

Abstract
Redox-active cysteine, a highly reactive sulfhydryl, is one of the major targets of ROS. Formation of disulfide bonds and other oxidative derivatives of cysteine including sulfenic, sulfinic, and sulfonic acids, regulates the biological function of various proteins. We identified novel low-abundant cysteine modifications in cellular GAPDH purified on 2-dimensional gel electrophoresis (2D-PAGE) by employing selectively excluded mass screening analysis for nano ultraperformance liquid chromatography-electrospray-quadrupole-time of flight tandem mass spectrometry, in conjunction with MODi and MODmap algorithm. We observed unexpected mass shifts (Δm = −16, −34, +64, +87, and +103 Da) at redox-active cysteine residue in cellular GAPDH purified on 2D-PAGE, in oxidized NDP kinase A, peroxiredoxin 6, and in various mitochondrial proteins. Mass differences of −16, −34, and +64 Da are presumed to reflect the conversion of cysteine to serine, dehydroalanine (DHA), and Cys-SO2-SH respectively. To determine the plausible pathways to the formation of these products, we prepared model compounds and examined the hydrolysis and hydration of thiosulfonate (Cys-S-SO2-Cys) either to DHA (Δm = −34 Da) or serine along with Cys-SO2-SH (Δm = +64 Da). We also detected acrylamide adducts of sulfenic and sulfinic acids (+87 and +103 Da). These findings suggest that oxidations take place at redox-active cysteine residues in cellular proteins, with the formation of thiosulfonate, Cys-SO2-SH, and DHA, and conversion of cysteine to serine, in addition to sulfenic, sulfinic and sulfonic acids of reactive cysteine.
Reactive oxygen species (ROS)1, generated from various external stimuli, cause nonenzymatic oxidative cysteine thiol modifications in proteins, and mediate cell proliferation, apoptosis, cell migration, and inflammation, among others (1, 2). Cysteine residues having a low pKa, called “redox-active Cys,” which are readily oxidized by ROS, thus play key roles in the regulation of protein function. The reversibility and the degree of oxidation state in redox-active Cys residues, is governed by the oxidative environment in the cell. Many reducing enzymes including families of peroxiredoxin (PRX), sulfiredoxin (SRX) and thioredoxin (TRX), also facilitate reversible cysteine oxidations by promoting reducing conditions. Sulfenic (Cys-SOH), sulfinic (Cys-SO2H), sulfonic acids (Cys-SO3H), and disulfide bond (Cys-SS-Cys), have long been recognized as products of Cys oxidation, but some novel Cys modifications hitherto unknown have been recently identified in peptide sequencing studies that employed sensitive tandem mass spectrometry (3, 4).
In most proteins, the first oxidation product of Cys (Cys-SH) by H2O2 is Cys sulfenic acid (Cys-SOH). Although sulfenic acids have been recently identified in cellular proteins under relatively stable environments (5–7), they are unstable and are readily oxidized to sulfinic (R-SO2H) and sulfonic acids, or to disulfide by condensation with free sulfhydryl. Sulfinic acid was considered to be nonreducible until the discovery of an ATP-dependent sulfinic acid reductase, SRX (8). SRX was shown to reduce sulfinic acid to sulfhydryl in PRX family by inducing local unfolding (9,10). Sulfinic acid is a relatively stable intermediate, but can be oxidized in vivo to sulfonic acid, the most highly oxidized and irreversible thiol (11–13). Reversible disulfide bond (Cys-SS-Cys) formed by condensation of sulfenic acid with free sulfhydryl, plays a key role in signaling pathways (14). Specific functional roles of some Cys modifications in numerous proteins have been characterized (10, 11). Identification of post-translational Cys modifications in proteins thus paves the way to understanding how protein function is regulated.
Mass spectrometry (MS), a highly efficient and sensitive technique (15), leads to the identification of new post-translational modifications (PTMs) caused by ROS in protein populations separated by 2D-PAGE including phosphorylation (16) and Cys oxidation (17). A newly developed algorithm, DBond, helped identification of disulfide bond formation using tandem MS (18). In the present study, we employed selectively excluded mass screening analysis (SEMSA) with nanoUPLC-q-TOF tandem MS for detecting low abundant protein modifications (17), and MODi and MODmap algorithm for searching for unknown modifications (19, 20). We characterized the nature as well as the abundances of these hitherto unknown Cys modifications in cellular GAPDH purified on 2D-PAGE. We found unexpected mass shifts at active site Cys residue (Δm = −16, −34, and +64 Da) in addition to those of previously known oxidation products including sulfinic and sulfonic acids, and disulfide bonds. Similar changes were also found in other ROS-sensitive proteins including nucleoside diphosphate kinase A (NDPK A), PRX6, and mitochondrial proteins. Mass differences of −16, −34, and +64 Da are presumed to reflect the conversion of Cys to Ser, DHA, and Cys-SO2-SH respectively. The plausible pathways leading to their formation from Cys were deduced from the distribution of the oxidative products and were confirmed in model systems by analyzing three-dimensional protein structures. Also sulfenic and sulfinic acids were detected as acylamide adducts (Δm = +87 and +103 Da) in samples on SDS-PAGE. These findings suggest that diverse Cys modifications of redox-active Cys can be generated by ROS. We are conducting further studies to characterize the biological regulation and functions of these modifications.
MATERIALS AND METHODS
Protein Samples
Human embryonic kidney epithelial cells (HEK293T) were grown and maintained in high glucose Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum at 37°C and 5% CO2. All experiments were performed on 50% confluent cell cultures. The cells (1.5 × 106) were seeded in 10-cm plates a day before transfection and transiently transfected with 6 μg of Flag-GAPDH expression plasmid by calcium phosphate method. The medium was refreshed 6 h following transfection, cultured for additional 19 h, and incubated with 1 mm H2O2 in Hanks balanced salts for 1 h at 37 °C. For immunoprecipitation studies, the cells were lysed in a buffer containing 50 mm Tris base, 150 mm NaCl, 2 mm EDTA, 0.5% Nonidet P-40, 1 mm phenylmethylsulfonyl fluoride, 0.5 mm dithiothreitol, 5 μg/ml aprotinin, 1 μg/ml leupeptin, 5 mm NaF, pH 8.0, for 30 min on ice. The lysates were centrifuged at 20,000 × g for 1 h and the supernatants incubated for 3 h at 4 °C with monoclonal anti-Flag M2-affinity agarose beads. The beads were washed three times each with 1 ml of lysis buffer (17). The precipitated immune complexes were left to stand for 30 min at room temperature in a sample buffer containing protease inhibitors (9.5 m urea, 2% Triton X-100, 5% β-mercaptomethanol, 1 mm phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 10 μg/ml pepstatin A, 10 μg/ml leupeptin, 1 mm EDTA, 10 mm Na3VO4, 10 mm NaF), applied by cup loading at the acidic end and electrofocused in 7 or 18 cm Immobiline DryStrips (pH 3–10) with the Amersham Biosciences IPGphor. The strips were then agitated for 15 min in an equilibration buffer (1.5 m Tris-Cl, pH 8.8, 6 m urea, 30% glycerol, 2% SDS, and 10 mg/ml dithiothreitol) and subjected to two-dimensional SDS-PAGE.
Recombinant nucleoside diphosphate kinase A (NDPK A) and its C4S mutant were isolated from the cytosolic fraction of E. coli strains BL21 (DE3) transformed with pET-3c expression plasmids containing nm23-H1 coding region, purified by ATP column chromatography (21, 22) as follows. The cytosolic fractions were each applied to 2 ∼ 4 ml of ATP-Sepharose column equilibrated with Buffer A (20 mm Tris acetate, 20 mm NaCl, 0.1 mm EDTA, 3 mm MgCl2, pH 7.4) at a flow rate of 3 ml/min. The columns were then washed with buffer A and then with the same buffer A containing 0.25 m NaCl, to remove nonspecifically bound proteins and eluted with Buffer A containing 1 mm ATP. The recombinant NDPK A was then treated with 1 or 5 mm H2O2 for 30 min followed by phosphate-buffered saline or the alkylating agent, 20 mm N-ethylmaleimide (NEM) for 30 min at 37 °C. NDPK A was finally subjected to 12% SDS-PAGE under nonreducing conditions, to confirm oxidation states and identified by staining with Coomassie blue.
Mitochondrial proteins from mouse livers were purified using a Subcellular Proteome Extract kit (Calbiochem) as previously reported (23). They were solubilized in a lysis buffer and separated on two-dimensional-PAGE as described previously (16, 17).
Analysis of PTMs using nanoUPLC-ESI-q-TOF Tandem Mass Spectrometry
The gel bands or spots were destained and digested with trypsin, or proteins in solution were digested with trypsin and the resulting peptides extracted as previously described (17). The extracts were evaporated to dryness in SpeedVac and redissolved in 10% acetonitrile containing 0.1% formic acid. The dissolved peptides were desalted on line prior to separation using trap column (i.d. 180 μm × 20 mm, Symmetry® C18) cartridge, and separated on a C18 reversed-phase 75 μm i.d. × 200 mm analytical column (1.7 μm particle size, BEH130 C18, Waters Co. UK) with integrated electrospray ionization PicoTipTM (±10 μm i.d., New Objective, USA) using nanoAcquityTM/ESI/MS (SYNAPTTM HDMSTM, Waters Co., UK) as previously described (24).
Database Search
The raw data files obtained from the mass spectrometry were converted to .pkl files using ProteinLynx Global Server data processing software (PLGSTM, version 2.3, Waters Co. UK). Tandem MS (MS/MS) spectra were matched against amino acid sequences in SwissProt human database (version 57.8., 20401 entries) using Mascot search (version 2.2.06). The search parameters were as follows: 0.3 Da tolerance for peptide and fragment ions; digestion with trypsin with up to one missed cleavage allowed. Acetylation (N-terminal), formylation (Lys), deamidation (Asn and Gln), oxidation (Met), phosphorylation (Ser, Thr, and Tyr), pyro-Glu modification (N-terminal Glu and N-terminal Gln), and propionamidation or cabamidomethylation (Cys) were the searched modifications. After establishing the number and types of potential PTMs by the Mascot search, additional searchs were performed by MODi, DBond and MODmap (http://prix.uos.ac.kr/) (18–20) against FASTA files of each protein, downloaded from NCBI (http://www.ncbi.nlm.nih.gov/protein/). Only significant hits, indicated by MASCOT probability analysis (probability based Mowse score p < 0.05) were considered. In addition, a minimum total score of 50, comprising at least a peptide match of ion score more than 20, was arbitrarily set as threshold for acceptance. All reported assignments were verified by automatic and manual interpretation of spectra. Each modification was assigned with an observed mass shift.
Model Building and Evaluation
Energy minimized models of human NDPK A, with Cys109 modified to Cys-SO2-SH were built based on its known structure (PDB ID: 1JXV), using MMFF94 parameters of the energy minimization protocol of sybylx1.1 (CERTARA, MO). The stereochemistry of the minimized structure was evaluated and assessed by PROCHECK (25).
RESULTS
We chose GAPDH (P04406) as a model for investigating ROS induced redox-active Cys modifications, because this enzyme is known as a major redox-sensitive protein having three Cys residues, two (152CxxxC156) in active site, and one (247Cys) on the surface (17, 26). HEK293 cells transiently transfected with Flag-tagged GAPDH, were exposed to 5 mm H2O2 for 1 h at 37 °C. Cellular GAPDH was purified by immunoprecipitation using flag-antibody, and the immune-complex was separated on 2D-PAGE (pH 3–10, Fig. 1C). We observed PI shifts to acidic region in oxidized GAPDH. We looked for less abundant modifications in Cys residues in each spot using peptide sequencing with nanoUPLC-ESI-q-TOF tandem MS, employing SEMSA for sensitive detection of low abundant PTMs (17) and searching for unknown PTMs using MODi and MODmap algorithm (19, 20). The observed Cys modifications are shown in Fig. 1A. These include modifications at Cys152 in the peptide 146IISNASCTTNCLAPLAK162 (m = 1718.8695 Da) containing active site 152CTTNC156, and at Cys247 in the peptide 235VPTANVSVVDLTCR248 (m = 1472.7656 Da). Most free sulfhydryls were easily labeled by generating acrylamide adduct (propionamide, Caa, Δm = +71.0359 Da) in SDS-PAGE. But some fractions of Cys152 were oxidatively modified with intradisulfide bond formation between Cys152 and Cys156 (Δm = −2 Da), sulfonic acid modification (152Cys-SO3H, Δm = +48.0380 Da), and unexpected mass shifts (Δm = −16 or +64 Da). Intriguingly, the only modification at Cys156 detected was the acrylamide adduct of free sulfhydryl, no oxidative modification.
Fig. 1.

Novel modifications of active site cysteine detected in GAPDH. A, Summary of cysteine modifications observed in cellular GAPDH purified from immuno-precipitation of HEK293 cells, by tandem MS. Peptide m/z and calculated mass, and detected nominal mass changes at 152C/156C and 247C residues were presented. B, MS/MS spectra of active site “CXXXC” containing unknown mass shift +64 and −16 Da. Caa indicates acrylamide adduct of Cys residue and star indicates fragment ion that lost H2O or NH3. Presumed chemical structure of each mass shift at Cys residue was presented on the right side. C, The immuno-precipitated GAPDH was separated on 2D-PAGE and detected with Coomassie staining. D, Quantitative analyses of modifications in peptide 146IISNASCTTNCLAPLAK162 based on precursor ion intensities in supplemental Fig. 1B using Glu-fibrinopeptide as an internal standard.
Other oxidative modifications including sulfinic, sulfonic acid, and dehydroalanine at Cys247 were also identified. These novel modifications at Cys152 and Cys247 were confirmed by sequencing with tandem MS, (Fig. 1B and supplemental Fig. 1A). Mass shifts of −15.94 Da, and −34 Da, at the Cys residue are presumed to indicate conversion of Cys to Ser and DHA respectively. A hitherto unknown mass shift, Δm = +63.97 Da, was observed in the MS/MS spectrum in Fig. 1B. We examined the MS/MS spectrum containing an unknown mass shift (Δm = +63.97 Da) of the precursor ion, and found the simultaneous existence of DHA fragment ions at Cys152 (Fig. 1B), neutral loss. This suggests that the mass shift +64 Da at Cys152 is an oxidative modification, because DHA fragment ions are readily generated in gas phase only from oxidized Cys modifications, and not from free sulfhydryl, as previously reported (27). We quantitatively analyzed these modifications based on precursor ion intensities (supplemental Fig. 1B) in spots 1 and 2 on 2D-PAGE (Fig. 1C). The relative intensities of each modification are presented in Fig. 1D. Discernible changes in Cys modifications were observed in each spot on 2D-PAGE. Acidic spot 2 contained more sulfonic acid and unknown Δm = +64 Da changes at Cys residue, and less free sulfhydryl as acrylamide adduct (AA) and Cys conversion to Ser (Δm = −16 Da) in a peptide including active site than control spot 1. This suggests that unknown mass change (Δm = +64 Da) moves GAPDH mobility toward acidic direction on 2D-PAGE.
Molecular ion mass increase of +63.97 Da at Cys residue can be deduced as SO2 and O4 within 5 ppm from same nominal mass candidates of SO2, O4, S2, C4O, CH4SO etc. because other candidates show more than 10 ppm deviation. Also O4 was ruled out because of the high reactivity of RSO2OOH (sulfonoperoxoic acid) (28). That leaves conversion of Cys-SH to Cys-SO2SH as the only plausible modification. Two possible routes can be suggested for this modification. In one possible route, disulfide bond (Cys-S-S-Cys) generated by condensation of sulfenic acid and sulfhydryl of Cys, is cleaved to DHA (R = CH, Δm = −34 Da) and persulfide (Cys-S-SH, Δm = +32 Da), which in turn is produced by cleavage of carbon and sulfur bond (C-S) of the disulfide bond by a basic residue in close proximity (29, 30), followed by further oxidation to Cys-SO2-SH as shown in Eq. 1. In the second possible route, thiosulfinate (Cys-S-SO-Cys) is generated from condensation of two sulfenic acids, then oxidized to thiosulfonate (Cys-S-SO2-Cys), with cleavage of the C-S bond to DHA (Δm = −34 Da) with the formation of Cys-SO2-SH (Δm = +64 Da) as shown in Eq. 2, because Cys-SO2-S− is a better leaving group than Cys-S-SO2−. Although DHA generated in Eq. 1 or Eq. 2 can be further modified to Ser under various cellular environments (Eq. 3), modification of Cys to Ser more likely occurs directly from thiosulfonate (Cys-S-SO2-Cys) as shown in Eq. 4.
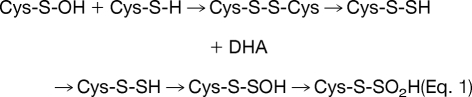 |
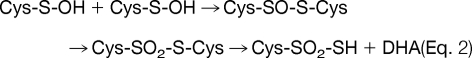 |
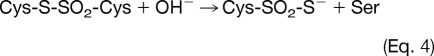 |
To further identify the oxidation pathway that produces Cys-SO2SH, DHA or Ser, from Cys, we employed recombinant NDPK A (P15531), as a model system. NDPK A is an oxidation-sensitive enzyme that plays key roles both as a tumor metastasis suppressor and as a house-keeping enzyme (22, 24). Redox active Cys109 of NDPK A, is easily oxidized, forming disulfide bonds with NDPK A and glutathione, and to sulfonic acid (24). This oxido-reduction regulates the biological activities of NDPK A as an enzyme and as a tumor metastasis suppressor. We analyzed various oxidation states of recombinant NDPK A with MS (Fig. 2A). Purified recombinant NDPK A was incubated with or without 20 mm NEM in phosphate-buffered saline for 30 min to block free Cys sulfhydryl residues, and separated under nonreducing SDS-PAGE. NEM treated NDPK A exists in one reduced form, whereas oxidized NDPK A exists in two populations including a band containing intradisulfide bond (Fig. 2B). Each population was subjected to tandem mass spectrometric analysis and then to a DBond algorithm (18) search for disulfide linked peptides. NEM-treated NDPK A showed mostly NEM-labeling on Cys residues. However, mildly oxidized NDPK A without NEM treatment, showed various modifications including intradisulfide bond (m = 1582.72 Da) between 106GDFCIQVGR114 and 2ANCER6 as identified by tandem MS (Fig. 2C). It also contained the oxidative products of this disulfide bond as precursor ions of thiosulfinate (Δm = +15.99 Da) and thiosulfonate (Δm = +31.98 Da) as shown in Fig. 2D and in the MS/MS spectrum of thiosulfonate (supplemental Fig. 2). This finding of thiosulfonate suggests that the +64 Da at Cys109 represents Cys-SO2-SH. Also DHA was simultaneously observed as the C-S cleavage product of thiosulfonate and Cys-S-CN (supplemental Fig. 3A).
Fig. 2.

The origins of novel Cys modifications analyzed using recombinant NDPK A. A, Summary of observed modifications at 109C in peptide 106GDFCIQVGR114 of purified recombinant NDPK A by MS/MS analysis. Peptide m/z and calculated mass, and detected nominal mass changes at 109C residues were presented. B, Samples analyzed in MS/MS analysis were separated on SDS-PAGE under nonreducing conditions and stained with Coomassie blue. This gel shows that NDPK A contains intradisulfide bonds. C, MS/MS spectrum of intradisulfide bond in a solution of recombinant NDPK A between 106GDFCIQVGR114 and 2ANCER6. Cs indicates cysteine persulfide fragment ion and star indicates fragment ion that lost H2O or NH3. D, MS spectra of thiosulfinate and thiosulfonate through further oxidation of disulfide bond in a solution of recombinant NDPK A. E, Quantitative MS analyses of modifications in peptide 106GDFCIQVGR114 of control and oxidized NDPK A in solution were carried out based on precursor ion intensities in supplemental Fig. 3B using Glu-fibrinopeptide as an internal standard. F, A new modification of Cys109 in the modeling structure of human NDPK A. NDPK A is a homohexameric protein. Each subunit was represented with different colors. The image shows a region around Cys109 located on the chain A of the NDPK A structure. The residues consisting of near Cys109 are represented by stick models. Yellow color represents sulfur, blue for nitrogen, red for oxygen, and green for carbon. The modified Cys109 is stabilized by positively charged environment mainly contributed by Arg18 and partly by two backbone nitrogen atoms of Ile110 and Gln111. There is a room to accommodate the extra sulfur and oxygen of modified Cys109 following energy minimization.
To ascertain the relation between unknown modifications and the disulfide bond, semiquantitative analysis of each species was performed in reduced NDPK A treated with NEM, and oxidized NDPK A, and integrated area of precursor ions using MS chromatograms obtained in response to precursor m/z with 0.2 Da. Digestion of reduced and oxidized NDPK A was performed in solution, not on the gel, because oxidized NDPK A exits as two populations on SDS-PAGE gel (Fig. 2B). Equivalent amounts of the reduced and oxidized NDPK As were loaded and analyzed. Glu-fibrinopeptide (GFP) was used as an internal standard, because this peptide does not have any oxidation residues and the ionization efficiency is high enough to allow quantitative analysis. MS chromatograms of various modifications at Cys109 of peptide 106GDFCIQVGR114 of NDPK A, including NEM labeled, intra disulfided with 2ANCER6, Cys-SO2-SH, DHA and cyano were extracted and presented in supplemental Fig. 3B. The extracted chromatogram area of each species was integrated and comparison was made between reduced and oxidized forms of NDPK A, as shown in Fig. 2E. The extraction peak areas do not reflect the absolute quantities of each species, because the ionization efficiencies of the peptides vary depending on their amino acid sequence and the degree of modification. But they indicate the relative abundance of each species in the oxidized and reduced forms. In control NDPK A (reduced monomer labeled with NEM), the peptide 106GDFCIQVGR114 was labeled with NEM, and there were negligible amounts of oxidation products. However, as shown in Fig. 2E, oxidized NDPK A, which has an intradisulfide bond, contained a significantly increased amount of peptide-containing disulfide bonds, Cys-SO2-SH (Δm = +64 Da), DHA, and Cys-S-CN. On the other hand, the amount of peptide containing Ser converted from Cys, is negligible and there was no discernible difference between the two forms in this regard. These results along with the GAPDH study clearly indicate that novel Cys modifications to Cys-SO2-SH (Δm = +64 Da), DHA, and Ser proceed via thiosulfonates, the oxidized products of disulfide bonds through pathways depicted in Eq. 2 and/or Eq. 4. The oxidative modification produces either Cys-SO2-SH/DHA or Cys-SO2-SH/Ser as pairs. It is presumed that when the redox-active Cys residues are oxidized to form disulfide species, further oxidation produces thiosulfonates, and the basic residues serve as bases to form DHA or Ser. The pathway in Eq. 2 is possible because sulfenic acid generated from redox-active Cys in the protein is relatively stable and long living, otherwise the reactive sulfenic acid would form a disulfide bond. Lack of thiosulfinate-driven modifications in the model synthetic peptide, also suggests that the predicted oxidative modification pathways can exist.
To confirm that the modification, Cys-SO2-SH (Δm = +64 Da) at Cys109 of NDPK A is derived from a disulfide bond, we examined the modifications at C109 of NDPK A mutant, C4S (Cys4 mutated to Ser). Purified recombinant wild-type and C4S mutant of NDPK A were treated with various concentrations of H2O2 followed by 20 mm NEM and separated on 12% SDS-PAGE under nonreducing conditions and modifications in each NDPK band were examined. As shown in Fig. 3A, C4S mutant of NDPK A cannot form intradisulfide bonds (lower band). Oxidative modifications including Cys-SO2-SH (Δm = +64 Da) of wild-type and mutant NDPK A were identified and quantitatively analyzed in the MS-chromatogram (Fig. 3B) using 529.69 Da precursor ion of Δm = +64 Da (Fig. 3C). Negligible amounts of Cys-SO2-SH (Δm = +64 Da) or other oxidation states at C109 were detected in this mutant (Fig. 3B and 3D). This confirms that Cys-SO2-SH (Δm = +64 Da) at Cys109 of NDPK A originated from the disulfide crosslinking between C4 and C109.
Fig. 3.

Quantitative analysis of Cys109 modifications in recombinant NDPK A and mutant (C4S) form. A, Wild-type NDPK A and its C4S mutant were treated with various concentrations of hydrogen peroxide and separated on 12% SDS-PAGE. Mutant C4S could not form intradisulfide bonds. B, Precursor mass (C) of peptide 106GDFCIQVGR114 containing mass shift +64 at Cys109 in MS chromatogram was extracted with a 0.2 Da windows and only show that Cys109 + 64 exist in intradisulfide containing line (b, c). D, Integrated chromatographic area of peptide 106GDFCIQVGR114 containing mass shift +64 at Cys109 was summarized.
To explore what other modifications are possible at the region around Cys109 of NDPK A, we constructed a model of Cys-SO2-SH at Cys109, based on the known crystal structure of native human NDPK A. NDPK A is known to form interdisulfide bonds between Cys109 and neighboring Cys109 (22) and intradisulfide between Cys4 and Cys109. Based on the energy minimized model of NDPK A, Cys-SO2-SH and Cys-SO3H could be accommodated in the region around Cys109 without clashes with surrounding residues and this negative charge at Cys109 is stabilized by forming a salt bridge with Arg18 within ∼1.8 Å (Fig. 2F). Because Arg18 is one of the residues necessary for the biological function of NDPK A, inactivation of NDPK A under oxidative conditions is partly explained by this finding with the energy minimized model (25).
To select the most plausible among the proposed pathways for Cys modification, other model systems were prepared and tested (supplemental Fig. 4). These model systems, which included N, N′-Di-Cbz-l-Cystine dimethylester (2) (31) and thiosulfonate (3) obtained from m-CPBA oxidation of 2 were used to test the plausibility pathways in Eq. 1 and Eq. 2. When 2 and 3 were subjected to various basic conditions, clear differences were noted in their ability to form DHA (Table I). Although both substrates were capable of generating DHA, thiosulfonate (3) showed much higher ability to form DHA under mild basic condition (entries 1 and 3). When the reaction was tested under physiologically relevant conditions, only thiosulfonate (3) produced DHA (entries 3, 4, and 5) along with a trace of Ser derivative as detected by MS (entry 5), suggesting that DHA and Ser can produced from thiosulfonate inside the cell.
Table I. DHA formation from Cys modification intermediates.

The leaving group ability of Cys-SO2-S− in conjunction with the inability of DHA to undergo direct hydration reaction with the hydroxide ion (Scheme 1), strongly suggests that the formation of Ser follows the pathway in the Eq. 4 rather than in Eq. 2.
Scheme 1.

Suggested model reactions for conversion of Cys to Ser.
A plausible mechanism for Ser formation is also depicted in Scheme 1. This mechanism is analogous to the known transformation of Cys to Ser under basic conditions. The hydroxide-assisted attack of carbonyl oxygen at the neighboring amide forms C-O bond while cleaving the C-S bond of thiosulfonate, and converting the intermediate into Ser (32). This explains why the Cys modification produces mostly either DHA with Cys-SO2-SH or Ser with Cys-SO2-SH instead of simultaneously forming all three modifications, presumably depending on the location of the relevant basic residue and the three-dimensional structure of the disulfide crosslinked proteins.
Newly Detected Modifications of Cysteine Include Formation of Sulfenic and Sulfinic Acid Derivatives
Scheme 2 summarizes the possible routes that produce novel oxidation products of redox-active Cys residue. These include, in addition to usual oxidation, alkylation of sulfenic and sulfinic acids or further oxidation of alkylated cysteine, because mass changes of +87 Da and +103 Da at Cys152 of GAPDH and Cys109 of NDPK A, respectively were observed (Figs. 4A–4C). The +87 Da reflects an elemental composition of C3H5NO2, as determined by MassLynx, and this is the sum of propionamide (C3H5NO, Δm = +71 Da, an acryl amide adduct), and oxygen (O, with Δm = +16 Da), corresponding to acrylamide adduct of sulfenic acid. The +103 Da shift also suggests a species that has been oxidized in one additional step at the cysteine residue, and causes a +87 Da shift as acrylamide adduct of sulfinic acid. This suggests that sulfenic and sulfinic acids can readily react with acrylamide as well as cysteine sulfhydryl. To confirm that sulfenic and sulfinic acid react with various alkylating agents as well as acrylamide, we examined the alkylation products of oxidized Cys residue in recombinant NDPK A using other alkylating agents. The alkylation products in oxidized Cys with iodoacetamide, were derivatives with Δm = +73 Da (16 (O) + 57 Da) from sulfenic acid and Δm = +89 Da (32 (2O) + 57 Da) from sulfinic acid. As shown in Figs. 4D and 4E, alkylation of sulfenic and sulfinic acids at C109 residue in NDPK A was readily detected with iodoacetamide (Data from another alkylation with NEM are presented in supplemental Fig. 5). It is not possible to infer the routes of formation of these products. Because these alkylation products of sulfenic and sulfinic acids are possibly produced by both pathways, we tried to determine whether the acrylamide adduct of Cys is oxidized to mass shift +87 and +103 Da during experimental procedures, or whether sulfenic and sulfinic acids reacted with acrylamide to generate the acrylamide adducts. If the first premise is right, mass shift +87 and +103 Da can occur in all cysteine residues. However, as mentioned above, in redox-active 152CXXXC156 of GAPDH, Cys156 was labeled with acrylamide but there was no +87 or +103 Da, and only Cys152 was readily modified to mass shift +87 Da and to sulfonic acid, the final product of sulfenic and sulfinic acids (Fig. 1A). This indicates that mass shifts of +87 and +103 Da are from acrylamide adducts of sulfenic and sulfinic acids produced on SDS-PAGE gel. To confirm that acrylamide adduct of sulfenic and sulfinic acids is readily generated from relatively stable sulfenic and sulfinic acids in redox-active cysteines, we examined the mass changes in NDPK A treated, first without or with 5 mm H2O2, and then with 20 mm NEM, and in NDPK A treated with 20 mm NEM first and then oxidized with 5 mm H2O2. As shown in supplemental Fig. 5, only negligible amounts of NEM adducts of sulfenic and sulfinic were detected in NEM treated NDPK A without oxidation; NEM adduct of sulfenic and sulfinic acids, and sulfonic acid were detected only in oxidized NDPK A; and NEM adduct of sulfenic acid, but not sulfinic acid, was detected in NDPK A treated with NEM first and then oxidized. These findings indicate that acrylamide adducts of sulfenic and sulfinic acid are generated by alkylation of sulfenic and sulfinic acids, and not as artifacts of the experimental procedure, even though the acrylamide adduct of free sulfhydryl can be oxidized to sulfenic acid. Also oxidation of the acrylamide adduct of free sulfhydryl can randomly occur without any site specificity. Unstable and highly reactive sulfenic and sulfinic acids have been reported as intermediates in the eventual formation of disulfide and sulfonic acids (5, 6). Mass spectrometric detection of sulfenic acid or sulfinic acid still remains a challenge, although some investigators provided evidence from tandem mass spectrometry that seems to suggest the existence of stable cysteine sulfenic acid in solution. There are also reports of the development of an immunochemical method for detecting sulfenic acid (5, 7). Thus, overall, there seems to be some evidence that sulfenic acids do exist in vivo and that they are stable to some degree in solution. However, it is not possible to totally exclude the idea that acrylamide adduct of sulfhydryl can be oxidized further. These results demonstrate that it is possible to detect sulfenic and sulfinic acids in proteins on gel using MS/MS.
Scheme 2.

A mechanism to explain novel cysteine modifications found in this study (blue). Possible pathways not detected in this study (green).
Fig. 4.

MS/MS spectra showing mass shifts of sum of alkylating agent and oxygen at Cys residues in GAPDH and NDPK A. A, Cys152 of GAPDH has +87 Da shift corresponding to sum of acrylamide adduct propionamide (+71 Da) and one oxygen (+16 Da). B, C, Cys109 of NDPK A have +87 and +103 Da shifts matching to sum of propionamide (+71 Da) and one (+16 Da) or two oxygens (+32 Da). D, E, Cys109 of NDPK A have +73 and +89 Da shifts of sum of iodoacetamide adduct acetamide (+57 Da) and one (+16 Da) or two oxygens (+32 Da). In MS/MS spectra, star indicates fragment ion that lost H2O or NH3.
The Novel Cys Modifications Observed in GAPDH Also Occur in Mouse PRX 6 and Mitochondrial Proteins
To explore whether the Cys modifications observed in GAPDH also generally occur in other proteins during cellular processes, we examined Cys modifications in mouse PRX 6, oxidoreductase (33, 34), and in purified mouse mitochondrial proteins, which are readily oxidized by H2O2 in the respiratory chain. B16F10 melanoma cells and mitochondrial proteins from mouse liver were separated on 2D-PAGE, and PTMs in each spot analyzed by MS/MS using SEMSA and MODi. PRX6 obtained from mouse melanoma B16F10 cells, was separated on 2D-PAGE and its peptide sequence analyzed for Cys PTMs using tandem MS. Mass shifts to Cys-SO2SH (Δm = +64 Da), DHA (Δm = −34 Da), sulfonic acid (Δm = +48 Da), and acrylamide adduct of sulfenic acid (Δm = +87 Da) and sulfinic acid (Δm = +103 Da) were detected at Cys47, a redox-reactive site (35), as shown In Figs. 5A–5C and supplemental Fig. 6. Formation of Cys-SO2-SH or Cys-SO3H (sulfonic acid) at the redox-active Cys47 in the PRX family is first identified in this study. To confirm that these modifications are from cellular oxidation, not artifacts of SDS-PAGE, we analyzed the modification of purified PRX6 in solution with tandem MS in solution, without separation on SDS-PAGE. As shown in supplemental Fig. 7, mass shift to Cys-SO2SH (Δm = +64 Da) was detected at Cys47, a redox-reactive site.
Fig. 5.

MS/MS spectra of peptide 42DFTPVCTTELGR63 of PRX6 separated on 2D-PAGE from B16F10 cells, showing unknown mass shifts of +64 (A), DHA (B), and sulfonic acid (C).
The novel Cys modifications we found in GAPDH were also found in 14 of 57 mitochondrial proteins examined. The modifications and their locations in these 14 proteins are summarized in supplemental Table 1. It is interesting that proteins functionally involved in cellular redox homeostasis such as sulfite oxidase (SOD, Q8R086), and protein disulfide-isomerase (PDI, P09103) especially known to generate disulfide bonds in response to ROS, were heavily modified in their redox-active cysteine residues. In the remaining 43 mitochondrial proteins, no novel Cys modifications were detected under our experimental conditions (supplemental Table 2), but it is possible that these proteins may be also modified similarly or otherwise in other cellular environments. Thus novel Cys modifications can occur widely in cellular processes influencing protein folding, protein-protein interactions, oxidation-reduction potentials, and other protein functions. This is an interesting area requiring further studies.
DISCUSSION
Redox-active Cys, and other cysteine residues that are readily oxidized, can play key roles in signaling pathways that regulate biological functions. Among Cys modifications, long recognized are the disulfide bond, and sulfenic-, sulfinic-, and sulfonic acids. The present studies describe and validate unexpected Cys modifications at redox-active Cys in various proteins. Employing high resolution UPLC-ESI-tandem MS combining selectively excluded mass screening analysis (SEMSA) (17) and MODi and MODmap algorithm (19, 20), to search for unknown PTMs, we identified some novel Cys modifications at redox-active Cys, including Cys-SO2SH (Δm = +64 Da), DHA (Δm = −34 Da), conversion of Cys to Ser, and acrylamide adducts (Δm = +87 and +103 Da) of sulfenic and sulfinic acid, in GAPDH, NDPK A, PRX6, and mitochondrial proteins. We proved, by elemental composition analysis, that the mass shift of +64 Da is sulfur dioxide (-SO2) and by hypothesis based quantitative analysis, that it originates from a disulfide bond. These results are summarized in Scheme 2. We also performed quantitative analysis of modified peptides at the reactive cysteine residue in reduced and oxidized conditions using model recombinant protein NDPK A. This model system was chosen because NDPK A is easily oxidized to form intra- and interdisulfide bonds (22, 24). Oxidized recombinant NDPK A and its mutants with and without disulfide bonds were analyzed using MS spectrometry. These studies prove the existence of disulfide bond associated-novel modifications. The NDPK A system, compared with GAPDH, contained DHA and Cys-S-CN. The amount of peptide containing Ser converted from Cys is negligible. From the results of our GAPDH study one could presume that the novel modification produces either Cys-SO2-SH/DHA or Cys-SO2-SH/Ser as pairs. Although cleavage of the disulfide bond is not easy because of the poor leaving group ability of Cys-S-S−, further oxidation of thiosulfonate can be ruled out because Cys-SO2-S− is a good leaving group. This reactivity pattern suggests that redox-active Cys oxidized to sulfenic acid in response to ROS, can generate a disulfide bond or thiosulfinate, depending on the protein environment. Although the disulfide bond can be reversibly reduced, thiosulfinate can be further oxidized to thiosulfonate, which can be readily cleaved to form Cys-SO2-SH and DHA, or substituted with hydroxide to form Ser and Cys-SO2-SH. Of these, thiosulfinate intermediate between peroxiredoxin and sulfiredoxin was demonstrated in the catalytic mechanism of sulfiredoxin (35, 36). The formation of thiosulfonate and its cleavage to Cys-SO2-SH, has not been reported prior to this study, possibly because the earlier studies did not employ highly sensitive techniques such as SEMSA or MODi for identifying unknown or less abundant PTMs.
The possible pathways for novel Cys modifications were tested using model systems. Our suggested pathway involves initial cleavage of carbon-sulfur bond in the disulfide bond or in thiosulfonate. The cleavage of disulfide bond would produce DHA and persulfide, which could be further oxidized to Cys-SO2-SH as shown in Eq. 1. Thiosulfinate is generated from condensation of two sulfenic acids, and then oxidized further to produce thiosulfonate, which then produces DHA and Cys-SO2-SH as shown in Eq.2. Eq. 1 and Eq. 2 were tested in model systems using N, N′-Di-Cbz-l-Cystine dimethylester (2) and m-CPBA oxidation derivative of 2 (3). When 2 and 3 were subjected to various basic conditions, they showed clear differences in their ability to form DHA with persulfide or Cys-SO2-SH (Scheme 1 Table). Thiosulfonate model compound 3 was cleaved much more efficiently than 2 whereas only a trace amount of Ser derivative was detected (entry 5). This confirms that the origin of novel modifications is the thiosulfonate.
The proposed pathway for the conversion of Cys to Ser indicated in Eq. 3 and Eq. 4 was based on the fact that the Cys modification produced either DHA/Cys-SO2-SH or Ser/Cys-SO2-SH. These results can only be explained by a Cys to Ser conversion mechanism that is independent of the formation of DHA. Otherwise, both Ser and DHA would be detected simultaneously.
These pathways were clearly demonstrated in model compounds 2 and 3. Thiosulfonate 3 showed much higher reactivity than disulfide 2 in Table 1. The lack of the reactivity of DHA with the hydroxide ion along with detection of Ser in minute amounts also supports the proposed mechanism for Cys to Ser transformation depicted in the Scheme 1.
The modifications we identified in redox-active Cys seem to occur quite commonly in many cellular proteins including PRX6, and various mitochondrial proteins that have reactive Cys. Because these Cys modifications irreversibly change protein sequence, they might have as yet unknown biological effects that need to be identified. Cys oxidation, sulfation, sulfite (SO32−), and sulfur dioxide (SO2) formation have all been reported as artifacts of the silver staining procedure (37, 38) used in the preparation of samples for SDS-PAGE gel. In this study, we avoided such artifacts by employing Coomassie blue staining as an alternate procedure and still detected chemical modifications of sulfenic and sulfinic acids (acrylamide adducts, +87 and +103 Da). These alkylations of sulfenic and sulfinic acids were also detected using other alkylation agents, such as iodoacetamide and NEM. There are two possible ways to generate these alkylation products: alkylation of sulfenic and sulfinic acids and alkylation of Cys residue followed by oxidation. We preferred the first possibility, because it is hard to detect +87 and +103 Da in reduced proteins. We believe that other unknown and low abundant modifications will possibly be revealed when the SEMSA method is used in conjunction with unrestricted search engines MODi and MODmap. Such an approach can be extended to proteins having catalytic redox-active cysteine residues identified by high throughput methodology (39).
Footnotes
* This work was supported through the Center for Cell Signaling Research and Drug Discovery Research (CCS and DDR, R15-2006-020) at Ewha Womans University from the National Core Research Center (NCRC) program, 21C Frontier Functional Proteomics Project from Korean Ministry of Education, Science and Technology (FPR-08-A1-020) and by WCU project (R31-2008-000-10010-0). J. Jeong, E. Lee, MS. Kim and S. Na were supported by Brain Korea 21 (BK21) Project and S Na was supported by Seoul Science Fellowship (SSF).
 This article contains supplemental Tables S1 and S2 and Figs. S1 to S7.
This article contains supplemental Tables S1 and S2 and Figs. S1 to S7.
1 The abbreviations used are:
- GAPDH
- Glyceraldehyde-3-phosphate Dehydrogenase
- NDPK A
- Nucleoside diphosphate kinase A
- PRX6
- Peroxiredoxin 6, Sulfenic acid
- RSOH
- Sulfinic acid
- RSO2H
- Sulfonic acids
- RSO3H
- Thiosulfinate
- RS-S(O)R'
- Thiosulfonate
- RS-S(O2)R'
- Cys-(S)-thiosulfonic acid
- RS(O2)-SH
- UPLC-ESI-q-TOF tandem MS; ultra performance liquid chromatography-electrospray-quadrupole-time of flight tandem mass spectrometer, 2D-PAGE; two-dimensional polyacrylamide gel electrophoresis.
REFERENCES
- 1. Berlett B. S., Stadtman E. R. (1997) Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 272, 20313–20316 [DOI] [PubMed] [Google Scholar]
- 2. Møller I. M., Kristensen B. K. (2006) Protein oxidation in plant mitochondria detected as oxidized tryptophan. Free Radic. Biol. Med. 40, 430–435 [DOI] [PubMed] [Google Scholar]
- 3. Orsatti L., Innocenti F., Lo, Surdo P., Talamo F., Barbato G. (2009) Mass spectrometry study of PRL-3 phosphatase inactivation by disulfide bond formation and cysteine into glycine conversion. Rapid Commun. Mass Spectrom. 23, 2733–2740 [DOI] [PubMed] [Google Scholar]
- 4. Bar-Or R., Rael L. T., Bar-Or D. (2008) Dehydroalanine derived from cysteine is a common post-translational modification in human serum albumin. Rapid Commun. Mass Spectrom. 22, 711–716 [DOI] [PubMed] [Google Scholar]
- 5. Seo Y. H., Carroll K. S. (2009) Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc. Natl. Acad. Sci. U.S.A. 106, 16163–16168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Salsbury F. R., Jr., Knutson S. T., Poole L. B., Fetrow J. S. (2008) Functional site profiling and electrostatic analysis of cysteines modifiable to cysteine sulfenic acid. Protein Sci. 17, 299–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shetty V., Neubert T. A. (2009) Characterization of novel oxidation products of cysteine in an active site motif peptide of PTP1B. J. Am. Soc. Mass Spectrom. 20, 1540–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Biteau B., Labarre J., Toledano M. B. (2003) ATP-dependent reduction of cysteine–sulphinic acid by S. cerevisiae sulphiredoxin. Nature 425, 980–984 [DOI] [PubMed] [Google Scholar]
- 9. Woo H. A., Jeong W., Chang T. S., Park K. J., Park S. J., Yang J. S., Rhee S. G. (2005) Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J. Biol. Chem. 280, 3125–3128 [DOI] [PubMed] [Google Scholar]
- 10. Jönsson T. J., Johnson L. C., Lowther W. T. (2008) Structure of the sulphiredoxin–peroxiredoxin complex reveals an essential repair embrace. Nature 451, 98–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aversa M. C., Barattucci A., Bonaccorsi P., Giannetto P. (2007) Recent advances and perspectives in the chemistry of sulfenic acids. Curr. Org. Chem. 11, 1034–1052 [Google Scholar]
- 12. Poole L. B., Nelson K. J. (2008) Discovering mechanisms of signaling mediated cysteine oxidation. Curr. Opin. Chem. Biol. 12, 18–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reddie K. G., Carroll K. S. (2008) Expanding the functional diversity of proteins through cysteine oxidation. Curr. Opin. Chem. Biol. 12, 746–754 [DOI] [PubMed] [Google Scholar]
- 14. Walsh C. T. (2006) Posttranslational modification of proteins expanding nature's inventory. Roberts and Company Greenwood Village, Co., pp. 95–120 [Google Scholar]
- 15. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 16. Kim Y. M., Seo J., Kim Y. H., Jeong J., Joo H. J., Lee D. H., Koh G. Y., Lee K. J. (2007) Systemic analysis of tyrosine phosphorylated proteins in angiopoietin-1 induced signaling pathway of endothelial cells. J. Proteome Res. 6, 3278–3290 [DOI] [PubMed] [Google Scholar]
- 17. Seo J., Jeong J., Kim Y. M., Hwang N., Paek E., Lee K. J. (2008) Strategy for comprehensive identification of post-translational modifications in cellular proteins, including low abundant modifications: application to glyceraldehyde-3-phosphate dehydrogenase. J. Proteome Res. 7, 587–602 [DOI] [PubMed] [Google Scholar]
- 18. Choi S., Jeong J., Na S., Lee H. S., Kim H. Y., Lee K. J., Paek E. (2010) New algorithm for the identification of intact disulfide linkages based on fragmentation characteristics in tandem mass spectra. J. Proteome Res. 9, 626–635 [DOI] [PubMed] [Google Scholar]
- 19. Na S., Jeong J., Park H., Lee K. J., Paek E. (2008) Unrestrictive identification of multiple post-translational modifications from tandem mass spectrometry using an error-tolerant algorithm based on an extended sequence tag approach. Mol. Cell. Proteomics 7, 2452–2463 [DOI] [PubMed] [Google Scholar]
- 20. Na S., Paek E. (2009) Prediction of novel modifications by unrestrictive search of tandem mass spectra. J. Proteome Res. 8, 4418–4427 [DOI] [PubMed] [Google Scholar]
- 21. Kim S. Y., Chang K. H., Doh H. J., Jung J. A., Kim E, Sim C. J., Lee K. J. (1997) Rapid purification and characterization of nucleoside diphosphate kinase isoforms using ATP-sepharose affinity column chromatography. Mol. Cells 7, 630–634 [PubMed] [Google Scholar]
- 22. Song E. J., Kim Y. S., Chung J. Y., Kim E., Chae S. K., Lee K. J. (2000) Oxidative modification of nucleoside diphosphate kinase and its identification by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Biochemistry 39, 10090–10097 [DOI] [PubMed] [Google Scholar]
- 23. Sabio G., Arthur J. S., Kuma Y., Peggie M., Carr J., Murray-Tait V., Centeno F., Goedert M., Morrice N. A., Cuenda A. (2005) p38γ regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP free Guadalupe. EMBO J. 24, 1134–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee E., Jeong J., Kim S. E., Song E. J., Kang S. W., Lee K. J. (2009) Multiple functions of Nm23-H1 are regulated by oxido-reduction system. PLoS ONE 4, e7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laskowski R. A., Macarthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 [Google Scholar]
- 26. Hwang N. R., Yim S. H., Kim Y. M., Jeong J., Song E. J., Lee Y., Lee J. H., Choi S., Lee K. J. (2009) Oxidative modifications of glyceraldehyde-3-phosphate dehydrogenase play a key role in its multiple cellular functions. Biochem. J. 423, 253–264 [DOI] [PubMed] [Google Scholar]
- 27. Steen H., Mann M. (2001) Similarity between condensed phase and gas phase chemistry: fragmentation of peptides containing oxidized cysteine residues and its implications for proteomics. J. Am. Soc. Mass Spectrom. 12, 228–232 [DOI] [PubMed] [Google Scholar]
- 28. Schulz M., Kluge R., Lipke M. (1993) Substitution of arylsulfonyl imidazolides by hydrogen peroxide: aryl sulfonic peracis as oxidants for olefins. Synlett. 915–918 [Google Scholar]
- 29. Wang H., Zang Z., Xian M. (2009) Facile formation of dehydroalanine from S-nitrosocysteins. J. Am. Chem. Soc. 131, 13238–13239 [DOI] [PubMed] [Google Scholar]
- 30. De Marco C., Coletta M., Cavallini D. (1963) Cystine cleavage in alkaline medium. Arch. Biochem. Biophys. 100, 51–55 [DOI] [PubMed] [Google Scholar]
- 31. Liu L., Tanke R. S., Miller M. J. (1986) Electrophilic sulfur transfer reactions in organic synthesis. Preparation of a diastereomer of the key macrocyclic component of griseoviridin. J. Org. Chem. 51, 5332–5337 [Google Scholar]
- 32. Okamoto R., Soumura S., Kajihara Y. (2009) Efficient substitution reaction from cysteine to the serine residue of glycosylated polypeptide: repetitive petide segment ligation strategy and the synthesis of glycosylated tetracontapeptide having acid libile sialyl-Tn antigens. J. Org. Chem. 74, 2494–2501 [DOI] [PubMed] [Google Scholar]
- 33. Chevallet M., Wagner E., Luche S., van Dorsselaer A., Leize-Wagner E., Rabilloud T. (2003) Regeneration of peroxiredoxins during recovery after oxidative stress: only some overoxidized peroxiredoxins can be reduced during recovery after oxidative stress. J. Biol. Chem. 278, 37146–37153 [DOI] [PubMed] [Google Scholar]
- 34. Wagner E., Luche S., Penna L., Chevallet M., Van Dorsselaer A., Leize-Wagner E., Rabilloud T. (2002) A method for detection of overoxidation of cysteines: peroxiredoxins are oxidized in vivo at the active-site cysteine during oxidative stress. Biochem. J. 366, 777–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roussel X., Béchade G., Kriznik A., Van Dorsselaer A., Sanglier-Cianferani S., Branlant G., Rahuel-Clermont S. (2008) Evidence for the formation of a covalent thiosulfinate intermediate with peroxiredoxin in the catalytic mechanism of sulfiredoxin. J. Biol. Chem. 283, 22371–22382 [DOI] [PubMed] [Google Scholar]
- 36. Roussel X., Kriznik A., Richard C., Rahuel-Clermont S., Branlant G. (2009) Catalytic mechanism of sulfiredoxin from Saccharomyces cerevisiae passes through an oxidized disulfide sulfiredoxin intermediate that is reduced by thioredoxin. J. Biol. Chem. 284, 33048–33055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gharib M., Marcantonio M., Lehmann S. G., Courcelles M., Meloche S., Verreault A., Thibault P. (2009) Artifactual sulfation of silver-stained proteins: Implications for the assignment of phosphorylation and sulfation sites. Mol. Cell. Proteomics 8, 506–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nekrassova O., Allen G. D., Lawrence N. S., Jiang L., Jones T. G. J., Comptona R. G. (2002) The oxidation of cysteine by aqueous ferricyanide. Electroanalysis. 14, 1464–1469 [Google Scholar]
- 39. Fomenko D. E., Xing W., Adair B. M., Thomas D. J., Gladyshev V. N. (2007) High-throughput identification of catalytic redox-active cysteine Residues. Science. 315, 387–389 [DOI] [PubMed] [Google Scholar]