published) [Google Scholar]
- 27.Lawton DG, Longstaff C, Wallace BA, Hill J, Leary SE, Titball RW, Brown KA. Interactions of the type III secretion pathway proteins LcrV and LcrG from Yersiniapestis are mediated by coiled-coil domains. J Biol Chem. 2002;277:38714–38722. doi: 10.1074/jbc.M203632200. [DOI] [PubMed] [Google Scholar]
- 28.Matson JS, Nilles ML. Interaction of the Yersinia pestis type III regulatory proteins LcrG and LcrV occurs at a hydrophobic interface. BMC Microbiol. 2002;2:16. doi: 10.1186/1471-2180-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matson JS, Nilles ML. LcrG-LcrV interaction is required for control of Yops secretion in Yersinia pestis. J Bacteriol. 2001;183:5082–5091. doi: 10.1128/JB.183.17.5082-5091.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee VT, Tam C, Schneewind O. LcrV, a substrate for Yersinia enterocolitica type III secretion, is required for toxin targeting into the cytosol of HeLa cells. J Biol Chem. 2000;275:36869–36875. doi: 10.1074/jbc.M002467200. [DOI] [PubMed] [Google Scholar]
- 31.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichiacoli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kenjale R, Wilson J, Zenk SF, Saurya S, Picking WL, Picking WD, Blocker A. The needle component of the type III secreton of Shigella regulates the activity of the secretion apparatus. J Biol Chem. 2005;280:42929–42937. doi: 10.1074/jbc.M508377200. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.Evans PR. Scaling and assessment of data quality. Acta Cryst D. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 35.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Cryst D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 36.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen SX, Jelloul MB, Long F, Vagin A, Knipscheer P, Lebbink J, Sixma TK, Lamzin VS, Murshudov GN, Perrakis A. ARP/wARP and molecular replacement: the next generation. Acta Cryst D. 2008;64:49–60. doi: 10.1107/S0907444907047580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 40.Vagin AA, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 41.Lovell SC, Davis IW, Arendall WB, III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Ca geometry: phi, psi and Cb deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 42.DeLano WL. The PyMOL molecular graphics system. California: DeLano Scientific; 2002. [Google Scholar]
- 43.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]