

Abstract
Thioredoxin (Trx) plays a critical role in the regulation of cellular redox homeostasis. Many disease causing pathogens rely on the Trx redox system for survival in conditions of environmental stress. The Trx redox system has been implicated in the resistance of Mycobacterium tuberculosis (Mtb) to phagocytosis. Trx is able to reduce a variety of target substrates and reactive oxygen species (ROS) through the cyclization of its active site dithiol to the oxidized disulphide Cys37–Cys40. Here we report the crystal structure of the Mtb Trx C active site mutant C40S (MtbTrxCC40S) in isolation and in complex with the hydroxycyclohexadienone inhibitor PMX464. We observe PMX464 is covalently bound to the active site residue Cys37 through Michael addition of the cyclohexadienone ring and also forms noncovalent contacts which mimic the binding of natural Trx ligands. In comparison with the ligand free MtbTrxCC40S structure a conformational change occurs in the PMX464 complex involving movement of helix α2 and the active site loop. These changes are almost identical to those observed for helix α2 in human Trx ligand complexes. Whereas the ligand free structure forms a homodimer the inhibitor complex unexpectedly forms a different dimer with one PMX464 molecule bound at the interface. This 2:1 MtbTrxCC40S-PMX464 complex is also observed using mass spectrometry measurements. This structure provides an unexpected scaffold for the design of improved Trx inhibitors targeted at developing treatments for tuberculosis.
Keywords: thioredoxin, inhibitor complex, Mycobacterium tuberculosis, quinol
Introduction
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis, a disease which latently affects over 1.7 billion people worldwide.1 The pathogenesis of Mtb still remains an expanding global health problem that compels new therapeutic and preventative measures, with the emergence of multi-drug-resistant strains creating a worldwide emergency.2 An obligate aerobe, Mtb resides within the well-aerated upper lobes of the lungs, where it is contained, but not necessarily eradicated, by mononuclear phagocytes.3 The pathogen endures phagocytosis by utilizing a variety of peroxidases and peroxiredoxins.4,5 These include, but are not limited to, the catalase-peroxidase enzyme, KatG, and alkyl hydroperoxidase, AhpC. The antituberculous prodrug Isoniazid is catalytically activated by KatG, resulting in a chemical that is a key component in the inhibition of ketoenoyl reductase.6 Resistance to Isoniazid was reported soon after use,7 pointing to mutations in the KatG gene and a consequent inactive catalase.8 Isoniazid resistant strains of the bacterium compensate for the loss of KatG by up-regulating AhpC.9 The thioredoxin (Trx) redox system has been implicated in the resistance of Mtb to phagocytosis.10,11 The role Trx plays in donating reducing equivalents to AhpC, along with its own inherent ability to reduce reactive oxygen species (ROS), suggests Trx is a potentially attractive target for therapeutic intervention.12
We recently reported the crystal structure of oxidized M. tuberculosis thioredoxin C (MtbTrxC).13 The overall structure of Trx remains conserved across all phyla and the sequence identity varies in the range 27–69%.14 The active site of Trx is located in a loop region between the strand β2 and helix α2. Trx uses a cyclic reduction-oxidation reaction, governed by an active site reduced dithiol (Cys37–Cys40) interaction, to reduce target substrates.15,16 Hydroxycyclohexadienone analogues, of which PMX464 is the parent compound, have encouraging results in inhibitory trials against Mtb H37Rv strains in vitro17 and studies showed that MtbTrxC catalyzed reduction of insulin is inhibited by PMX464 in a potent and dose-dependent manner consistent with irreversible inhibition of Trx (IC50 < 6 μM).18 To investigate how the inhibitor binds and thus provide a template for design and development we have determined the structure of PMX464 in complex with a catalytically inactive MtbTrxC.
Results
MtbTrxCC40S ligand free and PMX464 bound structures
Initial attempts at crystallization of the wild type MtbTrxC with PMX464 were not successful and only crystals of the oxidized unliganded form resulted.13 The normal recognition of substrates for Trx operates through a hydrophobic groove which extends either side of the reactive thiol forming pockets A and B. Trx-ligand complex crystal and NMR structures have been successfully determined with peptides and proteins binding into this groove utilizing the two pockets. In each case, an active site mutant was used to trap the complex and prevent formation of the oxidized Trx.19–21 We therefore adopted a similar approach and generated an active site mutant C40S of MtbTrxC (MtbTrxCC40S).
Recombinant MtbTrxCC40S protein samples were initially crystallized in the absence of PMX464, from 0.2M potassium thiocyanate, 2.0M ammonium sulfate and the structure determined by molecular replacement with a refined Rfactor of 18.6 to 1.6 Å resolution (Supporting Information Table S1). The asymmetric unit contains two molecules, which form a noncovalent dimer due to hydrophobic contacts in the region around the active site involving pockets A and B [Fig. 1(A)]. In this interaction Trp36, from one molecule sits in the hydrophobic Pocket B of the second molecule formed by Val65, Ala72, Val77, and Ile80 [Supporting Information Fig. S1(A)]. The MtbTrxCC40S dimer arrangement is surprisingly identical to that observed in crystals of human Trx [Supporting Information Fig. S1(B)].
Figure 1.
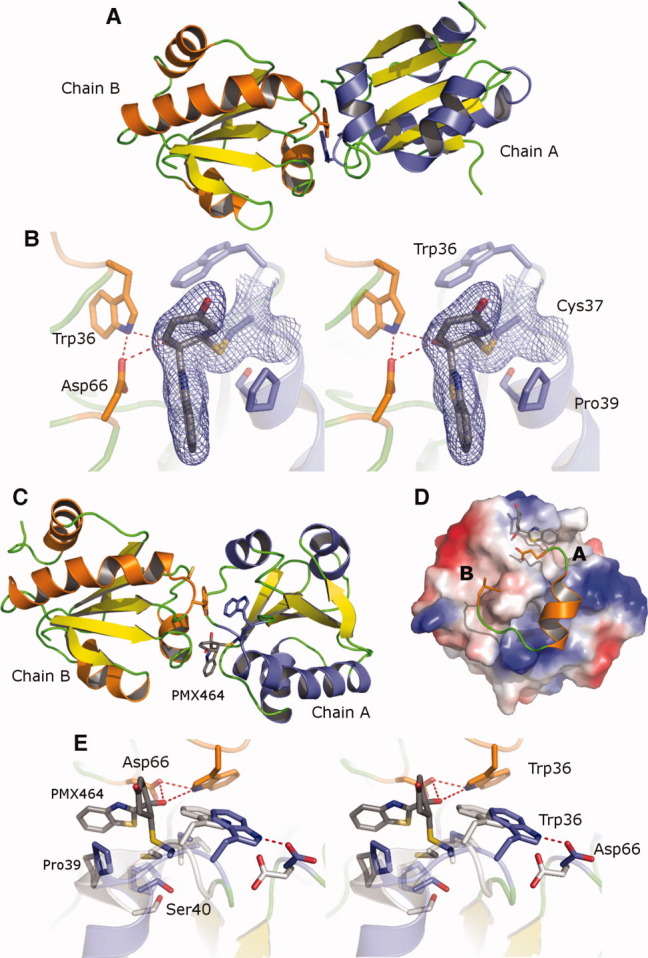
MtbTrxCC40S structures. (A) Cartoon diagram of the uncomplexed MtbTrxCC40S homodimer shown with chain A (helices blue) and chain B (helices orange). (B) Electron density map (blue) calculated using refined phases and 2Fo-Fc coefficients and contoured at 1σ, surrounding the active site Cys37 and bound PMX464 is shown as stick. (C) Cartoon diagram of the MtbTrxCC40S–PMX464 complex, chain A (blue) and chain B (orange). (D) Charged surface representation of MtbTrxCC40S with bound PMX464 shown as stick occupying pocket A and residues Asp66 to Ile80 (chain B) shown in cartoon. (E) Superposition of the active site residues of the MtbTrxCC40S–PMX464 complex (blue) with the unliganded structure (white). An interactive view is available in the electronic version of the article. PRO533 Figure 1
The MtbTrxCC40S–PMX464 inhibitor complex crystallized from a mixture of protein to inhibitor ratio of 1:2 with a final protein concentration of 5 mg/mL. Crystals were characterized as spacegroup C2 and the structure determined by molecular replacement using the oxidized MtbTrxC crystal structure and the refined Rfactor was 20.4 to 2.4 Å resolution (Supporting Information Table S1).13 In the asymmetric unit four MtbTrxCC40S and two PMX464 molecules are observed and PMX464 is covalently bound to the nucleophilic thiol Cys37 [Fig. 1(B)]. The Michael addition of Cys37 onto the cyclohexadienone ring leads to the formation of a half chair conformation, with the cysteine residue attacking from the benzothiazole side of the ring resulting in an anticonformer along the α-β bond and it is this conformation that is observed in the electron density [Supporting Information Fig. S1(C)].22 The carbonyl group of the cyclohexenone ring points away from the protein surface and forms no interactions, whereas the hydroxyl group points into pocket A of the hydrophobic groove. The heteroaromatic benzothiazole ring lies perpendicular to the cyclohexenone ring and forms a face-edge interaction between the benzothiazole ring and the Pro39 sidechain. The sulfur atom points into the peptide binding groove, packing against the Ile80 main chain carbonyl oxygen at a distance of 3.0 Å. This is a unique orientation and exchange of the nitrogen and sulfur atoms by 180° rotation of the benzothiazole ring results in a poorer fit of the density with additional positive and negative density appearing in the difference map. This orientation may be dictated by the loss of planarity in the cyclohexenone ring resulting in a steric conflict with the larger sulfur atom, rather than a favorable interaction with the Trx surface.
An unexpected observation from the crystal structure was that one PMX464 is bound at the interface of two molecules of MtbTrxCC40S and two identical copies of this ternary complex are defined in the asymmetric unit [Fig. 1(C)]. PMX464 covalently binds to the active site cysteine but does not occupy the breadth of the peptide binding groove and instead packs against Pro39 and in this structure one face of the aromatic benzathiozole ring structure is exposed and parallel to the groove. A second MtbTrxCC40S molecule interacts here using two loops, with residue Asp66 prominently contacting one side of the groove and inserting the sidechain carboxyl underneath PMX464, forming a hydrogen bond to the main-chain nitrogen of Ile80 [Fig. 1(D)]. The aliphatic Cβ and main-chain atoms of Asp66, in addition to the sidechain of Thr67, form further contacts burying the PMX464 benzothiazole ring in the ternary complex (Supporting Information Movie S1). Further extensive contacts are made with residues from continuation of the sequence from Asp66 around the α3 helix to residue Ile80. Perhaps more significantly, the Val78 sidechain from the α3β4 loop is inserted into Pocket B to form contacts with Ile80 and Val65 [Fig. 1(D), Supporting Information Fig. S1(D)]. The total surface area of the interaction is 1200 Å2, and yet the second subunit active site Cys37 does not make any contacts nor is any density for bound PMX464 evident. This contrasts with the MtbTrxCC40S homodimer where it is Trp36A, rather than Val78A that packs into pocket B. Figure 1(A) displays the homodimer left subunit (orange helices) with the equivalent orientation to the heterodimer left subunit in Figure 1(C) illustrating the large difference in angle between the subunits in each dimer (these are superposed and illustrated in Supporting Information Movie S2).
Mass spectrometry was used to further examine the stoichiometry of PMX464 binding to MtbTrxCC40S. A mass spectrum of the free protein was first obtained, with the significant peak at m/z 12,671, corresponding to MtbTrxCC40S. The mass spectrum from the MtbTrxCC40S–PMX464 sample showed a peak for the free protein together with an m/z 12,915, relating to the weight of a 1:1 MtbTrxCC40S–PMX464 complex. The largest peak had a molecular mass of m/z 25,588 corresponding to a 2:1 complex of MtbTrxCC40S and PMX464, which is consistent with the dimer complex observed in the crystal structure and no 2:2 species is evident (Supporting Information Fig. S2).
Conformational changes in MtbTrxCC40S
Superposition of the oxidized MtbTrxC structure with the reduced MtbTrxCC40S ligand free structure reveals they are essentially identical (r.m.s.d. 0.60 Å) with only a small movement of the position of the Cys37 SG atom. Superposition of the MtbTrxCC40S–PMX464 complex with the unliganded MtbTrxCC40S structure reveals extensive conformational changes, not just localized to the area of Cys37, but encompassing the backbone and side-chain atoms for the β2α2, β3α3, and α3β4 loops at the top of the fold, which form the borders of the peptide binding groove. Residues Thr35-Met42 show the largest changes with a two component switch in conformation observed such that (i) Trp36 and the β2α2 loop translates by 3.0 Å in the direction of Asp66; (ii) residues Gly38-Pro39 at the top of helix α2 shift by 2.2 Å, breaking two α-helical hydrogen bonds [Fig. 1(E), Supporting Information Movie S3]. This conformational alteration to the α2 helix is not recovered fully until Val43. The NMR solution structures of NF-κB (PDB:1MDI)20 and Ref-1 (PDB:1CQH)21 bound to human Trx and the crystal structure of 3′-phosphoadenosine-5′-phosphosulfate reductase in complex with E. coli Trx 1 (PDB:2O8V)19 have been reported. In each case a comparison with the unliganded structure reveals a conformational change in helix α2 similar to those observed in the MtbTrxCC40S upon binding PMX464. Furthermore, these bound ligands occupy both pockets A and B, with residues equivalent to Ile80 (Thr74, human; and Ile75, E. coli) each making conserved hydrogen bonds with the ligand, through a backbone carbonyl to the backbone amine which is also observed for PMX464 binding.
Discussion
Thioredoxins play a critical role the regulation of cellular redox homeostasis for many pathological micro-organisms. Mtb is an example of a micro-organism with a high dependence on redox homeostasis as it resides largely in mononuclear phagocytes, where it relies on redox proteins to mitigate the damaging effects of toxic ROS to survive. Unlike humans, there is a total absence of the glutathione system in the Mtb genome, and this implies a greater degree of dependence on the Trx system representing a lucrative target for selective therapy.23
We have previously reported the antitubercular properties of PMX464 and related substituted hydroxycyclohexadienones and shown in vitro that PMX464 inhibits MtbTrxCC40S.17 Here the crystal structure of MtbTrxCC40S in complex with PMX464 reveals this is mediated by both a covalent bond between the active site Cys37 thiol and the cyclohexadienone ring together with additional noncovalent contacts with the Trp36 sidechain and between the benzothiazole ring and Ile80 mainchain and Pro39 sidechain mimicking contacts made in other native Trx ligands. Comparison with the unliganded MtbTrxCC40S structure reveals conformational change in helix α2 and the Cys37 loop. Interestingly these changes are similar to those observed in other Trx structures that have a thiol containing ligand covalently attached to the active site and bound in the groove. Although PMX464 binds directly to Cys37, the peptide binding groove remains largely unoccupied allowing the unexpected formation of an active site occluded heterodimer with a second MtbTrxCC40S molecule contributing further interactions with a single PMX464 to form an inactivated ternary complex. The inhibitor complex structure and unexpected heterodimer described here represents a scaffold which can be utilized for future design of more potent and selective derivatives of PMX464. The area of the A pocket and the heterodimer interface has significant variability between human and MtbTrx and thus modification or substitution of the benzothiazole ring is one strategy that can be employed to achieve selectivity.
Materials and Methods
Protein expression and purification
The open reading frame for M. tuberculosis Trx C (MtbTrxC) wild-type was amplified from genomic DNA, using standard PCR protocols and cloned into the expression vector pGAT (EMBL). The mutation of Cys40Ser was carried out using the Quickchange site-directed mutagenesis kit (Stratagene). The resulting plasmid was transformed into E. coli BL-21 (DE3) pLysS cells for expression and induced at 295K overnight. The cells were harvested by centrifugation and resuspended in Tris-buffered saline, pH 7.4 and 5 mM imidazole before lysis by sonication (Branson). His-tagged MtbTrxCC40S was purified from the supernatant by Ni2+ affinity chromatography using a precharged nickel-chelate affinity column (GE Life Sciences). The His-tag was cleaved overnight at 4°C using thrombin (HTI) at an enzyme:substrate ratio of 1:100 (w:w), resulting in an extra two N-terminal residues Gly and Ser prior to the start of the native protein sequence. The protein was further purified using gelfiltration and concentrated to 5 mg/mL−1.
Crystallization, data collection, structure determination
Crystallization conditions were screened by sitting drop vapor diffusion at both 4 and 19°C, using Qiagen 96-well block screens. Optimal crystals of MtbTrxCC40S grew at 4°C in 0.2M potassium thiocyanate and 2.0M ammonium sulfate. Crystals of MtbTrxCC40S–PMX464 grew at 19°C in 0.1M Na MES buffer (pH 6.5), 1.6M ammonium sulfate and 4% (w/v) PEG 400. In both cases, a cryoprotectant consisting of 20% glycerol in mother liquor was used. X-ray diffraction data were collected at the E.S.R.F. (Grenoble) using beamlines ID14-3 and ID29. Both sets of data were indexed and integrated in MOSFLM and scaled using SCALA.24 All crystal structures were determined using the molecular replacement program PHASER,25 with the MtbTrxC wild-type structure (PDB:2I1U) as the search model. In each case, the model building and structural refinement was carried out with Coot,26 PHENIX,27 and REFMAC 5.0,28 using restrained refinement and isotropic B-factors.
Mass spectrometry
The MtbTrxCC40S and MtbTrxCC40S–PMX464 samples were desalted using C18 ZipTip® pipette tips (Millipore Co.) and eluted into 50% acetonitrile, 0.2% formic acid. An electrospray Q-TOF2 tandem mass spectrometer (Waters Co.) allowed the accurate mass of the whole protein and complexes to be determined. The resulting spectra were deconvoluted using the MaxEnt1 maximum entropy software.
Acknowledgments
We thank Manish Shah (Hauptman-Woodward Medical Research Institute, Buffalo) and Andrew Westwell (University of Cardiff) for providing the plasmid constructs and Pharminox Ltd, Nottingham, UK, for the PMX464 compound. We acknowledge the European Synchrotron Facility (ESRF) for providing access to X-ray sources. We also thank Susan Liddell (University of Nottingham) for the mass spectroscopy work she carried out for the group.
Glossary
Abbreviations:
- Trx
thioredoxin
- MtbTrxCC40S
Mycobacterium tuberculosis thioredoxin C active site C40S mutant
- ROS
reactive oxygen species
- AhpC
alkyl hydroperoxide reductase C, KatG, hemoprotein processing catalase-peroxidase
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells.
References
- 1.Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA. 1999;282:677–686. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 2.Glickman MS, Jacobs WR., Jr Microbial pathogenesis of Mycobacterium tuberculosis: dawn of a discipline. Cell. 2001;104:477–485. doi: 10.1016/s0092-8674(01)00236-7. [DOI] [PubMed] [Google Scholar]
- 3.Gordon AH, Hart PD. Stimulation or inhibition of the respiratory burst in cultured macrophages in a mycobacterium model: initial stimulation is followed by inhibition after phagocytosis. Infect Immun. 1994;62:4650–4651. doi: 10.1128/iai.62.10.4650-4651.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, Xie QW, Nathan C. Alkyl hydroperoxide reductase subunit C (AhpC) protects bacterial and human cells against reactive nitrogen intermediates. Mol Cell. 1998;1:795–805. doi: 10.1016/s1097-2765(00)80079-9. [DOI] [PubMed] [Google Scholar]
- 5.Shinnick TM, King CH, Quinn FD. Molecular biology, virulence, and pathogenicity of mycobacteria. Am J Med Sci. 1995;309:92–98. doi: 10.1097/00000441-199502000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Rouse DA, Morris SL. Molecular mechanisms of isoniazid resistance in Mycobacterium tuberculosis and Mycobacterium bovis. Infect Immun. 1995;63:1427–1433. doi: 10.1128/iai.63.4.1427-1433.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Middlebrook G. Sterilization of tubercle bacilli by isonicotinic acid hydrazide and the incidence of variants resistant to the drug in vitro. Am Rev Tuberc. 1952;65:765–767. [PubMed] [Google Scholar]
- 8.Wengenack NL, Rusnak F. Evidence for isoniazid-dependent free radical generation catalyzed by Mycobacterium tuberculosis KatG and the isoniazid- resistant mutant KatG(S315T) Biochemistry. 2001;40:8990–8996. doi: 10.1021/bi002614m. [DOI] [PubMed] [Google Scholar]
- 9.Guimaraes BG, Souchon H, Honore N, Saint-Joanis B, Brosch R, Shepard W, Cole ST, Alzari PM. Structure and mechanism of the alkyl hydroperoxidase AhpC, a key element of the Mycobacterium tuberculosis defense system against oxidative stress. J Biol Chem. 2005;280:25735–25742. doi: 10.1074/jbc.M503076200. [DOI] [PubMed] [Google Scholar]
- 10.Akif M, Khare G, Tyagi AK, Mande SC, Sardesai AA. Functional studies of multiple thioredoxins from Mycobacterium tuberculosis. J Bacteriol. 2008;190:7087–7095. doi: 10.1128/JB.00159-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Hillas PJ, Ortiz de Montellano PR. Reduction of peroxides and dinitrobenzenes by Mycobacterium tuberculosis thioredoxin and thioredoxin reductase. Arch Biochem Biophys. 1999;363:19–26. doi: 10.1006/abbi.1998.1056. [DOI] [PubMed] [Google Scholar]
- 12.Jaeger T, Budde H, Flohe L, Menge U, Singh M, Trujillo M, Radi R. Multiple thioredoxin-mediated routes to detoxify hydroperoxides in Mycobacterium tuberculosis. Arch Biochem Biophys. 2004;423:182–191. doi: 10.1016/j.abb.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 13.Hall G, Shah M, McEwan PA, Laughton C, Stevens M, Westwell A, Emsley J. Structure of Mycobacterium tuberculosis thioredoxin C. Acta Crystallogr D Biol Crystallogr. 2006;62:1453–1457. doi: 10.1107/S0907444906038212. [DOI] [PubMed] [Google Scholar]
- 14.Eklund H, Gleason FK, Holmgren A. Structural and functional relations among thioredoxins of different species. Proteins. 1991;11:13–28. doi: 10.1002/prot.340110103. [DOI] [PubMed] [Google Scholar]
- 15.Holmgren A. Thioredoxin. Annu Rev Biochem. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 16.Holmgren A. Thioredoxin and glutaredoxin systems. J Biol Chem. 1989;264:13963–13966. [PubMed] [Google Scholar]
- 17.Shah M, Wells G, Bradshaw TD, Laughton CA, Stevens MFG, Westwell AD. Antitubercular properties of substituted hydroxycyclohexadienones. Lett Drug Des Discov. 2006;3:419–423. [Google Scholar]
- 18.Bradshaw TD, Matthews CS, Cookson J, Chew EH, Shah M, Bailey K, Monks A, Harris E, Westwell AD, Wells G, Laughton CA, Stevens MF. Elucidation of thioredoxin as a molecular target for antitumor quinols. Cancer Res. 2005;65:3911–3919. doi: 10.1158/0008-5472.CAN-04-4141. [DOI] [PubMed] [Google Scholar]
- 19.Chartron J, Shiau C, Stout CD, Carroll KS. 3′-Phosphoadenosine-5′-phosphosulfate reductase in complex with thioredoxa structural snapshot in the catalytic cycle. Biochemistry. 2007;46:3942–3951. doi: 10.1021/bi700130e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qin J, Clore GM, Kennedy WM, Huth JR, Gronenborn AM. Solution structure of human thioredoxin in a mixed disulfide intermediate complex with its target peptide from the transcription factor NF kappa B. Structure. 1995;3:289–297. doi: 10.1016/s0969-2126(01)00159-9. [DOI] [PubMed] [Google Scholar]
- 21.Qin J, Clore GM, Kennedy WP, Kuszewski J, Gronenborn AM. The solution structure of human thioredoxin complexed with its target from Ref-1 reveals peptide chain reversal. Structure. 1996;4:613–620. doi: 10.1016/s0969-2126(96)00065-2. [DOI] [PubMed] [Google Scholar]
- 22.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 23.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 24.Leslie AGW. 1998. MOSFLM v.6.0., MRC Laboratory of Molecular Chemistry, Cambridge.
- 25.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 26.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 27.Afonine PV, Grosse-Kunstleve RW, Adams PD. 2005. Phenix. CCP4 Newsl. 42, contribution 8.
- 28.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]