

Abstract
Electron paramagnetic resonance (EPR) spectroscopy coupled with site-directed spin labeling (SDSL) is a valuable tool for characterizing the mobility and conformational changes of proteins but has seldom been applied to intrinsically disordered proteins (IDPs). Here, IA3 is used as a model system demonstrating SDSL-EPR characterization of conformational changes in small IDP systems. IA3 has 68 amino acids, is unstructured in solution, and becomes α-helical upon addition of the secondary structural stabilizer 2,2,2-trifluoroethanol (TFE). Two single cysteine substitutions, one in the N-terminus (S14C) and one in the C-terminus (N58C), were generated and labeled with three different nitroxide spin labels. The resultant EPR line shapes of each of the labels were compared and each reported changes in mobility upon addition of TFE. Specifically, the spectral line shape parameters h(+1)/h(0), the local tumbling volume (VL), and the percent change of the h(−1) intensity were utilized to quantitatively monitor TFE-induced conformational changes. The values of h(+1)/h(0) as a function of TFE titration varied in a sigmoidal manner and were fit to a two-state Boltzmann model that provided values for the midpoint of the transition, thus, reporting on the global conformational change of IA3. The other parameters provide site-specific information and show that S14C-SL undergoes a conformational change resulting in more restricted motion than N58C-SL, which is consistent with previously published results obtained by studies using NMR and circular dichroism spectroscopy indicating a higher degree of α-helical propensity of the N-terminal segment of IA3. Overall, the results provide a framework for data analyzes that can be used to study induced unstructured-to-helical conformations in IDPs by SDSL.
Keywords: intrinsically disordered proteins, site-directed spin labeling, SDSL, EPR, TFE, IA3
Introduction
Intrinsically disordered proteins (IDPs) are proteins or protein segments (>50 residues) that lack uniform secondary and tertiary structure under physiological conditions.1–7 Many proteins from higher eukaryotic systems contain regions of disorder in their structure,8 which often correlate to vital functional roles in biology such as transcriptional and translational regulation, signal transduction, and protein phosphorylation.4,9 In other instances, IDPs undergo coupled folding and binding, where conformational changes in segments of the IDP are induced upon binding to their target protein.6,10,11
IA3 is a 68 amino acid protein found in the cytoplasm of Saccharomyces cerevisiae that acts as a potent inhibitor of yeast proteinase A (YPRA). X-ray crystallography studies reveal that the first 34 amino acids of IA3 adopt an α-helical conformation upon binding in the active site of YPRA, whereas the 34 C-terminal amino acids have not been resolved in the X-ray data.12,13 Far-UV circular dichroism (CD) studies show that IA3 is unstructured in solution. In the presence of the secondary structural stabilizer 2,2,2-trifluoroethanol (TFE) a two-state transition from an unstructured-to-α-helix conformation is induced.13 The CD analysis cannot, however, establish site-specific information regarding the extent to which a specific residue within the IA3 sequence gained α-helical structure or whether the TFE-induced two-state transition was uniform throughout the protein. 2D15N 1H NMR spectroscopy coupled with singular-valued decomposition (SVD) analysis shows that the N-terminal residues adopt more pronounced α-helical structure with TFE than the C-terminal residues.14
Site-directed spin-labeling (SDSL)-electron paramagnetic resonance (EPR) has become a powerful technique for investigating macromolecular structure, conformational changes, and protein dynamics.6,7,15–19 In this method, an EPR-active reporter group, such as a nitroxide spin label, is introduced into the biological system to obtain site-specific information about structure and flexibility. Typically, a non-native cysteine (CYS) residue is incorporated into a desired location within the protein via site-directed mutagenesis, which is then chemically modified with a spin label.
The EPR spectral line shape generated from the spin label provides valuable structural information.6,7,15–17,19 When the overall motion of the spin label varies in the 0.1–50 ns regime, the motion has a dramatic effect on the observed EPR line shape.20 The following three primary modes of motion contribute to the EPR spectrum: the overall tumbling of the molecule (τR), the motion of the spin label about the bonds that connect it to the protein (τi), and the motion of the protein backbone to which the spin label is attached (τB). The backbone motion and the motion of the spin label about the tethering bonds provide the most structural information on biomolecules in SDSL studies.21 Although SDSL-EPR has been shown to be a useful technique to study ordered-to-disordered transitions in numerous proteins,18,22–25 it is not widely recognized as a tool to study IDP systems.9 One notable example found in the literature is the characterization of an induced folding event of the intrinsically disordered C-terminal domain of the measles virus nucleoprotein.6,7,26
The work presented here extends the applicability of SDSL-EPR to study conformational changes in IDPs. Here, we use SDSL-EPR spectroscopy to characterize the TFE-induced α-helical conformational change in IA3. Because of the relative simplicity of this protein and detailed characterization previously performed by CD and NMR spectroscopy, IA3 serves as an excellent model system for further development of the SDSL-EPR methodology for studying IDPs. Two cysteine substitutions were generated in IA3; one, in the N-terminus which has been shown by X-ray crystallography to undergo an unstructured-to-α-helical transition upon binding to YPRA, and one in the C-terminus that remained unresolved12 in the crystal structure but has been shown by NMR to adopt α-helical structure in the presence of TFE.14
Results
Effects of spin label moiety on monitoring the helical conformational change
The following three different spin labels were used in this study: methanethiosulfonate (MTSL), 4-maleimido-TEMPO (MSL), and 3-(2-iodoacetamido)-proxyl (IAP). The structures of each label and the resultant modified CYS residue are given in Figure 1(A–C). The effects of the CYS substitution and incorporation of the spin label on the secondary structure of IA3 were investigated with far-UV CD. Spectra were collected for 0 and 30% TFE for wild-type (WT) IA3 and for each CYS variant labeled with MTSL, MSL, or IAP. CD spectra of the modified IA3 constructs are nearly identical to the results obtained for the WT protein. The structure of the modified IA3 constructs are determined to be predominantly random coil in solution, indicated by a negative band near 200 nm and some weak bands between 220 and 230 nm. For 30% TFE, the conformation of the modified proteins becomes α-helical, as indicated by double minima at 222 nm and ∼210 nm and a maximum at 190 nm.13 The CD spectra of the spin-labeled constructs indicate that the amino acid substitution and spin labeling do not alter the disorder-to-α-helical transition in IA3 upon induction by TFE (amino acid sequences and CD spectra are given in the Supporting Information).
Figure 1.

Structures of the spin labels used in this study and the resulting chemical modification of the cysteine side chain. (A) MTSL, (1-oxyl-2,2,5,5-tetramethyl-D3-pyrroline-3-methyl) methanethiosulfonate; (B) MSL, 4-maleimido-TEMPO; (C) IAP, 3-(2-iodoacetamido)-PROXYL (D) 100G X-band CW- EPR spectrum of IA3 S14C-IAP with labeled transitions indicating the peak-to-peak intensities of the low-field, h(+1), center-field, h(0), and high-field, h(-1), resonances. (E and F) Stack plots of area normalized 100G X-band EPR spectra of IA3 variants S14C and N58C labeled with MTSL, MSL, and IAP in the presence of 0, 15, and 30% TFE.
Continuous wave (CW) X-band EPR spectra were collected for IA3 samples labeled at sites S14C and N58C with MTSL, MSL, or IAP in increasing concentrations of TFE that ranged from 0 to 40% in 5% increments. Figure 1(E and F) show stack plots of double-integral-(area)-normalized 100 G X-band EPR spectra for samples with 0, 15, and 30% TFE. The complete sets of EPR data are given in the Supporting Information.
Two general conclusions can be drawn from the data plotted in Figure 1(E and F). First, the spectral line shapes for the three spin labels follow the expected trend of mobility given their individual structures and tethering geometries. Mobility of the spin label is defined as both the rate of motion and the restricted/unrestricted conformations which it samples, often described as order of the spin label.15,19 The spectra are plotted with normalized areas; hence, the overall intensity of the signal is proportional to the mobility. The higher the intensity, the more motional averaging the spin probe undergoes. As mobility decreases, the normalized intensity will decrease, and the spectra will have an overall broadening. For both sites investigated, the line shapes have the following expected mobility trend: IAP > MTSL > MSL.
A second general observation is that upon addition of TFE, the overall intensity of all of the spectra decrease and broaden; indicative of a conformational change. To ensure the reduced mobility seen in the spectral line shapes did not arise from changes in solution viscosity, control experiments (data shown in Supporting Information) were performed for IA3 in solutions of 6% sucrose, which is iso-viscous to the 30% TFE at 27°C. Negligible changes were observed in comparison of the 0% TFE EPR spectra to those obtained with 6% sucrose; thus, indicating that the changes seen upon addition of TFE in the X-band spectra arise from conformational changes of the protein backbone, which lowers the overall mobility of the spin label.
Quantitative analysis of the TFE-induced change observed via SDSL
The previous section provides a qualitative approach in discussing the spectral line shapes shown in Figure 1(E and F) and Supporting Information Figures S8–S10. However, a more quantitative approach can be taken by analyzing various EPR spectral line shape parameters. Figure 1(D) shows a representative nitroxide EPR spectrum. The three observed transitions arise from hyperfine coupling of the electron with the magnetic moment of the nitrogen nucleus.27 As shown, the low-field transition is designated as h(+1), the center-field transition is designated as h(0), and the high-field transition is designated as h(−1). When spectra are plotted with normalized area, the intensities of the three transitions provide information about the spin label mobility.28
Although the high field transition of the X-band CW EPR line shape is often correlated to the most sensitive molecular motion,29 values of the h(+1)/h(0) ratio were utilized in a previous SDSL study of a highly dynamic protein; specifically, values of this parameter were used to monitor conformational changes in the measles virus nucleoprotein upon binding to its target.28 We also utilized this parameter, and Figure 2 plots values of h(+1)/h(0) as a function of %TFE for IA3 sites S14C and N58C labeled with MTSL and MSL at 27°C and with IAP at 27 and 5°C. All of the data sets have a sigmoid shape that could readily be fit to the two-state Boltzmann function [Eq. (1)], where the results from the best fits are given with solid lines through the data points. From fitting the data to Eq. (1), values of the %TFE transition midpoints were determined to be 17 ± 2%, 20 ± 1%, 20 ± 1%, 14 ± 1%, 19 ± 1%, and 18 ± 2% for S14C-MTSL, S14C-MSL, S14C-IAP, N58C-MTSL, N58C-MSL, and N58C-IAP, respectively. The average midpoint value of 18 ± 1% determined from the EPR data collected at 27°C, agrees well with the value of 18.3% obtained from previously reported NMR data analysis.14
Figure 2.
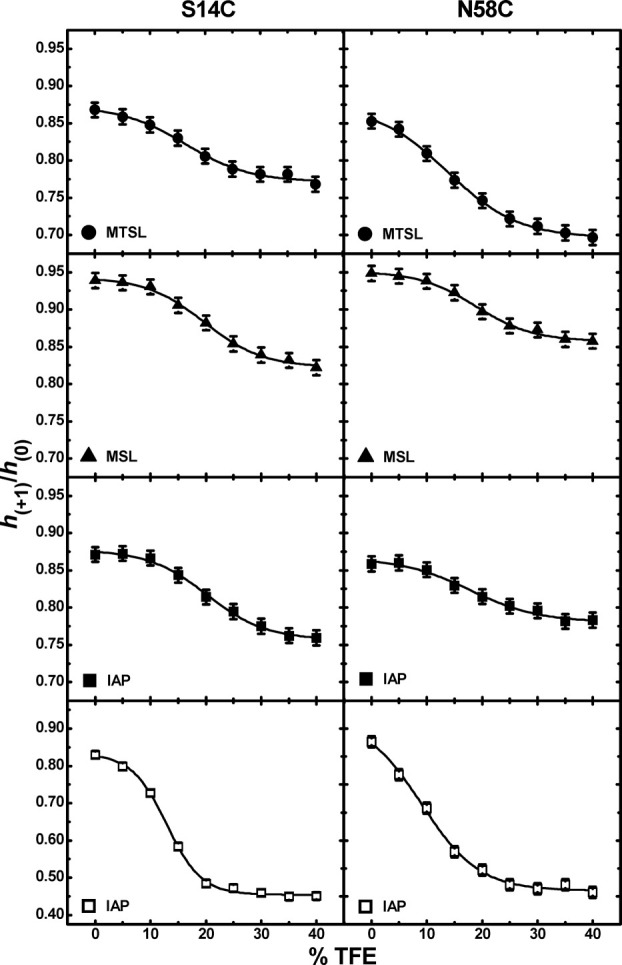
Plots of h(+1)/h(0) as a function of TFE for S14C (left) and N58C (right) labeled with either MTSL (•), MSL (▴), and IAP (▪) collected at 27°C and IAP (□) collected at 5°C. The sizes of the data points are larger than errors in the measurements.
The effects of temperature were also investigated. Spectra were also collected at 5°C for S14C-IAP and N58C-IAP, where the global tumbling of the protein should be reduced. Data for samples collected at 5°C are shown in the bottom panels of Figure 2 (only IAP labels were collected at the colder temperatures). The magnitude of the change in the values of this parameter are greater at the lower temperature, reflecting a more distinct conformational change, and possibly higher α-helical character, which is consistent with degree of helicity seen in CD spectra of IA3 with TFE at lower temperatures.30 As stated above the solid lines represent the best fits with Eq. (1), and the midpoints were found to be 12 ± 2% and 9 ± 2% for S14C-IAP and N58C-IAP, respectively, which are lower than the values obtained at 27°C. These results agree well with data obtained from previously reported CD analysis, which showed the transition occurs at a lower TFE percentage at colder temperatures, and those thermodynamic parameters predict a midpoint %TFE of 12% at 5°C.30
An additional way to analyze the EPR line shapes is by analyzing how the local tumbling volume, VL, of the spin label changes upon addition of TFE. Plots of VL as a function of %TFE for both S14C and N58C labeled with IAP at both 27 and 5°C are shown in Figure 3. In the unstructured state, the local tumbling volume is expected to be small. As the α-helical character is induced, the Stokes–Einstein radius of a rigid helix should increase, thus increasing the local tumbling volume of the spin label. For both S14C and N58C at 27°C, as expected, VL is seen to increase linearly upon addition of TFE to percentages below 25%. Above 25%, the slope flattens, indicating a change in the dependence of VL with %TFE. For the data collected at 5°C, a transition where the slopes of the linear regions differ can also be observed. The greater negative slope for 25–40% TFE at 5°C is believed to arise from inaccuracy of the simulation method for these more anisotropic line shapes (discussed below) and errors in viscosity measurements arising from salt precipitation at the lower temperature that occur for high %TFE. Note, no protein precipitation was observed in the EPR samples, but salt precipitation was observed over time in viscosity measurements. The intersection points for each site are found to be roughly 20 and 24% TFE for 27 and 5°C, respectively. These points of intersection represent the concentration of TFE for where the conformational change has completed and agree well with previous results from CD and NMR analysis. An interesting observation is that at both temperatures, the magnitude of the change of VL for site S14C-IAP is greater than that of site N58C-IAP, indicating that the N-terminal site is undergoing a larger induced conformational change than the N58C site. This finding is also in agreement with previously published NMR data.14
Figure 3.
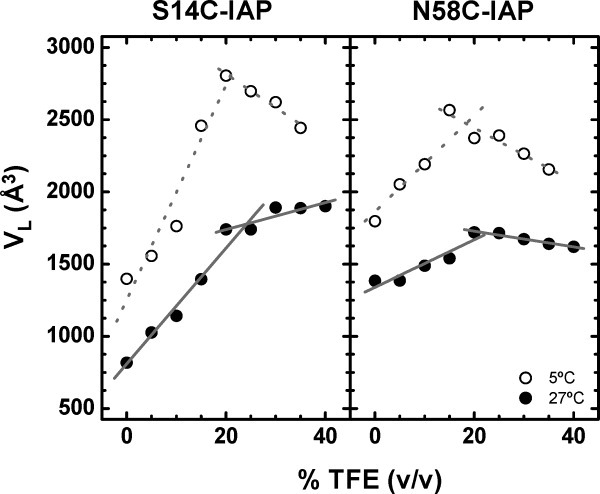
The local tumbling volume (VL) of the protein as a function of increasing percentage TFE for S14C-IAP (left) and N58C-IAP (right) collected at 27°C (•) and 5°C (○). Lines represent linear regression fits to the data. The sizes of the data points are larger than errors in the measurements.
The TFE-induced conformational change of IA3 was also monitored by observing the percentage change in the normalized intensity of the high field transition, h(−1), as a function of % TFE. Figure 4 plots the absolute value of this parameter upon increasing % TFE determined from spectra of both S14C and N58C IA3 in solution for the various spin labels used here. Analyzing the data in this fashion provides a similar trend as the results obtained from the local volume analysis given in Figure 3. For each sample, the dependence of the value of the percentage change in the normalized intensity of the high-field transition, h(−1), on %TFE can easily be seen to fall into two linear regions described as follows: for TFE < 20%, the value of this parameter is highly dependent on %TFE, and for TFE > 20%, this line shape parameter is nearly independent of TFE concentration. Again, the significance of 20% TFE is indicative of the concentration for which the conformational change is complete. At concentrations TFE > 20%, the values of h(−1) do not significantly change upon further increases in TFE concentration, indicating that the spin label mobility and, hence, protein structure remain constant above this concentration. For concentrations < 20% TFE, the EPR spectra broaden progressively as TFE is added, which is consistent with an increase in the relative percentage of α-helical conformation that would change the relative mobility of the spin label in a dose-dependent manner. The value of 20% TFE identified from this analysis is again consistent with prior CD analyses of IA3, which found that complete α-helical structure is obtained at 23% TFE.13 In addition, as noted above, the magnitude of the overall change in this line shape parameter, regardless of spin label used, is less for the C-terminal site than for the N-terminal site, reflecting a greater degree of conformational change in the N-terminus of IA3.
Figure 4.
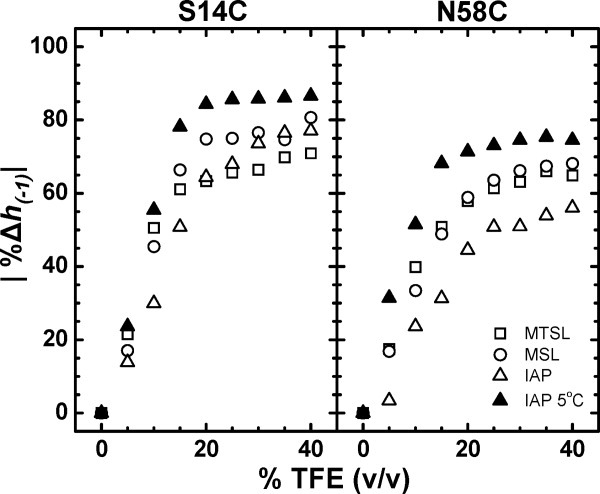
The absolute value of the percent change of the intensity of the high-field resonance, h(-1), as a function of increasing percentage TFE for S14C (left) and N58C (right) labeled with either MTSL (□), MSL (○), and IAP (Δ) collected at 27°C and IAP (▴) collected at 5°C. The percent change was calculated by subtracting the h(−1) intensity for spectra collected in the presence of TFE from the intensity of the spectrum in the absence of TFE (I0), dividing that quantity by I0 and multiplying by 100%. The sizes of the data points are larger than errors in the measurements.
Discussion
Effects of spin label choice on data analysis
In an effort to discern if various spin label structures report more readily on the induced unstructured to α-helical conformational change, three separate spin-labeled samples were prepared for each site in IA3. The structures of each spin label are shown in Figure 1. The MTSL probe [Fig. 1(A)], when attached to a protein, it is often considered to have restricted mobility around only two rotatable bonds described as the χ4/χ5 model31 and may report most effectively on backbone conformational changes. MTSL modifies cysteine sites by forming a disulfide bond, which is easily reduced in the presence of other reactive thiol groups or reducing agents, which may be a drawback of utilizing this spin label in highly dynamic unstructured proteins. In this study, it was observed that MTSL dissociated from the protein and dimerized with itself, as evident by the spin dimer peaks present in the N58C-MTSL spectra shown in Figure S9 of the Supporting Information. This phenomenon has been shown to occur in other systems as well, which lead to the necessity to investigate the use of other nitroxide spin labels with different binding chemistry.32 MSL [Fig. 1(B)] forms a nonreducible thioester bond with the cysteine side chain and is considered to be rigid with limited rotations arising from the bulkiness of the 4-maleimido-TEMPO group. IAP [Fig. 1(C)] also forms a nonreducible thioester bond but has an extended and flexible linker region in comparison with the other two labels; thus, having the greatest degree of motionally averaged line shape. Spin labeling was attempted both in air and under argon and no differences were detected in the labeling efficiency as determined by comparing the integrated EPR signal and total protein concentrations. We find for this system that labeling with IAP was more efficient that with either MSL or MTSL; most likely because the iodoacetemide chemistry of may be capable of labeling preformed disulfide bonds; whereas the maleimide or thiosulfonate need reduced cysteine side chains. Although the various labels showed differing degrees of labeling efficiencies, all line shape analyses shown here reveal that each of the three spin labels readily reports on the global conformational change of IA3 upon undergoing an unstructured to α-helical conformational change induced with TFE. From the data collected in this study, we concluded that IAP was the “best” spin label to work with for this study due to difficulties in labeling with MTSL due to the high propensity of this peptide to disulfide bond with itself during protein purification and subsequent labeling. The unrestricted mobility of the IAP label was quite useful for analysis of changes in local tumbling volume and reporting on the global conformational change from an unstructured random coil to a more rigid helical rod that had an altered Stokes–Einstein radius and slower global tumbling correlation time.
Data analysis via h(+1)/h(0)
In a previous SDSL study of a highly dynamic IDP, the h(+1)/h(0) parameter was used to monitor conformational changes occurring in the measles virus nucleoprotein.6,7 Here, we also utilized this parameter to characterize the conformational changes in IA3 upon the addition of TFE. Our results show that both the N and C termini of IA3 are undergoing a conformational change as the percentage of TFE increases. Control experiments in both urea and various concentrations of sucrose (Supporting Information) indicate that the changes in the spectral line shapes seen upon addition of TFE do not simply arise from changes in solution viscosity but are indeed indicative of the induced conformational change in the protein. Regardless of the choice of spin label used, all plots of h(+1)/ h(0) give a sigmoid shape that could be readily fit to a two-state Boltzmann function. Ganesh et al. observed by SVD analysis of their NMR data that the overall effect of TFE on the coil-to-helix transition of IA3 can be evaluated as a two-state transition, where they state “The first two components of the TFE dependence of both the 1HN and 15N chemical shifts exhibit sigmoidal trends with increasing amounts of TFE. These movements are strongly indicative of a two-state transition, where one state is progressively depopulated in favor of a second state as the TFE concentration rises.”14 The h(+1)/ h(0) data in this study also seem to be indicative of a conformational change from the unstructured state to the α-helical conformation. The h(+1)/h(0) method gave midpoint values that were in agreement with those previously reported in a detailed NMR study, where NMR coupled with singular value decomposition was used to determine the degree of helical conformation and the midpoint of the conformational transition for IA3.14 The h(+1)/h(0) analysis provides information regarding the global conformational change from a highly dynamic unstructured peptide segment to a more structured rigid protein that has changes in both local spin label correlation times and global protein tumbling, τR, where, τR is likely dominating the motional averaging of the nitroxide hyperfine tensor.
Comparison of VL to the percent change of h(−1)
An additional parameter that we used to characterize the unstructured to α-helical conformation of IA3 was the local tumbling volume parameter, VL, introduced by Freed and coworkers. Previously, this line shape parameter was also extensively utilized by Millhauser and coworkers to characterize cold-temperature-induced α-helical conformational changes in short alanine synthetic peptides, where as the temperature was decreased a more pronounced helix was formed.33,34 Values for VL represent the volume of the spin-labeled protein as the nitroxide reorients on the time scale of τR.34 The VL parameter provides structural information without the affect of external environmental factors such as viscosity and temperature, therefore providing information solely on the change in protein structure.33 Values of VL for IA3 increase as a function of %TFE indicating a conformational change from disordered-to-ordered system. When IA3 is unstructured the nitroxides ability to reorient back to its original starting position is fast, and therefore, gives a smaller value of VL. As α-helical structure is induced upon addition of TFE, the amount of time it takes for the nitroxide to reorient is slowed and results in a larger tumbling volume. The data shown in Figure 3 indicate that VL is providing site-specific information, where the volume in the S14C site undergoes a much greater change than the volume in the N58C site. VL does not report on the overall global change like the h(+1)/h(0) parameter and, therefore, plots as a function of %TFE do not have a sigmoid shape but instead change linearly until the conformational change is completed. One difficulty we faced, however, in utilizing VL, is the need to determine the rotational correlation time, which may not always be straightforward. As shown, the EPR data collected at 5°C have significant broadening of the high-field transition upon addition of TFE, indicating more anisotropic motion become concurrent with a correlation time that can no longer be accurately modeled by the simpler isotropic motion limit and would likely more accurately be described by simulations in the intermediate motion regime.15,35 This fact may contribute to the negative slopes of VL in Figure 3. Even the data obtained at higher temperatures have line shapes indicative of slower motion, outside the regime of the simple isotropic model used here.
Another parameter we found to report on site-specific changes in conformation in IA3 is the percentage change of the intensity of the h(−1) resonance. Both h(−1) and VL parameter profiles give similar results, where two distinct regions are identified (Figs. 3 and 4) and the point where the transition was complete can readily be determined from the intersection of the slopes of the two regions. Both parameters also provided information on the degree of change in each of the spin-labeled sites, and indicate that the N-terminal site is undergoing a larger conformational change than the C-terminal site. This finding is in agreement with the NMR data previously reported.14 The percent change in h(−1) has not been previously used as a parameter to gain structural information for IDPs. This study shows that the h(−1) parameter provides similar information as does calculating VL and does not require line shape simulations.
All three parameters used in this study provided valuable information on the induced conformational change occurring as a function of TFE. From this study, it appears that for small dynamic IDP systems that data analysis parameters that mainly rely on changes occurring in the high-field transition seem to be reporting on site-specific conformational changes, whereas parameters that rely on changes in the low- and center-field transitions provide information on the global conformational change. The local tumbling volume and the percent change of the h(−1) parameters appear to yield the TFE percentage needed to complete the conformational change. These parameters also indicate that the N-terminus is undergoing a larger conformational change than the C-terminus. The h(+1)/h(0) parameter appears to be reporting on the global conformational change, where it provides information on the overall global unstructured-to-α-helical transition that both the N- and C-terminal regions are experiencing. Although the h(+1)/h(0) parameter has been shown to be site-specific in a study on the C-terminal domain of the measles virus nucleoprotein,6,7,26 it does not appear to report the same information on IA3 which is significantly smaller, therefore, the EPR spectral line shapes are being dominated by τR. However, this parameter is still useful in investigating small IDP systems as it provides the transition midpoint of the conformational change.
Conclusions
SDSL-EPR spectroscopy was used to monitor the disordered-to-α-helical transition of IA3. Three different nitroxide probes were used, and it was shown that due to the tendency of MTSL to dissociate from the protein, it is not the best spin label choice for studying highly dynamic IDP systems. Although qualitative information can be obtained by studying the EPR line shapes, more information about the transition comes from analysis of various line shape parameters. To gain more quantitative information from the resultant line shapes, three techniques were used. The percent change in the h(−1) intensity and values of VL provide similar information, indicating at what point the conformational transition are complete and provide site-specific information, which indicate that N58C, in the C-terminus, does not undergo as large of a change as does site S14C in the N-terminus. The other parameter used was analysis of h(+1)/h(0), which has previously been used to monitor an induced conformational change in an IDP system.6 Here, we find upon titration of TFE, this parameter gave sigmoidal trend that could readily be fit with a two-state Boltzmann function providing information about the midpoint of the conformational change that agrees well with previous NMR and CD investigations.13,14,30 The results from this model system show that the SDSL-EPR data can be interpreted in either global or site-specific conformational changes and imply that SDSL-EPR methods should be applicable to other IDP systems. EPR methods may provide an advantage over NMR methods given that the SDSL-EPR has no upper size limitations and the h(+1)/h(0) parameter, and the percentage change in the h(−1) line shape analyses are straightforward.
Materials and Methods
Materials
(1-Oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl) methanethiosulfonate spin label (MTSL) was purchased from Toronto Research Chemicals, (North York, ON, Canada). 3-(2-iodoacetamido)-PROXYL (IAP) and 4-maleimido-2,2,6,6-tetramethyl-1-piperidinyloxy (4-maleimido-TEMPO, MSL) spin labels were purchased from Sigma Aldrich (St. Louis, MO). His-Bind resin was purchased from Novagen (Gibbstown, NJ). The QuikChange site-directed mutagenesis kit was purchased from Stratagene (La Jolla, CA). BL21(DE3) cells were purchased from Invitrogen (Carlsbad, CA). Unless otherwise stated, all other reagents were purchased from Fisher Scientific (Pittsburg, PA) and used as received.
Protein expression and purification of IA3 mutants
E. Coli codon-optimized DNA encoding for the IA3 gene was purchased from DNA2.0. The gene was cloned into the pET-22b vector (Novagen) generously provided by Art Edison. Cysteine substitutions for spin labeling were introduced using the QuikChange site-directed mutagenesis kit, and the sequence was confirmed by DNA sequencing. WT and cysteine variants were expressed via an E. Coli system modified from the original protocol.12 The bacterial host strain for expression was BL21(DE3) cells. Cells were grown at 37°C in LB medium to an OD600 of ∼0.6 before induction with 1 mM IPTG. After ∼2 h of expression, the cells were pelleted, resuspended, and lysed by sonication and three passes through a 35-mL French pressure cell (Thermo Scientific, Waltham, MA). After lysis, the supernatant was boiled for 5 min to precipitate structured proteins and assist in purification, and the supernatant was centrifuged to remove insoluble material (18,500 g, 20 min, 4°C). Boiling the lysate versus boiling the purified protein was a modification from the original protocol. It has been shown that boiling the lysate assists in protein purification of IDPs.36 The soluble recombinant protein was purified utilizing a C-terminal His-tag encoded by the pET-22b vector by affinity chromatography as described previously.12 Protein was estimated to be pure by a 16.5% Tris-Tricine SDS-page gel. EDTA (1 mM) was added to purified IA3 to remove any residual nickel that may have been leeched off the affinity column.
Spin labeling
Proteins containing the cysteine substitutions were buffer exchanged into 50 mM sodium phosphate, 300 mM sodium chloride, pH 7.4 using a HiPrep 26/10 desalting column (Amersham, Pittsburg, PA). Molar excess (100:1) of dithiothreitol (DTT) was added to the 50 μM protein solution and allowed to react overnight. DTT was removed using a desalting column (as described above) equilibrated and eluted with 50 mM sodium phosphate, 300 mM sodium chloride, pH 7.4 for both IAP or MSL, or pH 6.9 for MTSL. Protein was then spin labeled with 10x molar excess of IAP, MSL, or MTSL dissolved in ethanol, in the dark at room temp (22°C) for 4 h. Excess spin label was removed via the desalting procedure described above. Protein was eluted in 50 mM sodium phosphate, 300 mM sodium chloride, pH 7.4. Spin-labeling efficiency was evaluated by double integration of the EPR signal and comparison with a TEMPO standard curve. The labeling yields of both the S14C and N58C variants were estimated to be ∼30% for both MTSL and MSL and 50% for IAP.
EPR sample preparation
Samples were prepared by adding 0–40% TFE in 5% increments to ∼100 μM spin-labeled protein. Samples were prepared with IA3 either free in solution or bound to His-Bind resin charged with 0.1 mM nickel sulfate and equilibrated in 50 mM sodium phosphate, 300 mM sodium chloride, pH 7.4 buffer containing 0–40% TFE. Excess protein was rinsed away from the resin with the aforementioned buffer.
CW X-band EPR spectra
CW X-band EPR spectra were collected on a modified Bruker ER200 spectrometer with an ER023 M signal channel, an ER032 M field control unit, and a loop gap resonator (Medical Advances, Milwaukee, WI). Samples of ∼10 μL were loaded into 0.60 i.d. × 0.84 o.d. capillary tubes (Fiber Optic Center, New Bedford, MA). All CW EPR experiments were performed at 27°C unless otherwise stated as some spectra were collected at 5°C. All spectra were collected as 100 Gauss (G) scans with 2-mW incident microwave power. The 100 kHz field modulation amplitude and time constant of the detector were optimized to provide maximum signal-to-noise ratio with no line broadening. All spectra are reported as the average of 10 scans. Spectra are plotted with intensities scaled to normalized absorption area. LabVIEW software was used for baseline correction and double integral area normalization, which was generously provided by Drs. Christian Altenbach and Wayne Hubbell (UCLA).
Data analysis
The EPR line shapes were analyzed using various analysis techniques. The first technique used monitors h(+1)/h(0) as a function of TFE percentage. The data can be fit to a two-state Boltzmann function, and midpoints of transition are determined. The Boltzmann function is shown in Eq. (1), were A1 is the initial value of the curve, A2 is the final value of the curve, and the midpoint is given by the xo term.
| (1) |
Monitoring the local tumbling volume (VL) of the spin label is another analysis technique that can be used to investigate conformational changes in biomolecules.33,34 It has been shown that this parameter reports on structural information of the protein system with external environment-dependent quantities removed.33,34 The expression for VL is given by Eq. (2),
| (2) |
where k is the Boltzmann constant, T is the absolute temperature, τR is the rotational correlation time, and η is the solvent viscosity. Values of τR were determined from line shape simulations using a program generously provided by Alex Smirnov (NCSU). The simulation provides values for the Lorentzian line widths of the three transitions that are subsequently used in Eq. (3),
| (3) |
where M is the nuclear spin quantum number of the Mth hyperfine line, T2(M) is the spin–spin relaxation time (which is inversely proportional to the homogeneous line width) of that line, and A, B, and C are the line shape parameters. In the fast motional regime, assumptions can be made and B and C alone can be used to determine rotational correlation time.37 Values of τR were determined using Eq. (4),
| (4) |
where the value of B is obtained from Eq. (3).
Viscosity measurements
Viscosity measurements were performed using Cannon-Fenske Viscometer (size 50) suspended in a water bath at 6 and 27°C. TFE solutions of 0–40% (v/v) in increments of 5% were made by combining the appropriate amount of TFE, and the pH 7.4 buffer previously mentioned. Measurements were repeated four times to ensure reproducibility. The values collected are expressed as kinematic viscosity in units of centistokes (cS) and needed to be converted to dynamic viscosity (kg m−1 s−1) utilizing the densities of each of the solutions.
Acknowledgments
The authors would like to thank Stephen J. Hagen and Arthur S. Edison for helpful discussions about the data, Alex Smirnov for providing the software for line shape simulations, and Christian Altenbach for Labview software to analyze the EPR data.
References
- 1.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Zhang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graphics Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 2.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 3.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 5.Fink AL. Natively unfolded proteins. Curr Opin Struc Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Morin B, Bourhis JM, Belle V, Woudstra M, Carriere F, Guigliarelli B, Fournel A, Longhi S. Assessing induced folding of an intrinsically disordered protein by site-directed spin-labeling electron paramagnetic resonance spectroscopy. J Phys Chem B. 2006;110:20596–20608. doi: 10.1021/jp063708u. [DOI] [PubMed] [Google Scholar]
- 7.Belle V, Rouger S, Costanzo S, Liquiere E, Strancar J, Guigliarelli B, Fournel A, Longhi S. Mapping alpha-helical induced folding within the intrinsically disordered C-terminal domain of the measles virus nucleoprotein by site-directed spin-labeling EPR spectroscopy. Proteins. 2008;73:973–988. doi: 10.1002/prot.22125. [DOI] [PubMed] [Google Scholar]
- 8.Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struc Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 9.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. 2007;92:1439–1456. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuxreiter M, Simon I, Friedrich P, Tompa P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J Mol Biol. 2004;338:1015–1026. doi: 10.1016/j.jmb.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 11.Lacy ER, Filippov I, Lewis WS, Otieno S, Xiao L, Weiss S, Hengst L, Kriwacki RW. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat Struct Mol Biol. 2004;11:358–364. doi: 10.1038/nsmb746. [DOI] [PubMed] [Google Scholar]
- 12.Li M, Phylip LH, Lees WE, Winther JR, Dunn BM, Wlodaawer A, Kay J, Gustchina A. The aspartic proteinase from Saccharomyces cerevisiae folds its own inhibitor into a helix. Nat Struct Biol. 2000;7:113–117. doi: 10.1038/72378. [DOI] [PubMed] [Google Scholar]
- 13.Green TB, Ganesh O, Perry K, Smith L, Phylip LH, Logan TM, Hagen SJ, Dunn BM, Edison AS. IA(3), an aspartic proteinase inhibitor from Saccharomyces cerevisiae, is intrinsically unstructured in solution. Biochemistry. 2004;43:4071–4081. doi: 10.1021/bi034823n. [DOI] [PubMed] [Google Scholar]
- 14.Ganesh OK, Green TB, Edisono AS, Hagen SJ. Characterizing the residue level folding of the intrinsically unstructured IA(3) Biochemistry. 2006;45:13585–13596. doi: 10.1021/bi061358w. [DOI] [PubMed] [Google Scholar]
- 15.Columbus L, Hubbell WL. Mapping backbone dynamics in solution with site-directed spin labeling: GCN4–58 bZip free and bound to DNA. Biochemistry. 2004;43:7273–7287. doi: 10.1021/bi0497906. [DOI] [PubMed] [Google Scholar]
- 16.Galiano L, Blackburn ME, Veloro AM, Bonora M, Fanucci GE. Solute effects on spin labels at an aqueous-exposed site in the flap region of HIV-1 protease. J Phys Chem B. 2009;113:1673–1680. doi: 10.1021/jp8057788. [DOI] [PubMed] [Google Scholar]
- 17.Fanucci GE, Cafiso DS. Recent advances and applications of site-directed spin labeling. Curr Opin Struct Biol. 2006;16:644–653. doi: 10.1016/j.sbi.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 18.Qu KB, Vaughn JL, Sienkiewicz A, Scholes CP, Fetrow JS. Kinetics and motional dynamics of spin-labeled yeast iso-1-cytochrome c .1. Stopped-flow electron paramagnetic resonance as a probe for protein folding/unfolding of the C-terminal helix spin-labeled at cysteine 102. Biochemistry. 1997;36:2884–2897. doi: 10.1021/bi962155i. [DOI] [PubMed] [Google Scholar]
- 19.Hubbell WL, Gross A, Langen R, Lietzow MA. Recent advances in site-directed spin labeling of proteins. Curr Opin Struct Biol. 1998;8:649–656. doi: 10.1016/s0959-440x(98)80158-9. [DOI] [PubMed] [Google Scholar]
- 20.Hubbell WL, Altenbach C. Investigation of structure and dynamics in membrane-proteins using site-directed spin-labeling. Curr Opin Struc Biol. 1994;4:566–573. [Google Scholar]
- 21.Qin PZ, Iseri J, Oki A. A model system for investigating lineshape/structure correlations in RNA site-directed spin labeling. Biochem Biophys Res Comm. 2006;343:117–124. doi: 10.1016/j.bbrc.2006.02.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fanucci GE, Coggshall KA, Cadieux N, Kim M, Kadner RJ, Cafiso DS. Substrate-induced conformational changes of the perplasmic N-terminus of an outer-membrane transporter by site-directed spin labeling. Biochemistry. 2003;42:1391–1400. doi: 10.1021/bi027120z. [DOI] [PubMed] [Google Scholar]
- 23.Nelson WD, Blakely SE, Nesmelov YE, Thomas DD. Site-directed spin labeling reveals a conformational switch in the phosphorylation domain of smooth muscle myosin. Proc Natl Acad Sci USA. 2005;102:4000–4005. doi: 10.1073/pnas.0401664102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Z, DeSensi SC, Stein RA, Brandon S, Dixit M, McArdle EJ, Warren EM, Kroh HK, Song L, Cobb CE, Hustedt EJ, Beth AH. Solution structure of the cytoplasmic domain of erythrocyte membrane band 3 determined by site-directed spin labeling. Biochemistry. 2005;44:15115–15128. doi: 10.1021/bi050931t. [DOI] [PubMed] [Google Scholar]
- 25.Kim M, Fanucci GE, Cafiso DS. Substrate-dependent transmembrane signaling in TonB-dependent transporters is not conserved. Proc Natl Acad Sci USA. 2007;104:11975–11980. doi: 10.1073/pnas.0702172104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kavalenka A, Urbancic I, Belle V, Rouger S, Costanzo S, Kure S, Fournel A, Longhi S, Guigliarelli B, Strancar J. Conformational analysis of the partially disordered measles virus N(TAIL)-XD complex by SDSL EPR spectroscopy. Biophys J. 2010;98:1055–1064. doi: 10.1016/j.bpj.2009.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berliner LJ. Biological Magnetic Resonance, Spin Labeling: Theory and Applications. 1. New York: Plenum Press; 1978. pp. 5–52. [Google Scholar]
- 28.Morin B, Bourhis J-M, Belle V, Woudstra M, Carriere F, Guigliarelli B, Fournel A, Longhi S. Assessing induced folding of an intrinsically disordered protein by site-directed spin-labeling electron paramagnetic resonance spectroscopy. J Phys Chem B. 2006;110:20596–20608. doi: 10.1021/jp063708u. [DOI] [PubMed] [Google Scholar]
- 29.Oppenheim SF, Buettner GR, Rodgers VGJ. Relationship of rotational correlation time from EPR spectroscopy and protein-membrane interaction. J Memb Sci. 1996;118:133–139. [Google Scholar]
- 30.Narayanan R, Ganesh OK, Edison AS, Hagen SJ. Kinetics of folding and binding of an intrinsically disordered protethe inhibitor of yeast aspartic proteinase YPrA. J Am Chem Soc. 2008;130:11477–11485. doi: 10.1021/ja803221c. [DOI] [PubMed] [Google Scholar]
- 31.Langen R, Oh KJ, Cascio D, Hubbell WL. Crystal structures of spin labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry. 2000;39:8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- 32.Kirby TL, Karim CB, Thomas DD. Electron paramagnetic resonance reveals a large-scale conformational change in the cytoplasmic domain of phospholamban upon binding to the sarcoplasmic reticulum Ca-ATPase. Biochemistry. 2004;43:5842–5852. doi: 10.1021/bi035749b. [DOI] [PubMed] [Google Scholar]
- 33.Todd AP, Millhauser GL. ESR spectra reflect local and global mobility in a short spin-labeled peptide throughout the alpha-helix—coil transition. Biochemistry. 1991;30:5515–5523. doi: 10.1021/bi00236a026. [DOI] [PubMed] [Google Scholar]
- 34.Miick SM, Todd AP, Millhauser GL. Position-dependent local motions in spin-labeled analogues of a short alpha-helical peptide determined by electron spin resonance. Biochemistry. 1991;30:9498–9503. doi: 10.1021/bi00103a016. [DOI] [PubMed] [Google Scholar]
- 35.Budil DE, Lee S, Saxena S, Freed JH. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg-Marquardt algorithm. J Magn Reson. 1996;120:155–189. [Google Scholar]
- 36.Kalthoff C. A novel strategy for the purification of recombinantly expressed unstructured protein domains. J Chromatogr B. 2003;786:247–254. doi: 10.1016/s1570-0232(02)00908-x. [DOI] [PubMed] [Google Scholar]
- 37.Marsh D. In: Experimental Methods in Spin-Label Spectral Analysis. Berliner L, Reuben J, editors. New York and London: Plenum Press; 1989. [Google Scholar]