

Abstract
A new approach to predicting the ligand-binding sites of proteins was developed, using protein-ligand docking computation. In this method, many compounds in a random library are docked onto the whole protein surface. We assumed that the true ligand-binding site would exhibit stronger affinity to the compounds in the random library than the other sites, even if the random library did not include the ligand corresponding to the true binding site. We also assumed that the affinity of the true ligand-binding site would be correlated to the docking scores of the compounds in the random library, if the ligand-binding site was correctly predicted. We call this method the molecular-docking binding-site finding (MolSite) method. The MolSite method was applied to 89 known protein-ligand complex structures extracted from the Protein Data Bank, and it predicted the correct binding sites with about 80–99% accuracy, when only the single top-ranked site was adopted. In addition, the average docking score was weakly correlated to the experimental protein-ligand binding free energy, with a correlation coefficient of 0.44.
Keywords: protein pocket prediction, ligand pocket, ligand binding site prediction, protein-compound docking, protein-ligand binding free energy
Introduction
Finding functional sites on protein molecular surfaces is crucial for revealing the mechanisms of molecular signaling involving target proteins. Ligand-binding sites are among the most promising targets for drug candidates, whose actions depend upon the inhibition or regulation of the target protein functions. However, in some cases, such ligand-binding sites must be predicted, because little or no experimental information about the protein's functional sites exists. For example, such information is lacking for many of the proteins with tertiary structures that have been determined by structural genomics projects. In addition, non-native ligand-binding sites need to be searched because of putative protein structural changes, due to allosteric effects.
Many researchers have published studies on the prediction of ligand-binding sites.1–30 Some methods use the mathematical shapes of target proteins, and identify the corresponding concavities as potential ligand-binding sites.1–23 In these approaches, the volumes of the binding pocket can be determined, but the boundaries between the pocket and non-pocket regions are often unclear. Other methods use a spherical probe with a suitable radius, and calculate the pseudo energy between the protein and the probe.24–27 A site that is predicted to bind the probe strongly is selected as a ligand-binding site. Evolutionary information is also employed, under the assumption that the amino-acid sequence of the ligand-binding site must be conserved in the evolution process. Thus, conserved sequences are potential candidates for ligand-binding sites. The propensity of each amino-acid residue to exist at a ligand-binding site has also been calculated from a database of protein-ligand complex structures.2,28–30 Trp, Phe, Tyr, and Arg tend to form ligand-binding sites, while Gly, Ala and Pro do not. Methods combining these bodies of knowledge have also been examined,30 and suggested the presence of multiple potential ligand-binding sites, even if only one true binding site exists.
Besides ligand-binding site prediction, the functional analysis and classification of the pockets have been reported.31–33 As with the folds of the proteins, the structural variety of the ligand-binding sites should be finite, and common structural motifs of ligand-binding sites are frequently observed, even if the global folds or amino-acid sequences are quite different.31 These findings suggest that the structural variety of ligands, as well as the variety of ligand-binding pockets, may be limited, and thus a finite number of compounds could form a probe set for detecting a ligand-binding pocket by a protein-compound docking study.
Currently, the accuracy of the above predictions is roughly 50–80%, when the top-ranked predicted pocket is adopted as the candidate binding site.1 When the top three to five predicted sites are adopted, the probability of finding the true site among them increases to 70–90%.1,28 However, it is still necessary to improve the accuracy and to estimate the affinity of the pocket. Information about the pocket location and size is not very useful if the pocket lacks strong affinity to any compound. Smith et al.34 showed that the average IC50 of known drugs is 30 nM, and that 70% of known drugs have an IC50 < 50 nM.
In the current study, we proposed a new method for ligand-binding site prediction based on protein-compound docking simulation, and we tried to estimate the protein compound binding free energy without known ligands. In our method, actual compounds were randomly selected from a large compound library, and their three-dimensional (3D) structures were used as the probes, instead of a spherical probe. Each compound was then docked onto the protein surface by the ligand-flexible docking of in silico drug screening. We assumed that the true ligand-binding site would show stronger affinity than the other sites to the compounds in the random library, even if the random library did not include the true binders to the binding site. This assumption was based on a previous experiment, in which we applied an in silico structure-based drug screening method to more than ten target proteins, and found that the docking poses of almost all compounds were localized around their true ligand-binding sites. We also assumed that the affinity of the true ligand-binding site would be correlated to the docking scores of the compounds in the random library if the ligand-binding site was correctly predicted, because the affinity of a drug depends on the shape of the ligand-binding pocket. In general, a deep binding pocket shows stronger affinity than a shallow pocket. We call this method the molecular-docking binding-site finding (MolSite) method. The MolSite method was applied to 89 proteins, and it predicted the ligand-binding sites of these proteins fairly correctly. The MolSite method was also used to examine the protein-compound affinity for 50 proteins, but only weak correlations were found between the predicted and experimental binding free energies when the volumes of the pockets were small.
Results
Ligand-binding site prediction
The Cα carbons were selected as the centers of the scoring grids for the 89 target proteins, as mentioned in the previous section. On average, 29.3 Cα carbons were selected for each target protein, and a 3D mesh was generated around the Cα carbons. The minimum number of selected Cα carbons was 10, for 2pk4, and the maximum number was 53, for 1dbj. The 10,000 compounds of the C1, C2 and C3 probe sets were docked to the whole surfaces of the target proteins, using the above mesh potential (see Table I and Materials section). The protein-compound docking was flexible, allowing up to 100 conformers for each compound, and was performed by the Sievgene/myPresto program, with an average docking time of about 2 seconds per compound. The Savg, S1 and Stop scores were calculated for each scoring grid from these docking scores for the C1, C2 and C3 sets.
Table I.
Summary of the Number of Heavy Atoms in Each Compound Set
| Compound sets | Min. | Average | Max. |
|---|---|---|---|
| C1 | 7 | 20.37 | 26 |
| C2 | 9 | 20.36 | 26 |
| C3 | 8 | 20.36 | 26 |
| Liganda | 5 | 21.25 | 46 |
Min., Max., and Average represent the minimum, maximum and average numbers of heavy atoms of the compound sets, respectively.
Ligands of the protein-compound complex structures summarized in Appendix A.
Table II shows the probabilities of finding the true scoring grid among the selected scoring grids by the three different scores, when the C1 set was used as the probe set (see Fig. 1 and Method section). The definition of “true scoring grid” is the condition in which the selected scoring grid contains the average atomic coordinates of the experimentally determined bound ligand. If multiple grids contain the average atomic coordinates of the experimentally determined bound ligand, then the true scoring grid was selected among these grids as follows: the distance between the center Cα carbon of the true scoring grid and the average coordinates of the bound ligand must be the minimum value among the values obtained from these grids. When the Savg score was used for the selection, all true scoring grids were selected. If the scoring grid had been randomly selected, then the probability of finding the true scoring grid would have been 3.4%, since the average number of scoring grids was 29.3 (3.4% = 100%/29.3). The S1 score gave the second best prediction probability. The Stop score was inferior to both the Savg and S1 scores. When the C2 and C3 sets were used as the probe set, the results were almost the same as those obtained with the C1 set, as shown in Table II. The probability of finding the true scoring grid did not depend on the choice of the probe set. The difference in the probability was less than 3%. When the Savg score was used for the selection, the true scoring grids were selected at 100% probability.
Table II.
Probability that the Selected Scoring Grids Include the True Ligand-Binding Sites, Depending on the Scores Used for Selection
| Library | Savg | Stop | S1 |
|---|---|---|---|
| C1a | 98.89 | 68.18 | 89.77 |
| C1b | 100.00 | 68.63 | 90.20 |
| C2b | 100.00 | 70.59 | 88.24 |
| C3b | 100.00 | 80.39 | 88.24 |
Values in the table are in %. C1, C2, and C3 are the three different compound libraries.
All 88 proteins were used (Appendix A).
The 50 proteins with ΔG values were used (Appendix B).
Figure 1.
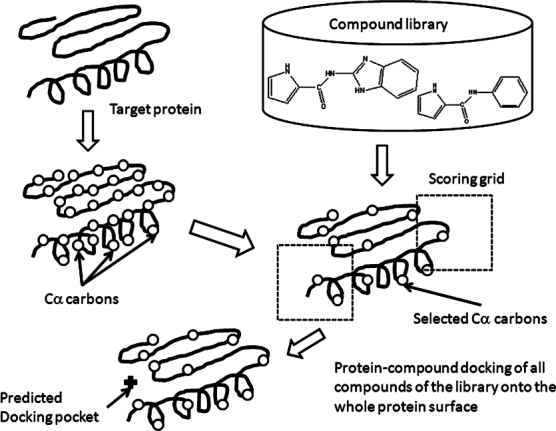
Schematic representation of the molecular-docking binding-site finding (MolSite) method.
Table III shows the prediction results for the centers of the ligand-binding sites, when the C1 set was used as the probe set. The scoring grids were selected using the Savg score. The distances between the center of the predicted ligand-binding site and the center of the bound ligand (Dc) were calculated for the 89 proteins. This distance was adopted as the measure of the prediction accuracy by Brylinski and Skolnick.28 The minimum distances between the center of each predicted binding site and any atom of the bound ligand (Dmin) were also calculated, as shown in Table III. This minimum distance was adopted as the measure of the prediction accuracy by Huang.1 The Dmin values are smaller than the Dc values in many cases, but there are some exceptions. The bound ligand of 1a6w adopts an “L” shape, and that of 1epo has a “C” shape. In these cases, the average coordinates of the ligand's atoms are outside of the molecule, and thus the Dc values are smaller than the Dmin values.
Table III.
Summary of the Dc and Dmin Values for All 89 Target Proteins
| Distance | |||
|---|---|---|---|
| Number | Target protein | Dc (Å) | Dmin (Å) |
| 1 | 1a6w | 0.50 | 0.82 |
| 2 | 1acj | 1.87 | 0.69 |
| 3 | 1bid | 3.79 | 1.72 |
| 4 | 1blh | 2.19 | 2.57 |
| 5 | 1byb | 2.12 | 1.78 |
| 6 | 1cdo | 1.65 | 1.01 |
| 7 | 1fbp | 1.10 | 0.78 |
| 8 | 1gca | 0.41 | 1.00 |
| 9 | 1hew | 5.28 | 1.02 |
| 10 | 1hfc | 2.03 | 0.95 |
| 11 | 1hyt | 5.74 | 1.17 |
| 12 | 1ida | 1.45 | 0.88 |
| 13 | 1igj | 5.11 | 1.77 |
| 14 | 1imb | 1.41 | 1.48 |
| 15 | 1inc | 4.89 | 1.04 |
| 16 | 1ivd | 1.50 | 0.88 |
| 17 | 1mrg | 3.08 | 1.56 |
| 18 | 1mtw | 4.08 | 0.73 |
| 19 | 1okm | 1.88 | 1.06 |
| 20 | 1pdz | 1.98 | 0.53 |
| 21 | 1phd | 2.62 | 1.30 |
| 22 | 1pso | 2.02 | 0.52 |
| 23 | 1qpe | 0.64 | 0.42 |
| 24 | 1rbp | 2.12 | 1.42 |
| 25 | 1rne | 1.58 | 0.68 |
| 26 | 1rob | 0.88 | 0.59 |
| 27 | 1snc | 0.87 | 0.48 |
| 28 | 1srf | 1.81 | 0.87 |
| 29 | 2ctc | 1.69 | 1.30 |
| 30 | 2h4n | 3.22 | 0.69 |
| 31 | 2pk4 | 3.22 | 0.69 |
| 32 | 2sim | 1.10 | 1.10 |
| 33 | 2tmn | 0.82 | 0.95 |
| 34 | 3gch | 2.87 | 0.90 |
| 35 | 3mth | 2.39 | 1.02 |
| 36 | 4phv | 39.98 | 27.29 |
| 37 | 5cna | 1.82 | 1.05 |
| 38 | 5p2p | 2.24 | 1.77 |
| 39 | 6rsa | 2.54 | 0.88 |
| 40 | 1dwd | 0.79 | 0.98 |
| 41 | 1stp | 1.17 | 1.18 |
| 42 | 1ulb | 3.37 | 1.18 |
| 43 | 2ifb | 0.41 | 2.03 |
| 44 | 3ptb | 3.60 | 1.37 |
| 45 | 2ypi | 1.56 | 1.17 |
| 46 | 4dfr | 3.69 | 0.53 |
| 47 | 7cpa | 3.67 | 0.73 |
| 48 | 1apu | 3.73 | 1.78 |
| 49 | 1abe1 | 7.15 | 9.64 |
| 50 | 1abe2 | 7.15 | 9.64 |
| 51 | 1abf1 | 5.78 | 7.39 |
| 52 | 1abf2 | 4.72 | 7.40 |
| 53 | 1cbx | 1.66 | 3.22 |
| 54 | 1dbb | 0.94 | 2.59 |
| 55 | 1dbj | 1.29 | 1.57 |
| 56 | 1dog | 0.96 | 2.70 |
| 57 | 1ebg | 0.59 | 1.51 |
| 58 | 1epo | 1.03 | 3.05 |
| 59 | 1etr | 1.12 | 2.51 |
| 60 | 1ets | 1.07 | 2.54 |
| 61 | 1ett | 1.58 | 3.06 |
| 62 | 1hpv | 1.47 | 0.44 |
| 63 | 1hsl | 0.76 | 1.12 |
| 64 | 1htf1 | 1.68 | 3.08 |
| 65 | 1htf2 | 1.40 | 4.14 |
| 66 | 1hvr | 0.62 | 1.33 |
| 67 | 1mbi | 4.05 | 4.92 |
| 68 | 1mnc | 1.26 | 5.23 |
| 69 | 1nsd | 1.23 | 2.40 |
| 70 | 1pgp | 4.01 | 5.43 |
| 71 | 1phf | 1.11 | 1.75 |
| 72 | 1phg | 1.04 | 0.85 |
| 73 | 1ppc | 1.80 | 2.05 |
| 74 | 1pph | 1.97 | 2.34 |
| 75 | 1rbp | 0.57 | 2.48 |
| 76 | 1tmn | 0.84 | 3.30 |
| 77 | 1tng | 2.21 | 4.19 |
| 78 | 1tnh | 0.81 | 3.78 |
| 79 | 2cgr | 0.43 | 3.05 |
| 80 | 2cpp | 0.68 | 0.81 |
| 81 | 2gbp | 1.96 | 2.95 |
| 82 | 2phh | 2.53 | 3.51 |
| 83 | 2r04 | 2.45 | 8.62 |
| 84 | 2tsc | 0.66 | 4.12 |
| 85 | 5abp1 | 5.18 | 7.48 |
| 86 | 5abp2 | 5.13 | 7.57 |
| 87 | 5cpp | 0.91 | 0.62 |
| 88 | 5tln | 0.96 | 2.96 |
| 89 | 6cpa | 0.69 | 3.60 |
| Average | 3.34 | 1.82 | |
This prediction is based on the Savg score. Dc: the distance between the center of the predicted ligand-binding site and the center of the bound ligand. Dmin: the minimum distance between the center of the predicted binding site and any atom of the bound ligand.
In the MolSite method, the center of the predicted docking pocket is given by the average coordinates of the docking poses of all compounds in the candidate scoring grid. In the current study, the center of the predicted docking pocket was the average coordinates of the 10,000 docking poses. The distribution (root-mean square deviation; RMSD) of the average coordinates of the 10,000 docked poses was calculated for each target protein. The average RMSD value for the 88 targets was 1.61 Å. This average RMSD value means that most of the docked poses were overlapped around the center of the predicted docking pocket for each target protein, when the compounds were selected randomly. With a greater number of compounds, we can expect the average coordinates of the docking pose to converge to a point.
For several targets (4phv, 1abe, 2r04, 1abf, etc.), the MolSite method failed in the prediction of the binding sites. Therefore, the probe-size dependence of the prediction was examined. Table IV shows the results obtained by the “Small” compound set, consisting of the ligands with HA < 13 atoms in the protein-ligand complex structures listed in Appendix C. The number of molecules was 21. After the scoring grid was determined using the C1 set, these 21 ligands were docked to the target proteins and the average coordinates of the docking poses were calculated. For 4phv, 1abe1, 1abe2, 1abf1, 1abf2, 5abp1 and 5abp2, the Dc and Dmin values were drastically decreased using the Small set, as compared to the results summarized in Table III. In actual ligand-binding site prediction, we usually do not know the ligand a priori. Thus, we did not use the results from the Small set for comparison with those obtained by other docking programs.
Table IV.
Summary of the Dc and Dmin Values for all 89 Target Proteins Obtained by the Small-Compound Set
| Distance | |||
|---|---|---|---|
| Number | Target protein | Dc (Å) | Dmin (Å) |
| 1 | 1a6w | 1.49 | 0.64 |
| 2 | 1acj | 2.18 | 1.00 |
| 3 | 1bid | 4.04 | 1.88 |
| 4 | 1blh | 2.50 | 1.84 |
| 5 | 1byb | 3.29 | 0.89 |
| 6 | 1cdo | 1.60 | 1.52 |
| 7 | 1fbp | 1.83 | 1.96 |
| 8 | 1gca | 0.73 | 0.80 |
| 9 | 1hew | 6.31 | 0.72 |
| 10 | 1hfc | 4.04 | 0.52 |
| 11 | 1hyt | 5.60 | 1.36 |
| 12 | 1ida | 1.53 | 0.46 |
| 13 | 1igj | 7.05 | 0.56 |
| 14 | 1imb | 3.00 | 0.73 |
| 15 | 1inc | 4.51 | 0.54 |
| 16 | 1ivd | 0.77 | 0.64 |
| 17 | 1mrg | 1.19 | 0.79 |
| 18 | 1mtw | 5.28 | 1.21 |
| 19 | 1okm | 3.10 | 0.65 |
| 20 | 1pdz | 1.12 | 0.65 |
| 21 | 1phd | 1.59 | 1.05 |
| 22 | 1pso | 1.81 | 0.86 |
| 23 | 1qpe | 3.68 | 0.83 |
| 24 | 1rbp | 2.45 | 1.48 |
| 25 | 1rne | 1.48 | 0.80 |
| 26 | 1rob | 1.33 | 1.44 |
| 27 | 1snc | 2.97 | 1.92 |
| 28 | 1srf | 2.94 | 0.65 |
| 29 | 2ctc | 0.80 | 1.48 |
| 30 | 2h4n | 1.75 | 0.59 |
| 31 | 2pk4 | 1.48 | 0.79 |
| 32 | 2sim | 1.71 | 1.05 |
| 33 | 2tmn | 1.16 | 1.21 |
| 34 | 3gch | 4.12 | 1.30 |
| 35 | 3mth | 3.86 | 2.61 |
| 36 | 4phv | 4.65 | 0.59 |
| 37 | 5cna | 0.73 | 1.24 |
| 38 | 5p2p | 4.04 | 1.10 |
| 39 | 6rsa | 2.28 | 0.77 |
| 40 | 1dwd | 5.46 | 2.95 |
| 41 | 1stp | 1.61 | 1.32 |
| 42 | 1ulb | 3.86 | 2.08 |
| 43 | 2ifb | 4.11 | 1.65 |
| 44 | 3ptb | 1.53 | 1.48 |
| 45 | 2ypi | 4.07 | 1.50 |
| 46 | 4dfr | 4.28 | 1.05 |
| 47 | 7cpa | 3.67 | 2.62 |
| 48 | 1apu | 3.10 | 1.40 |
| 49 | 1abe1 | 1.76 | 0.80 |
| 50 | 1abe2 | 1.76 | 0.80 |
| 51 | 1abf1 | 1.42 | 0.51 |
| 52 | 1abf2 | 1.49 | 0.67 |
| 53 | 1cbx | 2.42 | 1.34 |
| 54 | 1dbb | 3.53 | 0.75 |
| 55 | 1dbj | 3.14 | 0.67 |
| 56 | 1dog | 1.06 | 1.05 |
| 57 | 1ebg | 1.16 | 0.32 |
| 58 | 1epo | 2.30 | 0.74 |
| 59 | 1etr | 3.73 | 1.91 |
| 60 | 1ets | 9.06 | 3.42 |
| 61 | 1ett | 7.01 | 3.84 |
| 62 | 1hpv | 0.79 | 0.85 |
| 63 | 1hsl | 1.18 | 1.00 |
| 64 | 1htf1 | 3.19 | 1.15 |
| 65 | 1htf2 | 2.80 | 1.76 |
| 66 | 1hvr | 3.74 | 1.82 |
| 67 | 1mbi | 7.32 | 6.36 |
| 68 | 1mnc | 8.16 | 4.13 |
| 69 | 1nsd | 1.81 | 1.29 |
| 70 | 1pgp | 3.97 | 2.43 |
| 71 | 1phf | 0.78 | 0.52 |
| 72 | 1phg | 0.44 | 1.53 |
| 73 | 1ppc | 4.60 | 1.47 |
| 74 | 1pph | 3.42 | 1.54 |
| 75 | 1rbp | 4.28 | 1.55 |
| 76 | 1tmn | 4.39 | 1.12 |
| 77 | 1tng | 1.63 | 1.39 |
| 78 | 1tnh | 1.67 | 1.94 |
| 79 | 2cgr | 5.64 | 0.55 |
| 80 | 2cpp | 0.51 | 0.86 |
| 81 | 2gbp | 0.97 | 1.03 |
| 82 | 2phh | 1.48 | 1.04 |
| 83 | 2r04 | 7.80 | 3.08 |
| 84 | 2tsc | 5.87 | 1.98 |
| 85 | 5abp1 | 1.10 | 0.38 |
| 86 | 5abp2 | 1.29 | 0.51 |
| 87 | 5cpp | 0.49 | 1.36 |
| 88 | 5tln | 3.16 | 0.65 |
| 89 | 6cpa | 3.86 | 0.48 |
| Average | 3.12 | 1.53 | |
This prediction is based on the Savg score. Dc: the distance between the center of the predicted ligand-binding site and the center of the bound ligand. Dmin: the minimum distance between the center of the predicted binding site and any atom of the bound ligand.
The prediction accuracies of our MolSite method, obtained with the C1 library, are shown in Tables V and VI with the accuracies of other methods,1,28 where the 48 target proteins are the same ones used for the validation of LIGSITE, PASS, Q-site finder and SURFNET.1 The target protein set was different from the set used for the validation of FINDSITE.28 Since the results obtained from the C2 and C3 libraries were exactly the same as those obtained from the C1 set when the 50 protein set (protein numbers 40-89 in Tables III and IV) was employed, only the C1 library was used.
Table V.
Summary of the Pocket-Prediction Accuracies of Various Methods. Prediction Accuracy of MolSite for the 48 Target Proteins, Which were Used for the Validation of LIGSITE, PASS, Q-site Finder, and SURFNET, as Shown in Appendix A
| Method | MolSite | MolSite | FIND SITE | LIG SITE | Meta Pocket | LIG SITE | PASS | Q-site Finder | SURFNET |
|---|---|---|---|---|---|---|---|---|---|
| No of Pocketsa | Top 1 | Top 1 | Top 5 | Top 5 | Top 1 | Top 1 | Top 1 | Top 1 | Top 1 |
| Distance (D) | Dc | Dmin | Dc | Dc | Dmin | Dmin | Dmin | Dmin | Dmin |
| D < 8 Åb | 97.92 | 97.92 | — | — | — | — | —s | — | — |
| D < 6 Åb | 97.92 | 97.92 | — | — | — | — | — | — | — |
| D < 5 Åb | 91.67 | 97.92 | — | — | — | — | — | — | — |
| D < 4 Åb | 87.50 | 97.92 | 70.9c | 51.3c | 83.0d | 81.0d | 58.0d | 75.0d | 42.0d |
| D < 3 Åb | 68.75 | 97.92 | — | — | — | — | — | — | — |
The values in this table are in %. This prediction is based on the Savg score. Dc: the distance between the center of the predicted ligand-binding site and the center of the bound ligand. Dmin: the minimum distance between the center of the predicted binding site and any atom of the bound ligand.
Number of pockets indicates the number of predicted pockets used for the analysis. If one of the predicted pockets is correct, then the prediction is counted as a successful prediction.
The 48 target proteins were used.
Reference28.
Reference1.
Table VI.
Summary of the Pocket-Prediction Accuracies of Various Methods. Prediction accuracy of MolSite for the Total of 89 Target Proteins, as Shown in Appendixes A and B
| Method | MolSite | MolSite |
|---|---|---|
| No. of pocketsa | Top 1 | Top 1 |
| Method | Dc | Dmin |
| D < 8 Åb | 98.88 | 95.51 |
| D < 6 Åb | 98.88 | 91.01 |
| D < 5 Åb | 92.13 | 89.89 |
| D < 4 Åb | 88.76 | 85.40 |
| D < 3 Åb | 79.78 | 76.40 |
Number of pockets indicates the number of predicted pockets used for the analysis. If one of the predicted pockets is correct, then the prediction is counted as a successful prediction.
All 89 target proteins were used.
When we used the 48 proteins to evaluate the prediction accuracy of the current method, the probabilities of Dc < 4 Å and Dmin < 4 Å were 87.50% and 97.92%, respectively. When we used all 89 proteins (=50+48–9) to evaluate the current method, the probabilities of Dc < 4 Å and Dmin < 4 Å were 88.76% and 85.40%, respectively. Therefore, the prediction performance of the MolSite method was better than that of the other methods. The Dc values determined by the MolSite method in this table were obtained from the top-ranked prediction site. On the contrary, the Dc values for the top 5 predicted sites were the lowest for the FINDSITE and LIGSITE methods.28 The Dmin values were obtained from the top-ranked prediction site.28
We examined the breakdown of the docking scores and the differences between the docking scores at the true binding site and the other sites. The Sievgene docking score consists of an accessible surface term (mainly hydrophobic interaction), an electrostatic term, a hydrogen bonding term and a van der Waals term. The total score, and the accessible surface, electrostatic, hydrogen bonding and van der Waals terms at the true binding site were 1.29, 1.28, 0.78, 1.32, and 1.26 times larger than those values at the other sites, respectively. The contributions of the accessible surface, electrostatic, hydrogen bonding and van der Waals terms were 89.9%, 0.88%, 5.41% and 3.78%, respectively. On average, 65% of the total accessible surface of the compound was buried in the protein. At the true binding site, 78% of the total accessible surface of the compound was buried in the protein. These results suggest that the surface complementarity and the hydrophobic interactions between the protein and the compounds are important in distinguishing the true binding site from the other sites.
The MolSite method was also applied to the unbound (apo) structures of 20 target proteins, which were the (5*n + 1)th and (5*n + 3)th (where n=0,.., 9) proteins of Table IV of reference 2. The prediction results are summarized in Table VII. To calculate the Dc and Dmin values, the Cα carbons of the unbound protein were superimposed on those of the bound (holo) protein, and the Dc and Dmin values were calculated based on the ligand coordinates of the bound protein. The apo and holo proteins are summarized in Table VII. The C1, C2 and C3 sets were used as the probe set. The scoring grids were selected using the Savg score. The MolSite method worked well for both the unbound and bound structures. The prediction results did not depend on the choice of the probe set. The probabilities of Dc < 4 Å and Dmin < 4 Å were 80% and 100%, respectively. This prediction accuracy for the apo protein was almost the same as that for the holo protein.
Table VII.
Summary of the Dc and Dmin Values for 20 apo Proteins
| Library | C1 | C1 | C2 | C2 | C3 | C3 | |
|---|---|---|---|---|---|---|---|
| Distance | Dc | Dmin | Dc | Dmin | Dc | Dmin | |
| Apo | Holo | RMSD (Å) | RMSD (Å) | RMSD (Å) | RMSD (Å) | RMSD (Å) | RMSD (Å) |
| 1hel | 1hew | 4.85 | 1.04 | 4.86 | 1.03 | 4.86 | 1.03 |
| 1hsi | 1ida | 3.02 | 2.03 | 3.03 | 2.05 | 2.99 | 2.03 |
| 1krn | 2pk4 | 2.48 | 1.29 | 2.48 | 1.28 | 2.47 | 1.28 |
| 1pdy | 1pdz | 1.66 | 0.90 | 1.67 | 0.91 | 1.67 | 0.91 |
| 1stn | 1snc | 4.30 | 0.50 | 4.31 | 0.49 | 4.33 | 0.49 |
| 1swb | 1stp | 7.41 | 1.23 | 7.41 | 1.22 | 7.40 | 1.20 |
| 3app | 1apu | 3.08 | 1.11 | 3.08 | 1.11 | 3.05 | 1.13 |
| 3p2p | 5p2p | 0.50 | 0.98 | 0.52 | 0.97 | 0.52 | 0.97 |
| 3tms | 1bid | 0.60 | 0.99 | 0.62 | 0.98 | 0.57 | 1.00 |
| 5dfr | 4dfr | 2.00 | 1.72 | 1.97 | 1.72 | 2.03 | 1.70 |
| 1mrg | 1ahc | 2.59 | 1.37 | 2.58 | 1.37 | 2.58 | 1.36 |
| 1blh | 1djb | 3.26 | 1.90 | 3.24 | 1.90 | 3.24 | 1.91 |
| 1inc | 1esa | 3.51 | 0.88 | 3.47 | 0.86 | 3.51 | 0.88 |
| 1dwd | 1hxf | 4.15 | 0.66 | 4.17 | 0.66 | 4.16 | 0.68 |
| 2ifb | 1ifb | 1.15 | 1.90 | 1.16 | 1.91 | 1.15 | 1.92 |
| 2tmn | 1l3f | 2.36 | 0.95 | 2.38 | 0.96 | 2.38 | 0.97 |
| 1pso | 1psn | 3.17 | 0.55 | 3.19 | 0.56 | 3.18 | 0.57 |
| 5cna | 2ctv | 1.56 | 0.26 | 1.58 | 0.25 | 1.60 | 0.23 |
| 1stp | 2rta | 0.90 | 1.28 | 0.90 | 1.28 | 0.88 | 1.27 |
| 6rsa | 7rat | 2.45 | 0.90 | 2.46 | 0.89 | 2.45 | 0.89 |
| Average | 2.75 | 1.12 | 2.75 | 1.12 | 2.75 | 1.12 |
This prediction is based on the Savg score.
Figure 2(A,B) show the ligand-binding site prediction results obtained by the current MolSite method, summarizing the data in Tables III, IV, and VII. It is clear that the prediction results were only slightly dependent on the selected library, and that the prediction performance for apo proteins was similar to that for holo proteins. As compared with the performances of other methods (i.e., Fig. 2 of reference 28), the performance of MolSite is equivalent or superior to those of other methods, although the data sets were different.
Figure 2.
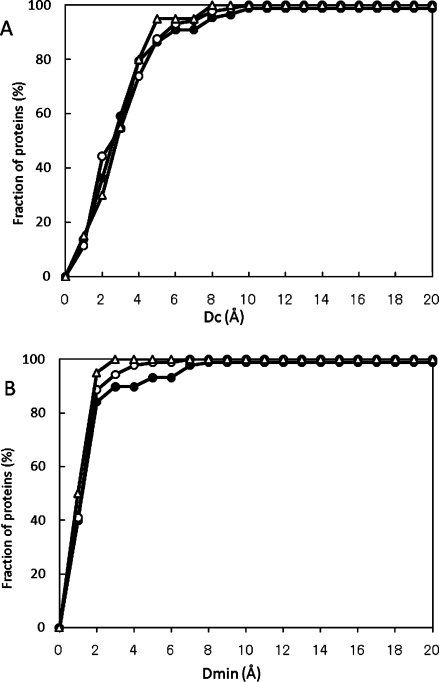
Performance of the MolSite method. The results are presented as the fraction of proteins with Dc and Dmin. Filled circles, open circles and open triangles represent the results of all 89 holo target proteins with the C1 library, those with the small compound set, and those of the 20 apo target proteins with the C1 library, respectively. (A) Fraction of proteins vs. Dc value, (B) fraction of proteins vs. Dmin value.
We also examined the dependence of the number of compounds on the prediction accuracy. For the 20 apo structures, 10, 100, and 1,000 randomly selected compounds were used, instead of the 10,000 compounds. The prediction accuracy was improved by increasing the number of compounds. Namely, the probabilities of Dc < 4 Å were 60%, 60%, 70% and 70% for 10, 100, 1,000 and 10,000 compounds, respectively. The probabilities of Dc < 5 Å were 70%, 80%, 80% and 90% for 10, 100, 1,000 and 10,000 compounds, respectively. The average Dc values were 3.34 Å, 3.52 Å, 3.42 Å and 2.99 Å for 10, 100, 1,000 and 10,000 compounds, respectively. The MolSite method could work with even 10 compounds, and the accuracy was not drastically improved with more compounds. Therefore, 104 compounds should be sufficient to use the MolSite method effectively.
When only one ligand of the target protein was used, the MolSite method still worked, but failed in some cases. The ligands were prepared for 16 apo structures (see Appendix D). The probabilities of Dc < 4 Å were 75% and Dmin < 4 Å were 81.25%. However, for three target proteins (1djb, 1esa and 1l3f), the MolSite method failed in prediction (Dc > 20 Å and Dmin > 18 Å).
Prediction of ligand-binding affinity
Figure 3 shows a comparison between the experimental ΔG values and the Savg values. The PDB identifiers of the 50 proteins used are summarized in Appendix B (protein numbers 40–89 in Tables III and IV). When all 50 results were used, there was almost no correlation between the experimental ΔG value and the Savg or S1 value. The correlation coefficients of Savg and S1 to the experimental ΔG values were 0.195 and 0.186, respectively. As shown in Tables III and IV, the prediction failed in several cases, but all of the results were used to calculate the correlation. The docking scores were distributed around 3–4, suggesting that the difference between the strongest and weakest affinities was about 30%. On the contrary, the experimental ΔG values ranged from −2 kcal/mol to −18 kcal/mol. Thus, the ΔG value cannot be predicted well by any score obtained by the MolSite method. The correlation coefficients of Savg and S1 were higher than those of the Stop score.
Figure 3.
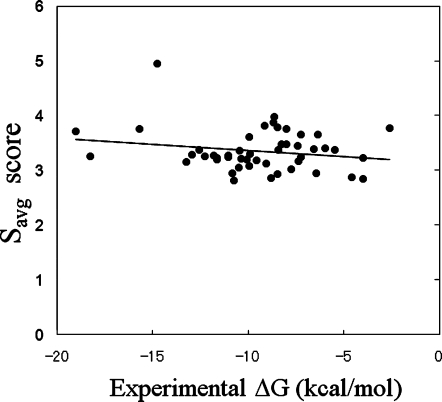
Correlation between the experimental ΔG value and the Savg value obtained by the Sievgene protein-compound docking program for all 50 target proteins. The solid line represents the linear regression result by the least-squares fit.
The volumes of the ligands in this dataset were widely distributed. As shown in Table I, the smallest number of heavy atoms in a ligand was only 5, and the largest number was 46. In contrast, the smallest number of heavy atoms in the compounds was 7-9, and the largest number of heavy atoms was 26. If the pocket is too large, as compared with the compounds of the decoy set, then those probes cannot estimate the protein-compound interaction of the whole pocket. Moreover, if the MolSite method fails in pocket prediction, then the docking score is not meaningful for estimating the protein-compound interaction of the true pocket. In this analysis, ligands with HA ≤ 26 were selected, and the prediction cases with Dc < 4 Å were chosen. Then, 22 target proteins (complex structures) were selected for the ΔG prediction.
Figure 4 shows a comparison between the experimental ΔG values and the Savg values for the 22 selected target proteins. A weak correlation appears between the experimental ΔG value and the Savg or S1 value. Namely, the correlation coefficient between ΔG and Savg was 0.441 (linear regression yielded ΔG = −3.429 Savg + 3.559), and that between ΔG and S1 was 0.425 (linear regression yielded ΔG = −3.306 S1 + 3.201). The Savg score was slightly better than the S1 score for the affinity prediction, even though the true binder was unknown. When the volume of the binding pocket was limited, the affinity of the binding pocket could be predicted, but the accuracy was not very high.
Figure 4.
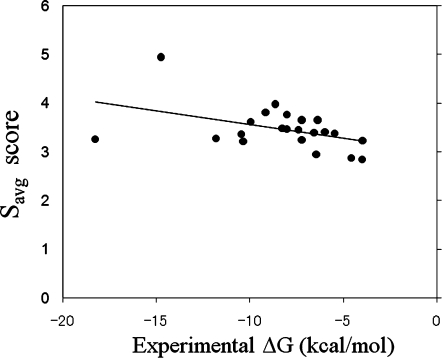
Correlation between the experimental ΔG value and the Savg value obtained by the Sievgene protein-compound docking program for the selected target proteins. The solid line represents the linear regression result by the least-squares fit.
Discussion
In Table III, the MolSite method failed to predict the binding sites of 4phv, 1abe, 2r04, 1abf and 5abp. Three of these—1abe, 1abf and 5abp—are sugar-binding proteins, with small binding pockets buried in the proteins. The ligands of 1abe, 1abf and 5abp have only 5–8 heavy atoms. The compounds of sets C1–C3 are much larger than sugars, and no members of our probe set could bind to the pocket correctly. All of the compounds were docked onto the protein surface, and no docking poses were generated for the actual buried binding pocket. We hypothesized that if the probe set had consisted of small compounds with sizes similar to those of sugars, then the MolSite method could have predicted the true binding pocket.
Therefore, we used the set of small compounds as the probe set, and the results are shown in Table IV. The small binding pockets in 1abe, 1abf, and 5abp were well predicted. In addition, the prediction of the buried pocket in 4phv was greatly improved. Therefore, when the ligand for a target protein is known to be small, we can use the Small set for better prediction accuracy.
For in silico screening, the conditions of Dc <4 Å and Dmin <4 Å proved too severe. As shown in Table II, 98.89% of the ligand-binding sites were included in the selected scoring grids by the Savg score. This prediction of the scoring grid should be sufficiently high for in silico screening. Such high accuracy should be required for docking pose analysis.
A comparison of Figure 3 with 4 reveals that the ΔG values show better correlation with the Savg or S1 values for smaller ligands with HA ≤ 26, rather than larger ligands with HA > 26. The molecular size of the C1–C3 sets was generally smaller than that of the ligands of the 50 protein-ligand complexes. Namely, the average number of heavy atoms of the C1–C3 sets was 20.4, and the range of the number of heavy atoms (HA) was 7–9 ≤ HA ≤ 26. If the probe set consisted of large compounds, then the Savg or S1 value would show better correlation to the experimental ΔG than that in the current study. The precise reproduction of these ΔG values would be difficult, since the ΔG value depends on the ligand that binds the same pocket. The ligand efficiency (LE) has been proposed as a measure of druggability, with LE = -ΔG /HA.35,36 The LE values of known drugs range from 0.1 kcal/mol/atom to 0.7 kcal/mol/atom, and the average LE value is 0.4 kcal/mol/atom.35,36 In the current study, the LE values ranged from 0.104 kcal/mol/atom to 1.640 kcal/mol/atom. Three LE values of the current data were extremely high (LE > 0.9 kcal/mol/atom). The ΔG value of a compound with an extremely low LE value could be improved by some chemical modifications, but we could not determine the maximum affinity. The maximum affinity of the pocket is not defined well enough to allow the prediction of the affinity without a known active compound.
Our previous work showed that the ΔG value predicted by our own docking program, Sievgene, is not highly accurate, with a correlation coefficient between the experimental and predicted ΔG values of about 0.7, which is the same as those of other docking programs.37 The previous work was based on exactly the same 50 protein-compound complex structures used in the current study. This accuracy is the upper limit by a naïve docking study. In the current study, the correlation coefficient between the experimental and predicted ΔG values was about 0.4, without using the true ligands (binders). A correlation coefficient of 0.4 is not very high, but it may not be so bad, considering the upper limit, 0.7.
One of the drawbacks of the MolSite method is the computational time. The MolSite method is a sort of ensemble docking study. When the protein surface is divided into 50 scoring grids and the library consists of 10,000 compounds, the total number of dockings is 50 × 10,000 dockings. Thus, the total CPU time is 1,000,000 seconds (=278 hours) for one target protein by one processor. However, completely parallel computation can easily be performed, by distributing the individual docking procedures to different CPUs. Therefore, despite this drawback, the MolSite method is useful for reliably predicting the possible ligand-binding sites for a target protein.
The MolSite method can easily be improved by increasing the compound database, and its prediction accuracy is high. Once the ligand-binding pocket is predicted, the subsequent in silico drug screening docks millions of compounds. In recent ensemble docking studies, multiple target-protein structures have been used, and the CPU time required for in silico screening is much longer than that needed for the MolSite method.
In the current study, Sievgene was adopted as the protein-compound docking program. Many protein-compound docking programs have been reported,38–40 and there is no clearly superior method.41 In the current study, we used the ordinary Sievgene score, rather than the special function. Thus, other protein-compound docking programs besides Sievgene could be used for the MolSite method.
Materials and Methods
Protein-compound docking on the whole protein surface was performed using a random compound library, and the ligand-binding sites were predicted based on the docking scores of these compounds. We assumed that the ligand-binding site would show stronger affinity to a compound than the other regions do, even if the compound was not the true binder of the site. Figure 1 provides a schematic representation of this MolSite method.
Since it is too time-consuming for a docking program to dock a compound on the whole protein surface, the entire protein surface was first divided into many small grid boxes for the docking procedure with the random compound library. The centers of these boxes were set to the positions of the Cα carbons. The grid boxes are called the “scoring grids” hereafter. The Cα carbons were selected to reduce the computational time. The minimum distance between two Cα carbons was set at 8 Å. The Cα carbon of the first residue was adopted first. If two Cα carbons were closer than 8 Å, then the Cα carbon belonging to the latter residue was neglected. As a result, 10–53 Cα carbons were selected for the target proteins.
The scoring grid was a cubic region with a cell size of 30 × 30 × 30 Å3, composed of a 3D mesh with grid points separated by 1 Å, each with the potential energy for ligand-binding to the target protein. Neighboring grids overlapped. All compounds of the random library were docked in these scoring grids.
To evaluate the “binding site likeness” of the grid, we prepared the following three scores.
Type1: Savg, The average value of all docking scores of compounds of the library.
Type 2: Stop, The best docking score among all docking scores of compounds of the library.
- Type 3: S1, where S1 = Savg + σ, where σ is the deviation of the docking scores, and
scorei and N are the docking score of the i-th compound and the number of compounds in the library, respectively.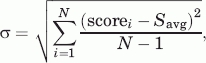
Usually, only one compound among the 10,000 randomly selected compounds is a truly active compound that experimentally shows strong affinity to the target protein. Thus, the protein-compound affinity is given by this truly active compound, among the many compounds. The highest docking score should be closer to the true docking score than the other scores, and docking scores lower than the highest score should be meaningless. Based on this logic, the Stop score should be the most reliable indicator.
However, the Stop score strongly depends on the choice of the random library. If the library contains a strong binder, then the Stop value should be very high. On the contrary, the Stop value may not be so high if the library lacks a strong binder. To reduce this library dependency, the average score value and deviation were introduced. Using the S1 or Savg values, the library dependency in the prediction of the ligand-binding sites could be reduced.
Each of these three scores (S1, Savg, and Stop) was calculated for each scoring grid, and the grid that yielded the best score was selected as the candidate scoring grid that contains the ligand-binding pocket. The center of the predicted docking pocket was then determined by the average coordinates of the docking poses of all compounds in the candidate scoring grid. The predicted affinity was given by each of the three scores (S1, Savg, and Stop) obtained in the candidate scoring grid.
To examine the MolSite method, we performed a protein-ligand docking simulation based on the known complex structures registered in the Protein Data Bank. The protein set consisted of two sets originating from the references. Here, 50 complexes with their experimental binding free energy values were selected from the database that was used for the determination of the ΔG scores of the PRO_ LEADS (protein numbers 40–89 in Tables III and IV).42 Forty-eight complexes were selected from the database that was used for the ligand-binding site prediction of the LIGSITE CSC (protein numbers 1–48 in Tables III and IV).2 Ten proteins were redundant (protein numbers 40–48 in Tables III and IV). Thus, a total of 89 (=50+48–9) proteins were used in the current study. The PDB identifiers are summarized in Appendix A. All water molecules were removed from the proteins, and all missing hydrogen atoms were added to form all-atom models of the proteins. For ligand-flexible docking, the Sievgene/myPresto program (protein-compound docking program) was used to generate up to 100 conformers for each compound.43 The Sievgene/myPresto program is available for free, from the web sites http://presto.protein.osaka-u.ac.jp/myPresto4/ and http://medals.jp/myPresto/index.html. Sievgene reconstructed 27.7%, 56.9%, and 66.2% of the total of 180 complexes that were adopted in the previous study43 with RMSDs < 1 Å, 2 Å, and 3 Å, respectively, and the average computational time was 2 CPU seconds.
Three compound libraries (C1, C2 and C3) were prepared. Each library consisted of 10,000 randomly selected compounds from the LigandBox compound database.44 The atomic charges of each compound were determined by the Mulliken charge, using MOPAC AM1 quantum chemical calculations (Quantum Chemistry Program Exchange, (QCPE), Indiana University, Bloomington, IN). The molecular weight (MW) of each compound was restricted to 150 Da < MW < 340 Da. The minimum, maximum and average numbers of heavy atoms (HA) are summarized in Table I.
The atomic charges of the proteins were the same as those in AMBER parm99.45 The minimum, maximum and average numbers of heavy atoms (HA) of the ligands of those protein-ligand complex structures are also summarized in Table I.
Conclusion
We developed the MolSite method for the prediction of the ligand-binding sites of a target protein. In this method, many compounds in a random library are docked over the whole surface of the target protein, and the ligand-binding site is predicted based on the resulting docking scores. We assumed that the actual ligand-binding sites would show statistically better docking scores and higher affinities to compounds in the random library than other sites do, even if the compounds are not the true ligands.
We applied the MolSite method to 89 known protein-ligand complex structures extracted from the PDB. The ligand-binding sites were predicted for the bound states of these target proteins. The center of the ligand-binding site was defined as the average coordinates of the bound ligand of the original complex structure. The center of the predicted ligand-binding site was defined as the average coordinates of all of the docked compounds of the probe set. The prediction accuracy was measured by the distance between the predicted center of the pocket and the actual center of the original complex structure.
The prediction accuracy of the MolSite method was higher than those of the other methods. Namely, the ligand-binding sites were predicted with 87.5% and 97.9% accuracies for the Dc value < 4 Å and the Dmin value < 4 Å for the bound structures, respectively, when only the single top-ranked site was adopted. The MolSite method worked well for both the unbound and bound structures. We also examined the prediction of the affinity of the ligand-binding site. When the pocket was small, the average docking score showed weak correlation to the experimental binding free energy. The results generated by the MolSite method did not depend on the choice of the compound data set.
Appendix A: Forty-Eight Proteins for Ligand-Binding Site Prediction and Binding Free Energy Estimation
The following PDB identifier complexes were used: 1bid, 1cdo, 1fbp, 1gca, 1hew, 1hyt, 1inc, 1rbp, 5cna, 1a6w, 1acj, 1blh, 1ivd, 1mtw, 1okm, 1phd, 1qpe, 1srf, 2h4n, 2sim, 3gch, 3mth, 5p2p, 1imb, 6rsa, 1rob, 4phv, 1byb, 1hfc, 1ida, 1igj, 1mrg, 1pdz, 1pso, 1rne, 1snc, 2ctc, 2pk4, 1apu, 1dwd, 1stp, 1ulb, 2ifb, 3ptb, 2ypi, 4dfr, 2tmn, and 7cpa.
Appendix B: Fifty Proteins for Binding Free Energy Estimation
The following PDB identifier complexes were used: 1abe, 1abf, 1apu, 1cbx, 1dbb, 1dbj, 1dog, 1dwd, 1ebg, 1epo, 1etr, 1ets, 1ett, 1hpv, 1hsl, 1htf, 1hvr, 1mnc, 1nsd, 1pgp, 1phf, 1phg, 1ppc, 1pph, 1rbp, 1stp, 1tmn, 1tng, 1tnh, 1ulb, 2cgr, 2cpp, 2gbp, 2ifb, 2phh, 2r04, 2tmn, 2tsc, 2ypi, 3ptb, 4dfr, 5abp, 5cpp, 5tln, 6cpa, and 7cpa. For 1abe, 1abf, 5abp, and 1htf, two receptor pockets were prepared, since each of these proteins binds two ligands.
Appendix C: Small Set
The small set consisted of the ligands of the following protein-ligand complex structures: 1mbi, 1tng, 1dwb, 1ebg, 1tnh, 2ypi, 3ptb, 1abe, 2phh, 1abf, 1dog, 1hsl, 1phf, 1ulb, 2cpp, 5cpp, 2gbp and 5abp. For 1abe, 1abf, 5abp, and 1htf, two receptor pockets were prepared, since each of these proteins binds the ligand with two different ligand-binding poses.
Appendix D: Sixteen Proteins for Ligand-Binding Site Prediction Using Only One Ligand Included in the Bound Complex Crystal
The following PDB identifier complexes were used: 1ahc, 1bid, 1djb, 1esa, 1hew, 1hxf, 1ida, 1ifb, 1l3f, 1pdz, 1snc, 2ctv, 2pk4, 2rta, 4dfr, and 5p2p. The used ligands for prediction of these proteins are the ligands of the holo structures summarized in Table VII.
References
- 1.Huang B. MetaPocket: a meta approach to improve protein ligand binding site prediction. OMICS. 2009;13:325–330. doi: 10.1089/omi.2009.0045. [DOI] [PubMed] [Google Scholar]
- 2.Huang B, Schroeder M. LIGSITEcsc: predicting ligand binding sites using the Connoly surface and degree of conservation. BMC Struct Biol. 2006;6:19–30. doi: 10.1186/1472-6807-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laskowski RA. SURFNET: a program for visualizing molecular surfaces, cavities and intermolecular interactions. J Mol Graph. 1995;13:323–330. doi: 10.1016/0263-7855(95)00073-9. [DOI] [PubMed] [Google Scholar]
- 4.Laskowski RA, Luscombe NM, Swindells MB, Thornton JM. Protein clefts in molecular recognition and function. Protein Sci. 1996;5:2438–2452. doi: 10.1002/pro.5560051206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brady GP, Jr, Stouten PFW. Fast prediction and visualization of protein binding pockets with PASS. J Comput Aided Mol Des. 2000;14:383–401. doi: 10.1023/a:1008124202956. [DOI] [PubMed] [Google Scholar]
- 6.Zhong S, MacKerell AD., Jr Binding response: a descriptor for selecting ligand binding site on protein surfaces. J Chem Inf Model. 2007;47:2303–2315. doi: 10.1021/ci700149k. [DOI] [PubMed] [Google Scholar]
- 7.Harris R, Olson AJ, Goodsell DS. Automated prediction of ligand-binding sites in proteins. Proteins Struct Funct Bioinf. 2008;70:1506–1517. doi: 10.1002/prot.21645. [DOI] [PubMed] [Google Scholar]
- 8.Cheng AC, Coleman RG, Smyth KT, Cao Q, Soulard P, Caffrey DR, Salzberg AC, Huang ES. Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotechnol. 2007;25:71–75. doi: 10.1038/nbt1273. [DOI] [PubMed] [Google Scholar]
- 9.Weisel M, Proschak E, Schneider G. PocketPicker: analysis of ligand binding-sites with shape descriptors. Chem Central J. 2007;1:7. doi: 10.1186/1752-153X-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peters KP, Fauck J, Frommel C. The automatic search for ligand binding sites in proteins of known three-dimensional structure using only geometric criteria. J Mol Biol. 1996;256:201–213. doi: 10.1006/jmbi.1996.0077. [DOI] [PubMed] [Google Scholar]
- 12.Xie L, Bourne PE. A robust and efficient algorithm for the shape description of protein structures and its application in predicting ligand binding sites. BMC Bioinformatics. 2007;8:S9. doi: 10.1186/1471-2105-8-S4-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hendlich M, Rippman F, Barnickel G. LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol Graph Model. 1997;15:359–363. doi: 10.1016/s1093-3263(98)00002-3. [DOI] [PubMed] [Google Scholar]
- 14.Kalidas Y, Chandra N. PocketDepth: a new depth based algorithm for identification of ligand binding sites in proteins. J Struct Biol. 2008;161:31–42. doi: 10.1016/j.jsb.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Coleman RG, Sharp KA. Protein pockets: inventory, shape, and comparison. J Chem Inf Model. 2010;50:689–603. doi: 10.1021/ci900397t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim D, Cho CH, Cho Y, Ryu J, Bhak J, Kim DS. Pocket extraction on proteins via the Voronoi diagram of spheres. J Mol Graphics Model. 2008;26:1104–1112. doi: 10.1016/j.jmgm.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Kahraman A, Morris RJ, Laskowski RA, Thornton JM. Shape variation in protein binding pockets and their ligands. J Mol Biol. 2007;368:283–301. doi: 10.1016/j.jmb.2007.01.086. [DOI] [PubMed] [Google Scholar]
- 18.Lichtarge O, Sowa ME. Evolutionary predictions of binding surfaces and interactions. Curr Opin Struct Biol. 2002;12:21–27. doi: 10.1016/s0959-440x(02)00284-1. [DOI] [PubMed] [Google Scholar]
- 19.Glaser F, Morris RJ, Najmanovich RJ, Laskowski RA, Thornton JM. A method for localizing ligand binding pockets in protein structures. Struct Funct Bioinf. 2006;62:479–288. doi: 10.1002/prot.20769. [DOI] [PubMed] [Google Scholar]
- 20.Chen BY, Bryant DH, Fofanov VY, Kristensen DM, Cruess AE, Kimmel M, Lichtarge O, Kavraki LE. Cavity scaling: automated refinement of cavity-aware motifs in protein function prediction. J Bioinf Comp Biol. 2007;5:353–382. doi: 10.1142/s021972000700276x. [DOI] [PubMed] [Google Scholar]
- 21.Joughin BA, Tidor B, Yaffe MB. A computational method for the analysis and prediction of protephosphopeptide-binding sites. Protein Sci. 2005;14:131–139. doi: 10.1110/ps.04964705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettit FK, Bare E, Tsai A, Bowie JU. HotPatch: a statistical approach to finding biologically relevant features on protein surfaces. J Mol Biol. 2007;369:863–879. doi: 10.1016/j.jmb.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawabata T. Detection of multiscale pockets on protein surfaces using mathematical morphology. Proteins Struct Funct Bioinf. 2010;78:1195–1211. doi: 10.1002/prot.22639. [DOI] [PubMed] [Google Scholar]
- 24.Kawabata T, Go N. Detection of pockets on protein surfaces using small and large probe spheres to find putative ligand binding sites. Proteins Struct Funct Bioinf. 2007;68:516–529. doi: 10.1002/prot.21283. [DOI] [PubMed] [Google Scholar]
- 25.Laurie ATR, Jackson RM. Q-siteFinder: an energy-based method for prediction of protein-ligand binding sites. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 26.Ming D, Cohn JD, Wall ME. Fast dynamics perturbation analysis for prediction of protein functional sites. BMC Struct Biol. 2008;8:5. doi: 10.1186/1472-6807-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coleman RG, Salzberg AC, Cheng AC. Structure-based identification of small molecule binding sites using a free energy model. J Chem Inf Model. 2006;46:2631–2637. doi: 10.1021/ci600229z. [DOI] [PubMed] [Google Scholar]
- 28.Brylinski M, Skolnick J. A threading-based method FINDSITE for ligand-binding site prediction and functional annotation. Proc Natl Acad Sci USA. 2008;105:129–134. doi: 10.1073/pnas.0707684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Capra JA, Laskowski RA, Thornton JM, Singh M, Funkhouser TA. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput Biol. 2009;5:e1000585. doi: 10.1371/journal.pcbi.1000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soga S, Shirai H, Kobori M, Hirayama N. Use of amino acid composition to predict ligand-binding sites. J Chem Inf Model. 2007;47:400–406. doi: 10.1021/ci6002202. [DOI] [PubMed] [Google Scholar]
- 31.Kinjo AR, Nakamura H. Comprehensive structural classification of ligand binding motifs in proteins. Structure. 2009;17:234–246. doi: 10.1016/j.str.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 32.Hoffmann B, Zaslavskiy M, Vert JP, Stoven V. A new protein binding pocket similarity measure based on comparison of clouds of atoms in 3D: application to ligand prediction. BMC Bioinformatics. 2010;11:99. doi: 10.1186/1471-2105-11-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abagyan R, Kufareva I. The flexible pocketome engine for structural chemogenomics. Methods Mol Biol. 2009;575:249–279. doi: 10.1007/978-1-60761-274-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith AJT, Zhang X, Leach AG, Houk KN. Beyond picomolar affinities: quantitative aspects of noncovalent and covalent binding of drugs to proteins. J Med Chem. 2009;52:225–233. doi: 10.1021/jm800498e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orita M, Ohno K, Niimi T. Two “Golden ratio” indices in fragment-based drug discovery. Drug Discov Today. 2009;14:321–328. doi: 10.1016/j.drudis.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Abad-Zapatero C, Metz JT. Ligand efficiency indices as guideposts for drug discovery. Drug Discov Today. 2005;10:464–469. doi: 10.1016/S1359-6446(05)03386-6. [DOI] [PubMed] [Google Scholar]
- 37.Fukunishi Y, Mikami Y, Kubota S, Nakamura H. Multiple target screening method for robust and accurate in silico ligand screening. J Mol Graph Model. 2005;25:61–70. doi: 10.1016/j.jmgm.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 38.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A Geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 39.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 40.Jones G, Willet P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 41.Warren GL, Webster Andrews C, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 42.Baxter CA, Murray CW, Clark DE, Westhead DR, Eldridge MD. Flexible docking using tabu search and an empirical estimate of binding affinity. Proteins. 1998;33:367–382. [PubMed] [Google Scholar]
- 43.Fukunishi Y, Mikami Y, Nakamura H. Similarities among receptor pockets and among compounds: analysis and application to in silico ligand screening. J Mol Graph Model. 2005;24:34–45. doi: 10.1016/j.jmgm.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 44.Fukunishi Y, Sugihara Y, Mikami Y, Sakai K, Kusudo H, Nakamura H. Advanced in-silico drug screening to achieve high hit ratio-development of 3D-compound database. Synthesiology. 2009;2:64–72. [Google Scholar]
- 45.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. 2004. AMBER 8, UCSF.