

Abstract
β-methylthiolation is a novel post-translational modification mapping to a universally conserved Asp 88 of the bacterial ribosomal protein S12. This S12 specific modification has been identified on orthologs from multiple bacterial species. The origin and functional significance was investigated with both a proteomic strategy to identify candidate S12 interactors and expression microarrays to search for phenotypes that result from targeted gene knockouts of select candidates. Utilizing an endogenous recombinant E. coli S12 protein with an affinity tag as bait, mass spectrometric analysis identified candidate S12 binding partners including RimO (previously shown to be required for this post-translational modification) and YcaO, a conserved protein of unknown function. Transcriptomic analysis of bacterial strains with deleted genes for RimO and YcaO identified an overlapping transcriptional phenotype suggesting that YcaO and RimO likely share a common function. As a follow up, quantitative mass spectrometry additionally indicated that both proteins dramatically impacted the modification status of S12. Collectively, these results indicate that the YcaO protein is involved in β-methylthiolation of S12 and its absence impairs the ability of RimO to modify S12. Additionally, the proteomic data from this study provides direct evidence that the E. coli specific β-methylthiolation likely occurs when S12 is assembled as part of a ribosomal subunit.
Several studies have indicated that structural features of the ribosome including post-transcriptional and post-translational modifications occur on RNA and protein components, respectively. Unlike RNA modifications, which have a wealth of genetic and biochemical evidence documenting their structure and function relationships (1, 2), post-translational modifications are presumed to be structurally or functionally important, based principally on indirect evidence, although the exact biological role for many of these modifications is unclear (3–5). Ribosomal protein S12 undergoes post-translational modification (PTM)1 of aspartic acid 88 (D88), a universally conserved residue of S12 within prokaryotes (6) (Fig. 1). This PTM has been identified in S12 orthologs from several phylogenetically distant bacteria and appears to be unique to bacteria as there have been no reports of this modification in ribosomal proteins from either eukarya or archaea (6–10). Site-directed mutagenesis studies in Thermus thermophilus revealed that substitutions at the D88 position are lethal whereas nearby substitutions are tolerated (11). Two such viable mutants, P90R and P90W, lacked the D88 modification, presumably because of steric hindrance preventing recognition by the modifying enzyme (12). Additionally, a genetic knockout of the rimO gene resulted in a viable strain that only contains unmodified S12 (13). Taken together this PTM is not essential even though D88 substitutions are apparently lethal suggesting that this modification is likely to be important under specific and yet to be defined conditions.
Fig. 1.
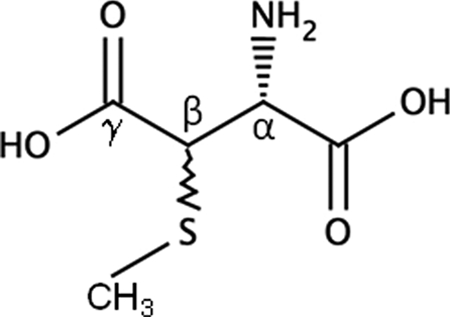
Structure of β-methylthioaspartic acid.
RimO has recently been identified to be required for the addition of this PTM (13–15). The highly conserved rimO gene displays striking similarity to the full-length gene sequence of the MiaB enzyme, a bifunctional system characterized in Escherichia coli that is involved in methylthiolation of transfer RNA (tRNA) (16–19). This tRNA modification involves addition of an S-methyl group to C2 of adenine 37 (A37) to produce 2-methylthio-N6-isopentenyl-adenosine (ms2i6A37) (1, 17). Recently, separate in vitro kinetic studies demonstrated that recombinant RimO from both E. coli and Thermotoga martima successfully methylthiolated a synthetic peptide substrate (mimicking the loop bearing D88) providing the first biochemical evidence showing S12 to be the probable substrate and the RimO mechanism to be consistent with a MiaB-like sulfur insertion (14, 15). However, the lack of efficient modification (both studies resulted in low yield of modified peptide) and the inability to utilize soluble S12 for these assays leaves questions unanswered about how RimO modifies S12 in vivo. For example, to date it is currently unknown if RimO requires other proteins to modify or recognize S12 and if the modification reaction in vivo occurs when S12 is in an isolated state or part of the ribosome.
Here we report a multidisciplinary strategy utilizing an endogenously expressed tagged S12 protein to capture S12 interacting protein(s). Our goal was to gain insight into how RimO modifies S12 in vivo and to identify a potential function of this PTM. Utilizing both proteomic and transcriptomic data we show that two candidate S12 interactors, RimO and YcaO, have a substantial effect on the methylthiolation status of the conserved Asp 88 in vivo and are likely to share a functional relationship. We also present preliminary data that suggests a link between β-methylthiolation of S12 and certain effects on gene transcription.
EXPERIMENTAL PROCEDURES
Construction of Recombinant E. coli SPA-S12
All bacterial strains used in this work are listed in Table I. Strain DY378 was used in the initial recombineering experiments to generate the SPA-S12 containing NCMS1 strain described below. The SPA (sequence peptide affinity) purification system involves adding a C-terminal tag consisting of three modified FLAG (3× FLAG) and a calmodulin binding peptide coding sequence to the chromosomal gene to produce an endogenously expressed bait protein for capturing native protein complexes from E. coli (20). The SPA cassette, containing the tag and a kanamycin selectable marker, was a generous gift from Jack Greenblatt (University of Toronto, Canada). Briefly, the SPA tag was inserted at the COOH end of the rspL chromosomal gene using λRed recombineering as previously described (21, 22). The tagged gene was transduced into W3110 using standard P1 techniques (21). The resulting SPA-S12 protein expression was confirmed by Western blot analysis. Additional strains were generated by either recombineering (as just described) or P1 transduction using standard media, methods, and selections (22).
Table I. Bacterial Strains used in this work.
| Strain | Genotype | Source/ref |
|---|---|---|
| W3110 | Wild type | |
| DY378 | W3110 IcI857D(cro-bioA) | Yu et. al., 2000 |
| NCMS1 | DY378 rpsL-SPA-kanR | Recombineering |
| NCMS2 | W3110 rpsL-SPA-kanR | P1 transduction |
| NCMS3 | DY378ΔrimO<>tet | Recombineering |
| NCMS4 | DY378ΔycaO<>tet | Recombineering |
| NCMS5 | W3110ΔrimO<>tet | P1 transduction |
| NCMS6 | W3110ΔycaO<>tet | P1 transduction |
| NCMS7 | W3110DrimO<>tetDycaO<>cat | P1 transduction |
| NCMS10 | NCMS2ΔrimO<>tet | P1 transduction |
| NCMS11 | NCMS2ΔycaO<>tet | P1 transduction |
| NCMS12 | DY378 fnr-SPA-kanR | Recombineering |
| NCMS13 | DY378 narL-SPA-kanR | Recombineering |
| NCMS14 | DY378 narP-SPA-kanR | Recombineering |
| NCMS15 | W3110 fnr-SPA-kanR | P1 transduction |
| NCMS16 | W3110 narL-SPA-kanR | P1 transduction |
| NCMS17 | W3110 narP-SPA-kanR | P1 transduction |
| NCMS18 | NCMS15ΔrimO<>tet | P1 transduction |
| NCMS19 | NCMS16ΔrimO<>tet | P1 transduction |
| NCMS20 | NCMS17ΔrimO<>tet | P1 transduction |
Isolation of SPA-S12 Complexes from Ribosome Depleted Supernatant
W3110 E. coli (negative control) and the W3110 strain in which an S12 bait protein had been SPA tagged were cultured in Terrific Broth media (Quality Biological, Inc. Gaithersburg, MD) at 37 °C to mid-log phase. Cells were harvested and pellets were frozen at −80 °C until purification. All SPA-S12 pull-downs were performed in parallel with a negative control lacking an SPA tag using the following conditions. E. coli cell pellets were resuspended in Rnase free Tris-buffered saline buffer (25 mm Tris pH 7.4, 137 mm NaCl, 3 mm KCl) supplemented with midi Complete EDTA free protease inhibitor mixture (Roche Diagnostics, Indianapolis, IN) and then lysed by sonication. DNase 1 (Sigma Chemical Company, St. Louis, MO) was added to the resultant lysate and incubated at ambient temperature for 1 h to degrade contaminant DNA. Cellular debris was removed by centrifuging the lysate at 16,000 × g for 15 min. Ribosomes (monosomes + polysomes) were depleted from samples prior to performing pull-downs by ultra-centrifuging the resulting supernatant at 100,000 × g for 4 h. In this work a single purification step utilizing the 3× FLAG epitope was performed for all experiments. To isolate SPA-S12, the resulting ribosome free supernatant (∼8 mg/ml) was then combined with 100 μg of washed Anti-FLAG agarose beads (Sigma Chemical Company) per ml of supernatant and incubated overnight at 4 °C using an orbital rotator. Following extensive washes with Tris-buffered saline buffer, SPA-S12 complexes were eluted using 100 μg/ml 3× FLAG peptides and then concentrated using an Ultracel YM-3 Centrifugal device (Millipore, Billerica, MA). These SPA-S12 and negative control eluates were quantified using the BCA assay with bovine serum albumin as a standard (Thermo, Rockford, IL). The eluted proteins were then heated at 70 °C for 7 min and separated by SDS-PAGE (10%) and the gel was stained with SimplyBlue Safestain (Invitrogen, Carlsbad, CA). All SPA-S12 pull-downs were repeated three times using three separate lysates. Following each SDS-PAGE the eluted proteins were analyzed by performing in-gel digestions and mass spectrometry as described below.
Mass Spectrometry of Peptides
Gel lanes were sliced into 20 ∼ 2-mm bands representing different molecular fractions. In-gel tryptic digestion and peptide extraction followed a modified version of a standard protocol recently described (23). For one-dimensional liquid chromatography tandem mass spectrometry (1D LC-MS/MS) extracted peptides were resuspended in 5% acetonitrile, 0.1% formic acid and then injected into a Nano LC one-dimensional Proteomics high performance liquid chromatography system (Eksigent, Dublin, Ca) coupled online to an LTQ-Orbitrap mass spectrometer (Thermo Fisher, San Jose, CA) equipped with a Nanomate nanoelectrospray ionization source (Advion, Ithaca, NY). Upon injection, all peptide samples were desalted and preconcentrated online with a nano-C18 cap trap (Micron technologies) and then separated using a 75 μm × 10 cm BetaBasic-18 IntegraFrit analytical column (New Objective, Woburn, MA) connected to the nanospray source. A linear gradient was developed using a 400 nl/min flow rate. LC mobile phases were A: 95% water/5% acetonirile/0.1% formic Acid (v/v/v), B: 20% water/80% acetonitrile/0.1% formic acid (v/v/v). Retained analyte was eluted by increasing the acetonitrile concentration to 60% at 1.25% per min. All 1D LC/MS/MS experiments were operated such that spectra were acquired for 60 min in the data dependent mode with dynamic exclusion enabled. The top five peaks in the 400–2000 m/z range of every MS survey scan were fragmented using a minimum threshold cutoff of 5000. Survey spectra were acquired with 60,000 resolution in the Orbitrap-mass analyzer and selected peptide ions were isolated and fragmented in the LTQ ion trap.
Informatics
All Thermo LTQ-Orbitrap Xcalibur raw files were processed by the automation client Mascot Daemon (version 2.3), which allows batch processing and analysis of data by Mascot. Mascot Daemon utilizes the Thermo utility extract_msn (version 5.0) to convert Xcalibur data into DTA format peak lists and submits these to a Mascot Server for searching (all DTA files for a given raw file are merged into an MGF file). Fragmentation spectra were searched using the Mascot version 2.3 search engine (Matrix Sciences, London, UK) against an E. coli database (created from the Swiss-Prot portion of the Uniprot Knowledgebase Release 15.15) supplemented with porcine trypsin and human keratin sequences. Alternative isoform sequences, where relevant, were retrieved from the varsplic annotation file in UniProtKB. This E. coli database contains all ECOLI records and is supplemented with ECOLX for completeness (contains 5,103 sequences; 1,603,703 residues). All data was searched against a decoy database composed of reverse translated sequences in order to determine false positive discovery rates. Search parameters were as follows: trypsin specificity, two missed cleavages, carbamidomethylation of cysteine as a static modification, methionine oxidation (+16 daltons) and aspartic acid methylthiolation (+46 daltons) as variable modifications, and +1 through +4 charge states. Subsequent database searches were conducted to consider a variety of common chemical modifications (e.g. pyroGlu from N-terminal Gln, and pyroCys from N-terminal carboxamidomethylCys) and PTMs of interest (e.g. N-acetylation of proteins). The precursor ion mass tolerance was ± 50 ppm and the fragment ion mass tolerance was ± 0.6 Da. A total of 3 experimental pull-downs and 2 negative controls were performed where 5 LTQ datasets were acquired (available publically via NCBI peptide data resource accession# PSE145). The experimental and negative control data were analyzed separately and the resultant output files were concatenated using the in-house software MassSieve (24). MassSieve parsimoniously reassembles experimentally based peptide identifications into protein identifications. MassSieve facilitates parsimonious comparisons of the concatenated experimental data with negative control data (resultant peptide and protein identifications are presented in supplementary tables). Proteins were considered true interactors if identified in at least two out of three experimental runs and were either absent in the negative control or substantially enriched in the SPA-S12 pull-down. Peptide identifications were the primary metric for inclusion of a protein; a minimum of three peptides were required for a protein to be considered sufficiently identified. Spectra matched to peptides were used as a secondary metric to access those proteins that were enriched in the SPA-S12 pull-downs relative to the control (at least four fold higher number of either peptide identifications or spectra matched to peptides). A protein could still be present at concentration levels below the detection limit required to trigger MS/MS. MassSieve filters were adjusted to include only peptide identifications with Mascot Ion Scores equal to or exceeding their identity scores (corresponds to ≥ 95% confidence). This resulted in a calculated false positive discovery rate of 2.91% and 2.82% for the control and pull-down data respectively. False positive discovery = 2 Nd/(Nd + Nt); where Nd and Nt are the number of matched decoy (resulting from the reversed translated sequence) and target peptides passing the above cutoff respectively.
Quantification of SPA-S12 Isoforms by Mass Spectrometry
Experiments were performed as described above with the following modifications. Two biological replicates were performed on gel tryptic digestions representing bands surrounding SPA-S12 purified from strains NCMS2 (wild type), NCMS10 (ΔrimO), and NCMS11 (ΔycaO) to ascertain reproducibility. The LTQ-Orbitrap mass spectrometer was configured to selectively isolate and fragment precursor ions representing the accurate m/z values for +2 and +1 charge states of the methylthiolated (465 m/z and 929 m/z) and unmodified forms (442 m/z and 883 m/z) of the tryptic peptide VKDLPGVR (representing residues 86–93). For these particular experiments the LTQ-Orbitrap was calibrated to a 400 ppm window. For preliminary analysis all data were searched against Mascot to identify the exact retention time for modified and unmodified forms of the peptide of interest. The integration of ion current intensities of peptide parent ions was calculated by MZmine 2 (25). In brief, m/z values and retention time were used to align corresponding peaks extracted from raw data and the integrated areas determined. Precursors of the modified and unmodified peptide VKDLPGVR that were not matched by Mascot were manually evaluated before use in the alignment process. From these data the total integrated peak areas (representing peptides from both replicates) were used to assess the relative abundance of all isoforms (available publically via NCBI peptide data resource accession number PSE145). The final abundance values reported in this study therefore represent the summed ion current from modified and unmodified forms from both replicates.
Transcriptome Analysis
Bacterial W3110 and MG1655 wild type, ΔrimO and ΔycaO deletion strains were grown either aerobically or anaerobically overnight in 5 mls of Luria Bertani broth at 37 °C with shaking (aerobic only). Overnight cultures were diluted 500-fold into fresh medium and incubated at 37 °C until O.D.600 of 0.3. The cultures were diluted 10-fold in fresh medium again and harvested at O.D.600 of ∼0.5 (exponential phase). Total RNA was extracted using the RNeasy mini kit (Qiagen, Valencia, CA). For DNA microarray hybridization and analysis, 10 μg of RNA was used to prepare biotinylated cDNA according to the GeneChip® Expression Analysis Technical Manual (Affymetrix, Santa Clara, CA). Following fragmentation and labeling, the biotin labeled cDNA (2.5 μg) was hybridized for 16 h at 45 °C on a GeneChip E. coli Genome 2.0 Array (Affymetrix). The arrays were then scanned using a GeneChip Scanner 3000 (Affymetrix). Data were preprocessed using RMA-sketch analysis in Affymetrix Power Tools. The gene expression fold changes were calculated for the mutants compared with the wild-type strain using a twofold cut off. A total of two biological replicates were performed for each MG1655 strain. Because the pull-downs were conducted in W3110, we also performed DNA microarray data analysis (These sequence data have been submitted to the GEO database under accession number: GSE21551) with the W3110 wild-type and ΔrimO strains and found no transcriptional differences that, in effect, resulted in a total of four independent biological replicates.
RNA Extraction from SPA-S12 Pull-Down Eluate
SPA-S12 eluates were mixed with an equal volume of Protinase K buffer (250 mm Tris pH 7.5, 300 mm NaCl, 25 mm EDTA, 50 units RNasin RNase inhibitors (Promega, Madison, WI) containing ∼100 μg of Protinase K (Roche, Indianapolis, IN) and digested for 45 min at 37 °C. To verify that 16S rRNA extracted from SPA-S12 was a result of co-isolation with small subunit ribonucleoprotein particles we performed RNA extractions on eluates from both SPA-S12 and negative control pull-down. Afterward, Protinase K treated samples were diluted to 200 mm monovalent cation Na+. Samples were then extracted twice with 1 volume of phenol/chloroform/isoamyl alcohol (25:24:1) and extracted once with an equal volume of cold chloroform. The resulting RNA in the aqueous phase was precipitated overnight by adding 1 μg glycogen and two volumes of 100% cold ethanol. Following spinning at 13,000 rpms at 4 °C degrees for 30 min the pellet was washed twice with 70% ethanol. To remove contaminant DNA prior to RT-PCR the pellets from both RNA extractions were resuspended in 5 mm Mg and incubated with ∼6 units RNase free DNase 1 (Sigma) at ambient temperature for 15 min. The reactions were quenched with 25 mm EDTA followed by heat treatment at 65 °C for 10 min.
RT-PCR of RNA Extracts from SPA-S12 Pull-Downs
The RT-PCR procedure using the Access RT-PCR system (Promega, Madison, WI) was performed on RNA extractions from SPA-S12 and negative control eluates according to the manufacturer's instructions. Oligonucleotide primers (5′- 3′) (Forward) GTCCTGCATGGCTGTCGTCA, (Reverse) CCATGAAGTCGGAATCGCTAGT were designed to amplify a 300 nucleotide length region. Equal volume of RT-PCR reactions were loaded on a 1.5% agarose gel, stained with ethidium bromide for visualization and analyzed using an LAS 1000 imager (Fujifilm, Valhalla, New York).
Quantitative Western Blot
The SPA tag was inserted at the COOH end of fnr, narL, and narP chromosomal genes using lambda Red recombineering as described above for rspL. This allowed the use of the 3× FLAG epitope for Western blot analysis. All Western blot analysis was performed in replicate to determine reproducibility. Briefly, 10 ml cultures of NCNS15-NCNS20 (see Table I) representing both wild-type and rimO deletion strains containing the above SPA tagged genes were grown to mid-log phase and then harvested. Cell pellets were resuspended, lysed, and the resulting crude protein lysate concentrations were quantified as described above for SPA-S12. Equal amounts of lysate (16 μg) were analyzed by Western blot as described previously with the modification of using polyvinylidene difluoride membranes (26). Monoclonal primary anti-FLAG (produced in mouse) and anti-mouse secondary horseradish peroxidase (HRP) conjugated antibodies (Sigma) were diluted 1:1000-fold and 1:15,000-fold, respectively. As an internal loading control, DnaK protein levels were analyzed with DnaK monoclonal antibodies diluted 1:15,000 (8E2/2 produced in mouse) (Assay Designs, Ann Arbor, MI). Antibody-antigen interactions were detected using the HRP sensitive chromogenic CN/DAB kit or SuperSignal West Pico chemiluminescent substrate kit (Pierce, Rockford, IL) and visualized using a LAS 1000 imager. Western blot band intensities were quantified using Image J software (open source Image J software available at http://rsb.info.nih.gov/ij/).
RESULTS
Creation and Utilization of SPA-S12 to Capture Molecular Complexes
In an effort to characterize the nature of β-methylthiolated S12 we adopted a sequence peptide affinity (SPA) strategy to identify candidate S12 interactors that are involved in β-methylthiolation or relevant to its biological function. A similar strategy has been employed to purify the yeast ribosome and its associated proteins (27). Using homologous recombination, an SPA tag was introduced at the 5′end of the chromosomal rspL gene (see Materials and Methods). The SPA method enhanced the likelihood that we analyzed biologically relevant complexes by generating a recombinant bait protein that is expressed at endogenous levels (20). The resulting E. coli strain (NCMS1) displayed a growth rate similar to wild type indicating the C-terminal tag had minimal effect on SPA-S12 function (data not shown).
To capture SPA-S12 associated complexes while preserving transient specific protein-protein interactions, a single anti-FLAG affinity chromatographic step was used. Initial pull-down experiments from whole cell lysates resulted in the copurification of a large number of proteins most likely associated with polysomes, including most of the ribosomal proteins and many translational components, (e.g. tRNA synthetases, initiation factors, elongation factors, etc.; data not shown). In order to reduce sample complexity, we performed subsequent affinity captures using polysome depleted extracts. Additionally, we performed parallel pull-downs using a wild-type strain lacking the tagged gene as a negative control to identify contaminants that bind nonspecifically to the affinity matrix or antibody. Proteins from SPA-S12 pull-downs and negative controls were separated by SDS-PAGE (Fig. 2A) and subjected to in-gel digestion. Following 1D-LC-MS/MS analysis the Mascot algorithm identified 2737 unique peptides (See supplemental Table S1A) representing proteins from any of three biological replicates (see supplemental protein Tables S1B and S1C). For this work, we considered only protein identifications based on three or more sibling peptides and detected in a least two of three biological replicates. In an effort to ensure that we identified components that form a stable complex and/or nonstoichiometric transient interactions, we considered as SPA-S12 specific only those proteins that were unique to or substantially enriched in the SPA-S12 pull downs. A total of 89 SPA-S12 specific proteins were identified this way (see supplemental Tables S1 and S2). Additionally, another 39 proteins were identified that were significantly enriched in the SPA-S12 pull-down.
Fig. 2.

SPA tagged S12 resulted in the enrichment of ribosomal small subunit associated complexes and the co-isolation of 16S rRNA. A, Polypeptides from eluates representing SPA-S12 (lane 2) and negative control (lane 3) pull-downs were separated by SDS-PAGE (10% polyacrylamide gels) prior to in-gel tryptic digestion and one-dimensional LC tandem mass spectrometry. Molecular weight standards are represented in lane 1. B, 1.5% agarose gel of 300-base-pair regions of 16S rRNA from SPA-S12 (lane 2) and negative control (lane 4) pull-downs. A DNA base pair ladder is represented in lane 1. Total RNA was isolated from pull-down eluates and RT-PCR analysis was performed (see Materials and Methods). Forward and reverse oligonucleotide primers were used to generate 300-base-pair cDNAs. As an additional control SPA-S12 eluates were incubated without reverse transcriptase prior to PCR (lane 3) to confirm that detectable cDNAs were because of the presence of 16S rRNA.
SPA-S12 Enrichment of Small Subunit Ribonucleoprotein Particles (RNPs) and Associated Proteins
The identified proteins fit into 3 major categories: first, those belonging to the 30S ribosomal subunit; second, translation specific, nonribosomal proteins; and third, apparent nonspecific binders.
Protein specificity is commonly associated with enrichment during purification as demonstrated with proteins belonging to the 30S ribosomal subunit (see Table II). However, care must be exercised as evidenced by an abundance of proteins with no known role in translation. In all biological replicates we consistently enriched for subunit proteins that were either primary binding proteins (S4, S7, S8, S15, and S20) or secondary binding proteins (S5, S6, S9, S11, S12, S13, and S18). The primary binding protein S17 and the secondary binding protein S19 were also identified but below the criteria cutoff (only two peptides were matched to each). Both primary and secondary binding protein types are categorized based on the order in which they are known to assemble during the early stages of 30S subunit biogenesis (28, 29) suggesting the possibility that ribosomal depleted extracts contain fully or partially assembled small subunit RNPs. To confirm the presence of RNPs, we performed RT-PCR to determine whether 16S rRNA was co-isolated with SPA-S12 complexes (Fig. 2B). The results from those experiments clearly indicated the co-isolation of 16S rRNA confirming the SPA-S12 specific enrichment of small subunit RNP complexes.
Table II. 30S subunit proteins enriched by SPA-S12 pull-downs.
| Accession | Protein name | Unique peptides | Matched spectra | % Sequence coverage |
|---|---|---|---|---|
| P0AG67 | S1 | 27 | 181 | 52% |
| P0A7V8 | S4 | 17 | 146 | 56% |
| P0A7W1 | S5 | 29 | 90 | 78% |
| P02358 | S6 | 9 | 79 | 67% |
| P02359 | S7 | 15 | 137 | 64% |
| P0A7W7 | S8 | 10 | 51 | 64% |
| P0A7X3 | S9 | 8 | 53 | 50% |
| P0A7R5 | S10 | 5 | 35 | 44% |
| P0A7R9 | S11 | 7 | 33 | 53% |
| P0A7S3 | S12 | 9 | 80 | 38% |
| P0A7S9 | S13 | 11 | 71 | 66% |
| P0ADZ4 | S15 | 8 | 103 | 66% |
| P0A7T7 | S18 | 5 | 67 | 51% |
| P0A7U7 | S20 | 4 | 19 | 44% |
The second group of identified binding proteins includes nonribosomal proteins, which were categorized as candidate S12 interactors using gene annotations (see Table III). Several candidates were assigned as specific interactors because they were known to associate with 30S subunits and had been recently implicated to interact with S12. We also included those proteins that were conserved without a known function. Two proteins from Table III were of particular interest; RimO and YcaO. RimO has been shown to be involved in modifying S12 (13–15). The conserved unknown protein YcaO was one of the most abundantly enriched as indicated by the number of spectra matched to peptides. Consequently, RimO and YcaO were therefore chosen for studies aimed at characterizing the S12 PTM.
Table III. Candidate S12 interactors enriched by SPA-S12 pull-downs.
| Accession | Interacting proteins | Unique peptides | Matched spectra | % Sequence coverage | Comment |
|---|---|---|---|---|---|
| P0AEI4 | RimO | 9 | 33 | 24 | MiaB like methyltransferase |
| P75838 | ycaO | 21 | 226 | 44 | Uncharacterized conserved proteina |
| P76116 | yncE | 8 | 40 | 39 | Uncharacterized conserved proteina |
| P33355 | yehs | 6 | 15 | 50 | Uncharacterized conserved proteina |
| P0AF93 | yjgf | 7 | 50 | 77 | Uncharacterized conserved proteina |
| P0CE47 | Elongation factor Tu | 34 | 1292 | 92 | Translation and peptide bond formation |
| P0A6P1 | Elongation factor Ts | 15 | 51 | 63 | Translation and peptide bond formation |
| P0A6M8 | Elongation factor G | 43 | 453 | 72 | Translation and peptide bond formation |
| P0A6N4 | Elongation factor P | 4 | 16 | 26 | Translation and peptide bond formation |
| P0A6N8 | Elongation factor P like protein | 3 | 18 | 17 | Translation and peptide bond formation |
| P0A6X3 | Protein hfq | 7 | 25 | 54 | Translational regulator |
| P21499 | Ribonuclease R | 20 | 63 | 41 | Degrades mRNA/rRNA: stress response |
| P0A805 | RRF | 6 | 17 | 47 | Ribosome recycling factor |
| P0A8A8 | RimP | 6 | 40 | 33 | Ribsome maturation factor |
a Uncharacterized according to the BioCyc Database collection (http://biocyc.org/).
RimO and YcaO Mutants Elicit Similar Transcriptional Phenotype
For the purpose of determining if YcaO was functionally related to RimO, a series of ΔrimO and ΔycaO genetic knockouts were constructed to screen for potential overlapping phenotypes. As a starting point we performed expression microarray analysis under both aerobic and anaerobic conditions to compare changes in transcript profiles for ΔrimO and ΔycaO mutants (NCMS5 and NCMS6) with the wild-type strain. Only under aerobic conditions did we observe a transcriptional difference. Both mutants exhibited ≥2 fold decrease in transcription levels for a subset of genes belonging to operons that require the fumarate and nitrate reductase regulator (FNR) for transcriptional activation and at least one of the nitrate and nitrite transcriptional response regulators NarL and NarP (see Table IV) (30, 31). However mRNA levels of these regulatory proteins remained unaffected. Supplementary Table S3 lists all genes that were affected by one or both mutants and the corresponding fold changes. Of all genes requiring FNR for transcriptional activation (32–39) 60% changed relative to the wild-type strain; a majority of these decreased in both knockout datasets although the ΔrimO mutant had a more pronounced impact (larger degree of change in transcription).
Table IV. DNA Microarray comparisons for ΔrimO and ΔycaO genetic knockouts with wild-type strain.
| Genes of same operon with transcriptional changes | Function | ΔrimO |
ΔycaO |
Transcriptional Regulators |
|---|---|---|---|---|
| Averaged fold diff.a | Averaged fold diff.a | |||
| nirBDC | Anaerobic reduction and transport of nitrite | 19 | 9 | Fnr, NarP, NarL, IHF |
| narGH | Anaerobic nitrate reduction | 10 | 6 | Fnr, NarL, IHF |
| narK | Anaerobic nitrate/nitrite transporter | 11 | 2 | Fnr, NarL, IHF |
| fdnG | Formate dehydrogenase-N alpha subunit | 5 | N/A | Fnr, NarL, NarP |
| nrfABD | Formate-dependent nitrite reduction, | 8 | 5 | Fnr, NarP, NarL, IHF |
| nikABCDE | Nickel ABC transporter | 9 | 3 | Fnr, NarL, NiKR |
| dmsABC | Anaerobic dimethyl sulfoxide reduction | 9 | 5 | Fnr, NarL, Fis |
| hypBCDE | Anaerobic hydrogenase formation | 5 | 3 | Fnr, NarL |
| napAD | Periplasmic nitrate reductase | 8 | 4 | Fnr, NarP, NarL |
| ansB | l-asparaginase II | 12 | N/A | Fnr, NarL |
| dcuC | Anaerobic C4-dicarboxylate transport | 12 | 4 | Fnr, NarL |
a Averaged fold change value for genes in operon with transcriptional changes. See Supplementary Table III for individual values.
To test the validity of these microarray data we SPA tagged all three transcriptional activators and performed quantitative western-blot analysis utilizing the wild-type and ΔrimO strain (this mutant had more pronounced transcriptional change over the ΔycaO mutant). We performed replicate experiments from extracts grown to mid-log phase. In both cases we observed consistent quantitative results indicating equivalent levels of DnaK as an internal loading control and reduced levels of FNR and NarL proteins in the ΔrimO strain (see Fig. 3). Approximately 80% less FNR protein and ∼15-fold less NarL protein were observed in the ΔrimO strain from two separate biological replicates; NarL levels were nearly undetectable in this mutant strain. A decrease in FNR and NarL protein levels correlated with the observed transcriptional changes (the mRNA levels for these proteins was not affected) for the ΔrimO strain supporting the probability that the overlapping microarray data reflect a functional linkage between RimO and YcaO. Previously published RimO studies therefore prompted us to test if this relationship involved the β-methylthiolation of S12.
Fig. 3.

FNR and NarL expression is decreased significantly in the rimO mutant. E. coli strains containing SPA tagged FNR, NarL, and NarP genes were grown to mid-log phase and lysed. No changes were observed for NarP. Sixteen micrograms of total protein were loaded in each lane. Expressions of SPA-tagged proteins were detected by Western blot analysis using anti-FLAG primary antibodies, DnaK monoclonal antibodies, and anti-mouse secondary horseradish peroxidase (HRP) conjugated antibodies. A, Representative Western blot of lysates from rimO mutant and wild type E. coli. B, Quantification of Western blots. Relative intensities of ECL bands were quantified using the National Institutes of Health Image software. Imunoreactivities were normalized to that of DnaK in the same sample and the samples were compared. A total of two biological replicates were performed. Error bars represent mean ± S.E.
RimO and YcaO Function in Modifying S12 at Asp 88
ΔrimO and ΔycaO genetic knockouts in the SPA-S12 background (NCMS10 and NCMS11) were next studied to facilitate the use of mass spectrometry to detect and quantify differences in the presence of SPA-S12 isoforms with the wild-type strain. In-gel digestion results in the release of the D88 containing tryptic peptide VKDLPGVR, corresponding to residues 86–93 in S12. Methylthiolated (+46 daltons m/z shift) and unmodified forms of the tryptic peptide VKDLPGVR were quantified by ion current intensities (see Table V). Although it is unknown how the modification affects ionization efficiency we were primarily concerned with ratio changes between the knockout and wild-type strains. We performed two biological replicates to ascertain reproducibility; all three strains gave consistent results. In particular we did not detect unmodified S12 in one of the wildtype replicates and barely detected it in the second; wild-type SPA-S12 was almost entirely modified as indicated by the 121:1 ratio difference in total integrated ion current for modified and unmodified forms. SPA-S12 from the ΔrimO mutant strain was completely unmodified in agreement with the recent RimO report (13) whereas the ΔycaO mutant exhibited a 5:1 (methylthiolated versus unmodified) difference in the first replicate followed by a 3:1 difference in the second. The wild-type MS/MS spectrum (Fig. 4A) clearly showed singly charged product ions (y6, y7, and b3) exhibiting mass values of 46 daltons heavier compared with the same fragment ions for the RimO MS/MS spectrum (Fig. 4B). The ΔycaO mutant had an intermediate impact on the modification status resulting in a 23-fold decrease in the ratio of methylthiolated to unmodified isoform in comparison to the wild-type. The more severe reduction in the levels of modified S12 in the ΔrimO mutant compared with the ΔycaO mutant correlates with the degree of transcriptional changes in both mutants. These independent sources of data collectively suggest that YcaO is functionally linked to RimO and its absence has a dramatic impact on the ability of RimO to add the modification.
Table V. Integrated ion current intensity and ratio values reflecting stoichiometric changes.
| Strain | Sum of numerically Integrated ion currenta |
Ion current ratio |
|
|---|---|---|---|
| Modified S12 | Unmodified S12 | Modified/ unmodified | |
| Wild type | 2.44 × 108 | 2.02 × 106 | 121: 1 |
| ΔycaO | 5.57 × 107 | 1.03 × 107 | 5.4: 1 |
| ΔrimO | Not present | 1.5 × 108 | 0 |
a values reported are from 2 biological replicates.
Fig. 4.

Fragmentation spectra of (A) the modified and (B) unmodified forms of the +2 charged tryptic peptide representing residues 86–93. The modified peptide was not identified in samples from the rimO mutant. Both spectra show singly charged y and b fragment ions. y6, y7 and b3 ions in the modified spectrum are 46 daltons (mass of the methylthiolation modification) higher than the same fragment ions for the unmodified spectrum.
DISCUSSION
The current understanding of the ribosomal protein S12 has been primarily focused on its functional and structural role in translation and tRNA decoding at the A site (40–43). Therefore, our goal was to begin to elucidate the regulatory mechanisms governing S12 D88 methylthiolation and its potential role in mediating S12 function. We applied a coordinated approach that included proteomic and transcriptomic discovery employing targeted gene knockouts studies to identify two functionally linked proteins involved in modifying S12. Although additional questions remain to be answered the data here provide new insight into how RimO modifies D88 and reveals a previously uncharacterized protein linked to S12 methylthiolation. We also present preliminary data that suggests S12 D88 methylthiolation might be linked to transcriptional regulation of a subset of genes.
S12 is Likely Modified When Assembled in an RNP Complex
Proteins involved in PTM reactions most often transiently interact with their substrate and are difficult to identify (44). It was, therefore, fortuitous to reproducibly capture the association of the methyltransferase RimO with SPA-S12 complexes. This research provides additional insight into the RimO modification of D88. The first report by Anton and coworkers involved a bioinformatic approach to identify the gene followed by preliminary experimental validation using a rimO genetic knockout to show that intact S12 lacked the PTM. Our proteomic data agree with these results and additionally show that SPA-S12, in the wild-type background, is almost entirely modified indicating the C-terminal tag minimally, if at all, perturbs RimO function (see Table V). By virtue of these MS/MS data we also provide evidence displaying RimO interactions with the small subunit suggesting that RimO likely modifies S12 in vivo when assembled in a small subunit RNP complex although RimO could also modify S12 in an isolated state. Previous experiments have been limited to in vitro assays using peptide substrates (mimicking the D88 bearing loop) and therefore could not address this detail (2, 28). Although it cannot be completely ruled out that we purified a mixture of free and assembled SPA-S12 (one or both potentially associated with RimO) separate attempts to isolate SPA-S12 alone and in a soluble form were unsuccessful (data not shown). It is therefore most probable that SPA-S12, under the affinity purification conditions presented in this work, represents only the assembled form as free SPA-S12 is structurally unstable in our hands.
We also identified a previously uncharacterized conserved hypothetical protein YcaO that interacts with the small subunit and is functionally linked to methylthiolation of D88. Analysis of the YcaO primary sequence using BlastP (http://blast.ncbi.nlm.nih.gov) and the JCVI comprehensive microbial resource (http://cmr.jcvi.org) indicate that, like RimO, the YcaO protein is conserved in phylogenetically distinct bacteria implying a biologically important role. Indeed, the absence of this protein has a dramatic impact on RimO function. YcaO is also captured in SPA-S12 enriched RNPs in the ΔrimO genetic knockout (strain NCMS10) demonstrating this protein interacts with the small subunit complex even when RimO is absent (data not shown). Perhaps YcaO function involves binding to the small ribosomal subunit as a chaperone that optimizes or facilitates RimO-S12 interactions. This putative YcaO function is consistent with a model where RimO modifies S12 assembled in the subunit. Finally, the apparent enrichment of YcaO and its functional linkage to the PTM, suggests it directly interacts with S12. However, YcaO may also interact with other components of the small subunit. Further studies will therefore be needed to elucidate the structural role this protein has with the small subunit and how that relationship involves RimO dependent methylthiolation of D88 on S12.
ΔrimO and ΔycaO Genetic Knockout Data Suggests a PTM-Specific Impact on Transcription
The presence of overlapping phenotypes, determined from two separate approaches, was crucial toward linking a functional relationship to RimO and YcaO. Regarding the microarray data, attributing the cause of a transcriptomic effect to a single ΔrimO knockout dataset could be misleading because RimO may have additional (yet undiscovered) substrates that inflict a nonspecific pleiotropic effect. Considering that over 4200 genes in E. coli are predicted to encode proteins, it is highly improbable that both genetic knockouts would randomly exhibit overlapping transcriptional changes unless the proteins were functionally linked. This is further accentuated by the fact that the ΔycaO knockout data resulted in an intermediate decrease in transcription (compared with ΔrimO) that correlates well with the observed change in SPA-S12 isoform stoichiometry. We also performed microarray analysis on a strain where both rimO and ycaO genes were knocked out and obtained microarray results similar to the ΔrimO mutant suggesting at the genetic level that rimO is indeed epistatic with ycaO (data not shown). Thus, when taken in concert the correlation between the mass spectrometry and microarray data suggests that the transcriptomic changes can be attributed to a loss or reduction of S12 methylthiolation in the absence of RimO and/or YcaO.
Connecting β-methylthiolation to a specific phenotype will require further investigation that is out of the scope of this report. However the data do provide an interesting insight inasmuch as FNR is an oxygen responsive global transcriptional regulator that controls expression of a large number of genes involved in anaerobic functions (45). In the absence of oxygen FNR up-regulates transcription of genes necessary for anaerobiosis. One would therefore expect a more profound transcriptomic effect under anaerobic conditions instead of a complete loss of the phenotype as we observed. The results presented in this work essentially indicate that the RimO/YcaO-mediated PTM is not essential for anaerobic induction of this FNR dependent subset of genes. In this regard, we also failed to observe any growth defect associated with the mutation under either anaerobic or aerobic conditions or any lag associated with an anaerobic-aerobic switch (data not shown). Interestingly, the quantitative Western blot data suggests that β-methylthio-aspartic acid 88 could affect the translational status of FNR and NarL mRNAs (under aerobic conditions) although future research will be required to understand the molecular details. The microarray data presented here are, therefore, suitable for assigning a functional relationship between RimO and YcaO but are incomplete for a functional assessment.
Additional Binding Partners Likely Form Direct Contacts with S12
Finally, it is worth noting that our SPA-S12 pull-down data included several additional binding proteins that have been previously implicated to form direct contacts with S12 (see Table III). Among these include the elongation factor Ef-Tu, which has recently been revealed using genetic studies to be involved in a signal relay with S12 during decoding of mRNA (40). The experimental data presented in this work indicate EF-Tu to be among the most abundant protein identified from our affinity capture. These results therefore support the above genetic study suggesting the highly enriched EF-Tu likely interacts with S12 or alternatively interacts with other/additional proteins within the small subunit. The ribosome recycling factor along with elongation factor G are required for recycling subunits for the next round of translation. Ribosome recycling factor has also been implicated to be involved in direct interactions with S12 (46) and our data further support this. We have recently acquired preliminary data (manuscript in preparation) indicating the proteins Rnase R (involved in mRNA turnover) and Hfq-1 (translational regulation) specifically interact with a biotinylated peptide mimic of the S12 D88 conserved second loop region. Although further experiments are in progress these results suggest that both proteins also make direct contacts with S12 via the second loop region. β-methylthiolated D88 may therefore be linked to binding specificity and a loss of the PTM could possibly alter this interaction.
Concluding Remarks
In summary, we present work combining proteomic and expression microarray data to further characterize the S12 PTM β-methylthiolation. First, we provide additional insight into the RimO mediated modification of S12 by providing biochemical evidence that RimO performs this reaction when S12 is part of a small subunit RNP. Second, we identified a previously uncharacterized protein YcaO that is also functionally linked to the methylthiolation of S12. Two points of significance remain to be elucidated from this work. First, a follow-up on YcaO-small subunit interactions and functional characterization will be required to identify the exact role this protein plays with regard to RimO mediated β-methylthiolation of D88. Finally the overlapping transcriptomic phenotype observed from these data are exciting and future experiments aimed at understanding the molecular details should provide the information necessary for understanding the biological significance of this PTM.
Acknowledgments
We would like to thank Jack Greenblatt (University of Toronto, Canada) for generously providing the SPA cassette used for generating SPA tagged genes. We would like to thank Mikhail Bunenko for his valuable comments and suggestions (National Cancer Institute/Frederick Cancer Research and Development Center, Frederick, MD). We would like to thank Dr. Lisa Renee Olano for her valuable comments and suggestions (Laboratory of Neurotoxicology, NIMH/NIH Bethesda, MD) We would also like to thank Dr. Ayca Akal-Strader (Biology Department, Georgetown University, Washington DC) for her valuable comments and suggestions during the editing of this manuscript. We also thank Brian Kennedy (2008 NIH summer student) for his contributions toward experiments involving 16S RNA extraction. M. B. Strader contributed as primary author and researcher. N. Costantino, C. Chen, I. Patel and A. Makusky participated as supporting researchers. C. Elkins, J. Choy, D. Court, S. Markey and J. Kowalak participated in the writing and/or editing of this manuscript.
Footnotes
* This work was supported by the Intramural Research Program of the National Institute of Mental Health(1ZIAMH000274).
 This article contains supplemental Tables S1 to S3.
This article contains supplemental Tables S1 to S3.
1 The abbreviations used are:
- SPA
- sequence peptide affinity
- PTM
- post-translational modification
- SPA-S12
- SPA tagged ribosomal protein S12
- 1D-LC/MS/MS
- one-dimensional liquid chromatography tandem mass spectrometry
- RNP
- ribonucleoprotein
- FNR
- fumarate and nitrate reductase.
REFERENCES
- 1. Decatur W. A., Fournier M. J. (2002) rRNA modifications and ribosome function. Trends Biochem. Sci. 27, 344–351 [DOI] [PubMed] [Google Scholar]
- 2. Gustilo E. M., Vendeix F. A., Agris P. F. (2008) tRNA's modifications bring order to gene expression. Curr. Opin. Microbiol. 11, 134–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cameron D. M., Gregory S. T., Thompson J., Suh M. J., Limbach P. A., Dahlberg A. E. (2004) Thermus thermophilus L11 methyltransferase, PrmA, is dispensable for growth and preferentially modifies free ribosomal protein L11 prior to ribosome assembly. J Bacteriol. 186, 5819–5825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Polevoda B., Sherman F. (2007) Methylation of proteins involved in translation. Mol. Microbiol. 65, 590–606 [DOI] [PubMed] [Google Scholar]
- 5. Wittmann H. G. (1982) Components of bacterial ribosomes. Annu. Rev. Biochem. 51, 155–183 [DOI] [PubMed] [Google Scholar]
- 6. Kowalak J. A., Walsh K. A. (1996) Beta-methylthio-aspartic acid: identification of a novel posttranslational modification in ribosomal protein S12 from Escherichia coli. Protein Sci. 5, 1625–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Suh M. J., Hamburg D. M., Gregory S. T., Dahlberg A. E., Limbach P. A. (2005) Extending ribosomal protein identifications to unsequenced bacterial strains using matrix-assisted laser desorption/ionization mass spectrometry. Proteomics 5, 4818–4831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Strader M. B., Verberkmoes N. C., Tabb D. L., Connelly H. M., Barton J. W., Bruce B. D., Pelletier D. A., Davison B. H., Hettich R. L., Larimer F. W., Hurst G. B. (2004) Characterization of the 70S Ribosome from Rhodopseudomonas palustris using an integrated “top-down” and “bottom-up” mass spectrometric approach. J. Proteome Res. 3, 965–978 [DOI] [PubMed] [Google Scholar]
- 9. Gupta N., Tanner S., Jaitly N., Adkins J. N., Lipton M., Edwards R., Romine M., Osterman A., Bafna V., Smith R. D., Pevzner P. A. (2007) Whole proteome analysis of post-translational modifications: applications of mass-spectrometry for proteogenomic annotation. Genome Res. 17, 1362–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Running W. E., Reilly J. P. (2009) Ribosomal proteins of Deinococcus radiodurans: their solvent accessibility and reactivity. J. Proteome Res. 8, 1228–1246 [DOI] [PubMed] [Google Scholar]
- 11. Carr J. F., Hamburg D. M., Gregory S. T., Limbach P. A., Dahlberg A. E. (2006) Effects of streptomycin resistance mutations on posttranslational modification of ribosomal protein S12. J. Bacteriol. 188, 2020–2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carr J. F., Gregory S. T., Dahlberg A. E. (2005) Severity of the streptomycin resistance and streptomycin dependence phenotypes of ribosomal protein S12 of Thermus thermophilus depends on the identity of highly conserved amino acid residues. J. Bacteriol. 187, 3548–3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anton B. P., Saleh L., Benner J. S., Raleigh E. A., Kasif S., Roberts R. J. (2008) RimO, a MiaB-like enzyme, methylthiolates the universally conserved Asp88 residue of ribosomal protein S12 in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 105, 1826–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee K. H., Saleh L., Anton B. P., Madinger C. L., Benner J. S., Iwig D. F., Roberts R. J., Krebs C., Booker S. J. (2009) Characterization of RimO, a new member of the methylthiotransferase subclass of the radical SAM superfamily. Biochemistry 48, 10162–10174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arragain S., Garcia-Serres R., Blondin G., Douki T., Clemancey M., Latour J. M., Forouhar F., Neely H., Montelione G. T., Hunt J. F., Mulliez E., Fontecave M., Atta M. (2010) Post-translational modification of ribosomal proteins: structural and functional characterization of RimO from Thermotoga maritima, a radical S-adenosylmethionine methylthiotransferase. J. Biol. Chem. 285, 5792–5801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pierrel F., Hernandez H. L., Johnson M. K., Fontecave M., Atta M. (2003) MiaB protein from Thermotoga maritima. Characterization of an extremely thermophilic tRNA-methylthiotransferase. J. Biol. Chem. 278, 29515–29524 [DOI] [PubMed] [Google Scholar]
- 17. Pierrel F., Douki T., Fontecave M., Atta M. (2004) MiaB protein is a bifunctional radical-S-adenosylmethionine enzyme involved in thiolation and methylation of tRNA. J. Biol. Chem. 279, 47555–47563 [DOI] [PubMed] [Google Scholar]
- 18. Hernández H. L., Pierrel F., Elleingand E., Garcia-Serres R., Huynh B. H., Johnson M. K., Fontecave M., Atta M. (2007) MiaB, a bifunctional radical-S-adenosylmethionine enzyme involved in the thiolation and methylation of tRNA, contains two essential [4Fe-4S] clusters. Biochemistry 46, 5140–5147 [DOI] [PubMed] [Google Scholar]
- 19. Booker S. J., Cicchillo R. M., Grove T. L. (2007) Self-sacrifice in radical S-adenosylmethionine proteins. Curr. Opin. Chem. Biol. 11, 543–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zeghouf M., Li J., Butland G., Borkowska A., Canadien V., Richards D., Beattie B., Emili A., Greenblatt J. F. (2004) Sequential Peptide Affinity (SPA) system for the identification of mammalian and bacterial protein complexes. J. Proteome Res. 3, 463–468 [DOI] [PubMed] [Google Scholar]
- 21. Sawitzke J. A., Thomason L. C., Costantino N., Bubunenko M., Datta S., Court D. L. (2007) Recombineering: in vivo genetic engineering in E. coli, S. enterica, and beyond. Methods Enzymol. 421, 171–199 [DOI] [PubMed] [Google Scholar]
- 22. Miller J. (1972) Experimental in Molecular Genetics, Cold Spring Harbor Laboratory Press, Plainview, N.Y [Google Scholar]
- 23. McFarland M. A., Ellis C. E., Markey S. P., Nussbaum R. L. (2008) Proteomics analysis identifies phosphorylation-dependent alpha-synuclein protein interactions. Mol. Cell Proteomics 7, 2123–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Slotta D. J., McFarland M. A., Markey S. P. (2010) MassSieve: panning MS/MS peptide data for proteins. Proteomics 10, 3035–3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pluskal T., Castillo S., Villar-Briones A., Oresic M. (2010) MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Towbin H., Staehelin T., Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Inada T., Winstall E., Tarun S. Z., Jr., Yates J. R., 3rd, Schieltz D., Sachs A. B. (2002) One-step affinity purification of the yeast ribosome and its associated proteins and mRNAs. RNA 8, 948–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mizushima S., Nomura M. (1970) Assembly mapping of 30S ribosomal proteins from E. coli. Nature 226, 1214. [DOI] [PubMed] [Google Scholar]
- 29. Held W. A., Ballou B., Mizushima S., Nomura M. (1974) Assembly mapping of 30 S ribosomal proteins from Escherichia coli. Further studies. J. Biol. Chem. 249, 3103–3111 [PubMed] [Google Scholar]
- 30. Kang Y., Weber K. D., Qiu Y., Kiley P. J., Blattner F. R. (2005) Genome-wide expression analysis indicates that FNR of Escherichia coli K-12 regulates a large number of genes of unknown function. J. Bacteriol. 187, 1135–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peters J. E., Thate T. E., Craig N. L. (2003) Definition of the Escherichia coli MC4100 genome by use of a DNA array. J. Bacteriol. 185, 2017–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Darwin A. J., Ziegelhoffer E. C., Kiley P. J., Stewart V. (1998) Fnr, NarP, and NarL regulation of Escherichia coli K-12 napF (periplasmic nitrate reductase) operon transcription in vitro. J. Bacteriol. 180, 4192–4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lamberg K. E., Kiley P. J. (2000) FNR-dependent activation of the class II dmsA and narG promoters of Escherichia coli requires FNR-activating regions 1 and 3. Mol. Microbiol. 38, 817–827 [DOI] [PubMed] [Google Scholar]
- 34. Page L., Griffiths L., Cole J. A. (1990) Different physiological roles of two independent pathways for nitrite reduction to ammonia by enteric bacteria. Arch. Microbiol. 154, 349–354 [DOI] [PubMed] [Google Scholar]
- 35. Cotter P. A., Gunsalus R. P. (1989) Oxygen, nitrate, and molybdenum regulation of dmsABC gene expression in Escherichia coli. J. Bacteriol 171, 3817–3823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lubitz S. P., Weiner J. H. (2003) The Escherichia coli ynfEFGHI operon encodes polypeptides which are paralogues of dimethyl sulfoxide reductase (DmsABC). Arch. Biochem. Biophys. 418, 205–216 [DOI] [PubMed] [Google Scholar]
- 37. Wu L. F., Mandrand-Berthelot M. A., Waugh R., Edmonds C. J., Holt S. E., Boxer D. H. (1989) Nickel deficiency gives rise to the defective hydrogenase phenotype of hydC and fnr mutants in Escherichia coli. Mol. Microbiol. 3, 1709–1718 [DOI] [PubMed] [Google Scholar]
- 38. Rowe J. L., Starnes G. L., Chivers P. T. (2005) Complex transcriptional control links NikABCDE-dependent nickel transport with hydrogenase expression in Escherichia coli. J. Bacteriol. 187, 6317–6323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Six S., Andrews S. C., Unden G., Guest J. R. (1994) Escherichia coli possesses two homologous anaerobic C4-dicarboxylate membrane transporters (DcuA and DcuB) distinct from the aerobic dicarboxylate transport system (Dct). J. Bacteriol. 176, 6470–6478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gregory S. T., Carr J. F., Dahlberg A. E. (2009) A signal relay between ribosomal protein S12 and elongation factor EF-Tu during decoding of mRNA. RNA 15, 208–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lodmell J. S., Dahlberg A. E. (1997) A conformational switch in Escherichia coli 16S ribosomal RNA during decoding of messenger RNA. Science 277, 1262–1267 [DOI] [PubMed] [Google Scholar]
- 42. Ogle J. M., Murphy F. V., Tarry M. J., Ramakrishnan V. (2002) Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111, 721–732 [DOI] [PubMed] [Google Scholar]
- 43. Ogle J. M., Ramakrishnan V. (2005) Structural insights into translational fidelity. Annu. Rev. Biochem. 74, 129–177 [DOI] [PubMed] [Google Scholar]
- 44. Rigaut G., Shevchenko A., Rutz B., Wilm M., Mann M., Séraphin B. (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17, 1030–1032 [DOI] [PubMed] [Google Scholar]
- 45. Green J., Trageser M., Six S., Unden G., Guest J. R. (1991) Characterization of the FNR protein of Escherichia coli, an iron-binding transcriptional regulator. Proc. Biol. Sci. 244, 137–144 [DOI] [PubMed] [Google Scholar]
- 46. Pai R. D., Zhang W., Schuwirth B. S., Hirokawa G., Kaji H., Kaji A., Cate J. H. (2008) Structural Insights into ribosome recycling factor interactions with the 70S ribosome. J. Mol. Biol. 376, 1334–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]