

Abstract
Oxidative stress is a critical route of damage in various psychological stress-induced disorders, such as depression. Antidepressants are widely prescribed to treat these conditions; however, few animal studies have investigated the effect of these drugs on endogenous antioxidant status in the brain. The present study employed a 3 weeks chronic regimen of random exposure to chronic mild stress (CMS) to induce oxidative stress in brain, and behavioural aberrations (anhedonia), in rats. The sucrose preference test was used to identify depression-like phenotypes, and reversal in these indices indicated the effectiveness of treatment with escitalopram 2.5mg/kg daily orally following CMS. The level of superoxide dismutase enzyme(SOD) as an antioxidant markers in erythrocyte lysates was reduced in CMS control group while it was elevated in CMS group treated with escitalopram. Also escitalopram significantly reduce the thiobarbituric acid reactive substance(TBARS) levels in selected brain areas homogenates to a level comparable to control group. Catalase activity and GABA levels in these brain areas were also increase in escitlopram treated group. In conclusion, escitalopram is suggested to have antioxidant effect associated with an increase in GABA level in frontal cortices and nucleus accumbens homogenates from rats exposed to CMS.
Keywords: Escitalopram, chronic mild stress, antioxidant effect, SOD, GABA levels
Introduction
Chronic mild stress (CMS), a rodent model of depression was developed to mimic anhedonia (the reduced responsiveness to pleasurable stimuli) and reduced activity level according to American Psychiatric Association [1] and Willner et al [2]. Behavioral changes are induced via a combination of seemingly mild annoyances or stressors presented in an unpredictable manner. This animal model has also been used to study behavioral and physiological changes associated with depression [3, 4], central nervous system mechanisms [5], and treatment of depressive disorders [6].
The protective role of nitric oxide [NO] against oxidative stress is supported by super oxide dismutase (SOD), an enzyme that has an important regulatory role against NO inactivation by reactive oxygen species [ROS] and the prevention of lipid per oxidative damage of nerve cells [7]. Exposure to stress induces damage in prefrontal cortex in rats probably caused by increase of ROS, extra cellular glutamate levels and excessive Ca2+ influx, Moreover, stress induces formation of ROS [8] which leads to oxidative injury in various tissues including blood [9] and brain [10]. Escitalopram (ESC) is an antidepressant drug. It is the S-enantiomer of citalopram and both are SSRIs[ll].
The present study, was conducted to evaluate the effect of escitalopram (ESC) on the SOD enzyme level in erythrocyte lysates , GABA level, , TBARS level and catalase enzyme activity in both frontal cortex and nucleus accumbens homogenates as an index of the GABAergic and antioxidant effect of ESC against stress induced by CMS exposure.
Materials and methods
Drugs
Escitalopram (Cipralex; Lund beck, Denmark available as powder), Super oxide dismutase (RANSOD, by Randox Laboratories). All other chemicals were purchased from Sigma chemicals Co. (Sigma, St Louis, MO, USA),
Animals
Thirty-six male albino rats, weighing 180-200 gm, were used in this study, randomly allocated into 3 groups, 12 rats each. Rats were allowed one week to acclimate to the surroundings before beginning of experiments and housed in individual plastic cages. Standard rat chow and tap water were available ad libitum for the duration of the experiments unless otherwise noted. Sucrose solution (2%) was available ad libitum for one week preceding the experimental procedures to allow adaptation to the taste of sucrose. The temperature was maintained at 22±2°C. The light-dark cycle was on a 12 h light/dark cycle with lights on at 06:00 a.m. and off at 06:00 p.m during the stress procedure (6 weeks). All experiments were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
Experimental protocol
Rats were weighed; each was placed in an individual cage. To introduce the rat to sucrose solution and to obtain baseline data on sucrose consumption, rats were given bottle of 2% sucrose. Twenty-four hours later, the bottles were removed and the amount of sucrose solution in mL consumed by each rat was measured. Sucrose solution intake was measured again for an hour period every Tuesday/week from 6 pm to 7 pm in the dark period. On the basis of body weight and sucrose solution intake (during the 24 and an hour periods), rats were assigned to experimental or control groups (n=12 rats each). Body weight, in addition to sucrose consumption, was used to separate animals in an effort to minimize future changes in sucrose intake caused by differences in body size. Group 2, 3 were exposed to 6 weeks of chronic mild stress. Escitalopram was administered alone in a dose of 2.5 mg/Kg p.o. daily in group 3 starting from the end of the 3rd week up to the end of the 6th week of CMS. The control animals were left undisturbed during the 6 weeks-period, except for scheduled daily oral intake of saline in the last 3 week simulating the test group of treated animals, in addition to cleaning, feeding and weighing procedures. An hour sucrose test was given to all animals once a week.
Chronic stress procedure
The chronic stress procedure [12, 13] was applied; the protocol consisted of the following stressors: 1: 16-h water deprivation (water bottles were removed from cage during this time); 2: 5 min.-tail suspension (animals were held upside down by their tail with metal tongs); 3: l-to2-h restraint (animals were placed in a 50 ml conical tube with breathing holes); 4: 30-45 min. paired housing (the rat was placed in the cage of another rat of the stress group, each week the home cage is alternated); 5: Soiled cage (100 ml 16-18°C water was poured into the cage); 6: 5-min forced swim in cold water (16-18°C). The stressors were presented weekly in a different order and given at different times of the day.
Sucrose test
Animals were given bottles of 2% sucrose for an hour once a week [every Tuesday], this occurs at 6 pm to 7 pm (because the pilot study revealed that rats consumed more fluid during their active dark period), thereby, enhancing the chance of seeing a difference in sucrose solution consumption. After an hour later, the bottles were removed and amount of sucrose solution consumption was measured. At the end of the 6th week, all rats were decapitated and their brains were dissected [14).
Determinations of GABA
GABA level in the tissue homogenates of the prefrontal cortex and nucleus accumbens was determined according to Gunawan et al. [15]. The method using The High Performance Liquid Chromatography (HPLC) method with pre-column phenyl-iso-thio-cyanate (PITC) derivatization for the determination of GABA levels in the homogenate of the prefrontal cortex and nucleus accumbens of the brain of rats of different groups. The measurement scale of the data will be in nmol/mg tissue protein.
Either the prefrontal cortex or nucleus accumbens obtained from each rat was homogenized, then, samples are centrifuged in a cooling (4°C) centrifuge at 10,000 Xg for 10 minutes. The supernatant is aspirated and transferred to an Eppendorff tube, while the pellet was kept at -70°C until assayed for its total protein content.
Each sample is derivatized via drying 100 μl of the aspirated supernatant in the centrivap, under vacuum (A centrivap is a centrifuge made by Backman industry that done centrifugation under vaccum during the whole period of centrifugation to evaporate any fluid and the sample, containing amino acids, remains as a powder in the bottom of the centrifuge tube.) The residue is dissolved in 20 μl of etnanol-water-triethylamine (2:2:1) and evaporate to dryness under vacuum. A 30 μl of ethanol-water-triethylamine phenylisothiocyanate [PITC] (7:1:1:1) is added to the residue and allowed to react for 20 min. at room temperature to form the PITC-derivatives of the amino acids. Excess reagent is then evaporated under vacuum. The mobile phase of HPLC consists of solvents A&B: Solvent A: 0.1 M sodium acetate buffer (pH = 5.8), solvent B: acetonitrile: water (60:40, v: v). A mixture of 80% solvent A and 20% solvent B is adjusted for the “isocratic” HPLC separations. Flow rate is set at 0.6 ml/min. The injected sample is 20 μl. The peaks are detected at 254 nm wave length. Standard curves for GABA and norvaline are plotted using norvaline 2 nmol/ 20 μl as an internal standard. The ratio of the peak area of each concentration of each standard to the peak area of the internal standard is determined and entered against the concentration of the standard, in a simple regression procedure.
Determination of SOD enzyme level in erythrocyte lysates
At the end of the 6th week, blood samples were collected from rats of all groups for measurement of SOD levels in erythrocyte lysates using commercially available colorimetric assay kits based on an indirect xanthine-xanthine oxidase method [16], and results were expressed in IU/ml.
Measurement of frontal cortex thiobarbituric acid-reactive substances (TBARS) as a marker of lipid per oxidation:
Another parts of frontal cortices and nucleus accumbens homogenates were rinsed with cold 0.14 M NaCI and homogenized in 25% ice cold 50 mM Tris-HCI buffer, pH 7.4 [17]. 150 μl of the tissue supernatant of samples were diluted to 500 μl with deionized water. 250 μl of 1.34% thiobarbituric acid will be added to all the tubes followed by addition of equal volume of 40% trichloroacetic acid. The mixture were shaken and incubated for 30 min in a boiling water bath. Tubes were allowed to cool to room temperature and the absorbance read at 532 nm using zero concentration as blank [18].
Catalase enzyme activity in frontal cortex and nucleus accumbens homogenates
Parts of frontal cortices and nucleus accumbens homogenates of different groups were homogenized in 1% Triton X-100 in the assay buffer at about 20,000 x g for 30 sec. on ice. The homogenates were centrifuged at 6000 x g at 4°C for 20 min. The supernatant was diluted with 1.5 volumes of the assay buffer (50 mM KH 2P04/50 mM Na2HPO4, pH 7.0). The enzyme activity was determined by the method described by Aebi [19]. Briefly, in a 5 mL cuvette, 2ml of sample was added and the reaction was initiated by adding 1 mL 30 mM H2O2, and the change in absorbance at 240 nm was monitored at 25°C for one min. A portion of the remaining sample was used for protein determination. Specific activity is expressed as μmol H2O2/min/mg of protein.
Protein determination
The protein content of frontal cortex and nucleus accumbens homogenates were determined by spectrophotometer [Beckman, UK] according to the method of Bradford [20]. The aim is to relate the GABA and oxidative markers concentrations to the total tissue protein.
Statistical analysis
The results are presented as means±SD of mean and evaluated using one-way AN0VA, followed by Bonferroni's post hoc determination, using Graph Pad Prism version 3.00 for Windows 97 (Graph Pad Software, San Diego, CA, USA).
Results
The mean±SD in rats exposed to escitalopram on CMS-induced anhedonia in mice
Figure 1 demonstrates the reversal of anhedonia after daily oral administration of escitalopram for 3 weeks. Sucrose consumption in mL of the different groups (control, CMS, CMS+escitalopram) was calculated. Compared with the control group, the CMS group was associated with a decrease in sucrose consumption. This decrease was significantly (P< 0.05) restored to control level in escitalopram treated group.
Figure 1.

reversal of anhedonia after daily oral administration of escitalopram for 3 weeks in mice.
Effect of escitalopram administration (for 3 weeks) on the GABA level in the prefrontal cortex of rats exposed to CMS protocol
Figure 2A represents the changes in GABA concentration in the prefrontal cortex of the control, CMS-untreated and CMS-treated rats. CMS decreased significantly (p<0.05) the GABA concentration in the prefrontal cortex. GABA concentration of rats significantly (P< 0.05) restored to control level by escitalopram administration (3 weeks) with continuous exposure to CMS protocol.
Figure 2.
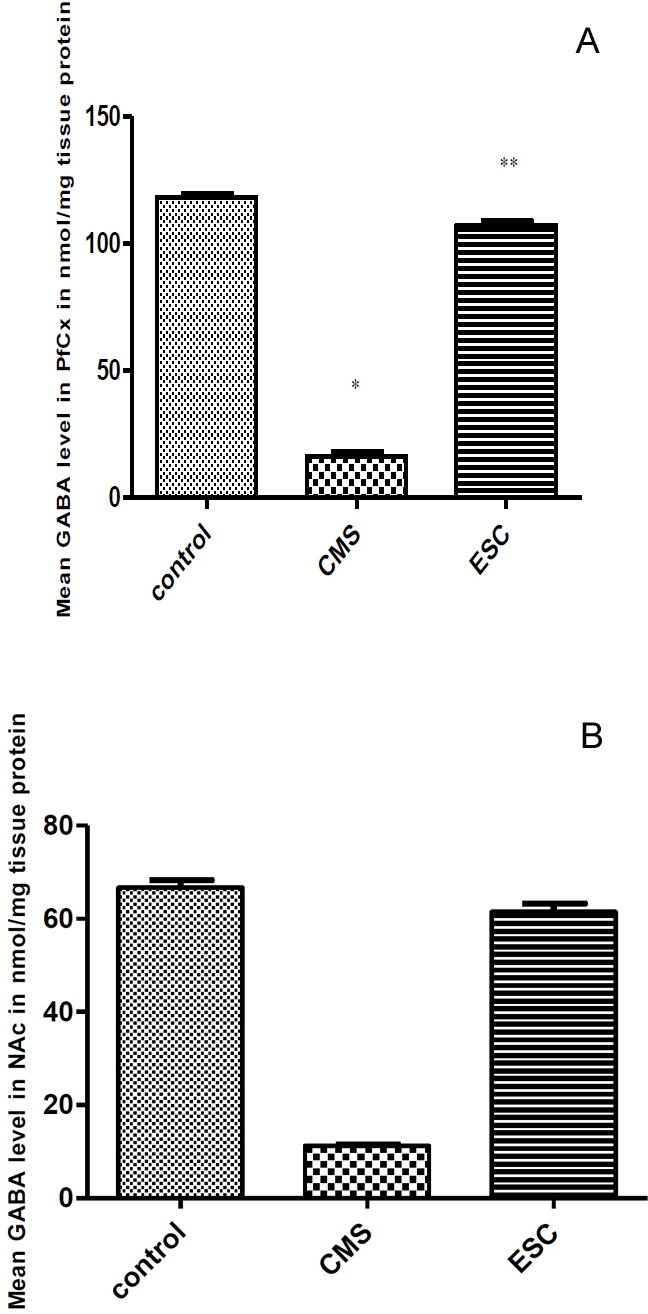
(A}. Effect of escitalopram administration (3 weeks) of on the GABA level in the prefrontal cortex of rats exposed to CMS protocol. B. Effect of escitalopram administration (3 weeks) on the GABA level in the nucleus accumbens [NAc] of rats exposed to CMS protocol.
Effect of escitalopram administration (for 3 weeks) on the GABA level in the nucleus accumbens of rats exposed to CMS protocol
Figure 2B represents the changes in GABA concentration in the nucleus accumbens of the control, CMS-untreated and CMS- treated rats. CMS significantly decreased (p<0.05) the GABA concentration in the nucleus accumbens that significantly (P< 0.05) restored to control level by escitalopram administration.
The mean±SD of escitalopram on SOD levels in erythrocyte lysates
Exposure to CMS model showed significant (p < 0.05) decrease in SOD in erythrocyte lysates (0.18 ± 0.23 IU/mg tissue protein compared with 22.65 ± 1.46 lU/mL in control group). Escitalopram administration in the last 3 weeks of exposure to CMS model significantly (P< 0.05) restored SOD level compared to control level (17.3± 5.20 lU/mL compared with 22.65 ± 1.46 lU/mL in control group).
The mean±SD of escitalopram on TBARS and catalase enzyme activity in frontal cortices and nucleus accumbens homogenates (Table 1).
Table 1.
Effect of escitalopram on TBARS in nmol/mg tissue protein and Catalase enzyme activity in μmol/min/mgtissue protein in frontal cortices and nucleus accumbens homogenates.
| Substance | CONTROL | CMS WITHOUT TREATMENT | CMS+ ESC | |||
|---|---|---|---|---|---|---|
| PfCx | NAc | PfCx | NAc | PfCx | NAc | |
| TBARS in nmol/mg tissue protein | 1.9±0.51 | 1.17±0.17 | 6.2±0.69* | 7.53±0.82* | 1.2±0.3** | 1.08±0.31** |
| Catalase enzyme activity in μmol/min/mgtissue protein | 225.5±8.3 | 110.3± 3.36 | 12.0±0.7* | 0.11 ± 0.02* | 300.4±27.8** | 111.5 ± 0.85** |
The results are expressed as means ± S.D. (N = 12 for each group).
Prefrontal cortices and nucleus accumbens homogenates of escitalopram treated group showed significant (P< 0.05) restoration of TBARS to control level compared to CMS-untreated group. Catalase enzyme activity in both areas homogenates was significantly (P< 0.05**) restored to control level in escitalopram treated group compared to the significant (*P< 0.05) decreased in its activity in CMS untreated group.
Discussion
The current study is undertaken to determine whether escitalopram administered to rats with CMS-induced anhedonia can protect against oxidative stress and can produce an effect on GABA level in selected brain areas compared with anhedonic group of rats without treatment. For depression, a solid foundation for oxidative stress hypotheses has been provided by biochemical, genetic, pharmacological, preclinical therapeutic studies and one clinical trial [21].
Sucrose intake was reduced in rats exposed to 6 weeks of CMS. The reduced sucrose intake in the CMS group is a specific indication of decreased responsiveness to a pleasurable stimulus corresponding to Willner et al. [12] and Grippo et al. [22]. Also the study showed that anhedonic rats displayed lower GABA level in brain homogenates as well as increased TBARS level associated with decreased catalase enzyme activity in homogenates and decreased level of SOD enzyme in erythrocyte lysates. These results were reversed with ESC treatment to a level comparable to that recorded with control rats.
The brain is very vulnerable to oxidative stress because of its elevated consumption of oxygen and the consequent generation of large amounts of ROS. Interestingly, our results evidenced a parallel increase in both SOD and CAT, the most important antioxidant enzymes, in response to ESC to stressed animals. Enhancement of their activities may provide an effective defense from the damaging effects of not only superoxide anion and hydrogen peroxide (neutralized by these enzymes), but also from the highly reactive and damaging hydroxyl radical, generated by Haber-Weiss or Fenton reactions of the former radicals [23]. Therefore, ESC may possibly act to avert extensive damage to different types of molecules, including proteins, nucleic acids, and lipids, thus playing a critical role in maintaining the integrity of the cell.
Enhanced SOD activity, as observed here by ESC treatment, is also an important neuronal defense mechanism against apoptosis, thereby preventing atrophy of brain regions in stress-induced depression [24]. Such cellular changes as a consequence of lowered antioxidant defense during prolonged stress contribute to the neuroanatomical abnormalities observed in mood disorders, including volume loss of certain brain regions in chronically depressed patients and other stress-related disorders [25].
Antidepressant therapy or other forms of treatment that upregulate SOD may also prevent worsening of the affective symptoms that are either directly or indirectly related to neurodegeneration [26]. Thus, any possible symptomatic improvement in this context may be related to the enhanced neuroprotection. Since antidepressants have been found to exert neurotrophic effects leading to increased neurogenesis [27] while oxidative stress existing in depression and other mood disorders may impair these processes [28], it may be possible that improvement of the antioxidant status by antidepressants could partially account for neuroprotective effects as well.
Furthermore, a significant reversal of stress-induced lipid peroxidation by antidepressants also reduces the secondary effects of oxidative stress which have great potential to adversely affect several key processes in the brain. The brain has a high content of oxidisable polyunsaturated fatty acids and prevention of lipid peroxidation by ESC probably serves to maintain membrane integrity by protecting membrane phospholipids from damage, which is the result of a complex cascade involving impairment of membrane-transport protein functions and ion channels that, in turn, mediates membrane lipid peroxidation-induced disruption of neuronal ion homeostasis [29].
The destruction of membrane phospholipids alters membrane viscosity and is implicated to influence several stages in biogenic amine function, such as receptor density or function of serotonergic/catecholaminergic receptors [30].
By reducing lipid peroxidation, antidepressants may possibly prevent the inhibitory effects which MDA directly exerts on serotonin-binding areas of the receptor [31]. Furthermore, membrane lipid peroxidation also modifies neurotransmitter release and uptake, ion-channel activity, the function of ATPases and glucose transporters, and the coupling of cell-surface receptors to GTP-binding proteins, to impair mitochondrial function and promote a cascade of events that culminates in apoptotic cell death (29). Prevention of these potentially damaging factors during stress-induced lipid peroxidation may possibly be a target of antidepressant action, relevant to their therapeutic benefits.
The amelioration in GABA level by escitalopram administration could be explained on the basis of serotonergic neurotransmission that enhance GABAergic neurotransmission that could potentiate its antidepressant activity [32].
Conclusion
In conclusion, the present study showed that escitalopram induced a reduction in susceptibility to oxidative stress and caused an increase in GABA level in selected brain areas in the presence of anhedonia after a prolonged period of exposure to mild stressors. This possible protective effect against stress with escitalopram may aid in the development of beneficial treatments for depressed patients.
Acknowledgments
Authors acknowledge the facilities provided by the Ain shams University.
References
- 1.American Psychiatric Association, editor. 4th Ed. Washington, DC: American Psychiatric Association; 1994. Diagnostic and Statistical Manual of Mental Disorders; pp. 317–391. [Google Scholar]
- 2.Willner P, Sampson D, Papp M, Phillips G, Muscat R. Animal models of anhedonia. In: Soubrié P, editor. Anxiety, Depression, and Mania. Basel, Switzerland: Karger; 1991. pp. 71–99. Animal Models of Psychiatric Disorders, edited by. [Google Scholar]
- 3.Willner P, Muscat R, Papp M. Chronic mild stress-induced anhedonia: a realistic animal model of depression. Neurosci Biobehav Rev. 1992;16:525–534. doi: 10.1016/s0149-7634(05)80194-0. [DOI] [PubMed] [Google Scholar]
- 4.Cheeta S, Ruigt G, van Proosdij J, Willner P. Changes in sleep architecture following chronic mild stress. Biol Psychiatry. 1997;41:419–427. doi: 10.1016/S0006-3223(96)00058-3. [DOI] [PubMed] [Google Scholar]
- 5.Muscat R, Towell A, Willner P. Changes in dopamine auto receptor sensitivity in an animal model of depression. Psychopharmacology. 1988;94:545–550. doi: 10.1007/BF00212853. [DOI] [PubMed] [Google Scholar]
- 6.Muscat R, Papp M, Willner P. Reversal of stress-induced anhedonia by the atypical antidepressants, fluoxetine and maprotiline. Psychopharmacology. 1992;109:433–438. doi: 10.1007/BF02247719. [DOI] [PubMed] [Google Scholar]
- 7.Pablos RM, Villaran RF, Arguelles S, Herrera AJ, Venero JL, Ayala A, Cano J, Machado A. Stress Increases Vulnerability to Inflammation in the Rat Prefrontal Cortex. J Neurosc. 2006;26(21):5709–5719. doi: 10.1523/JNEUROSCI.0802-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mclntosh LJ, Sapolsky RM. Glucocorticoids may enhance oxygen radical-mediated neurotoxicity. Neurotoxicology. 1996;17:873–882. [PubMed] [Google Scholar]
- 9.Oishi K, Yokoi M, Maekawa S, Sodeyama C, Shiraishi T, Kondo R, Kuriyama T, Machida K. Oxidative stress and haematological changes in immobilized rats. Acta Physiol Scand. 1999;165:65–69. doi: 10.1046/j.1365-201x.1999.00482.x. [DOI] [PubMed] [Google Scholar]
- 10.Liu J, Wang X, Shigenaga MK, Yeo HC, Mori A, Ames BN. Immobilization stress causes oxidative damage to lipid, protein, and DNA in the brain of rats. FASEB J. 1996;10:1532–1538. [PubMed] [Google Scholar]
- 11.Ceglia I, Acconia S, Fracasso C, Colovic M, Caccia S, Invernizzi R. Effect of chronic treatment of escitalopram and citalopram on extracellular 5-HT in the prefrontal cortex of rats: role of 5-HT1A receptors. Br J Pharmacol. 2004;142:469–478. doi: 10.1038/sj.bjp.0705800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willner P, Towell A, Sampson D, Sophokleous S, Muscat R. Reduction of sucrose preference by chronic unpredictable mild stress and its restoration by a tricyclic antidepressant. Psychopharmacol (Berl.) 1987;93:358–364. doi: 10.1007/BF00187257. [DOI] [PubMed] [Google Scholar]
- 13.Solberg LC, Horton TH, Tarek FW. Circadian rhythms and depression: effects of exercise in an animal model. Am J Physiol Regul Integr Comp Physiol. 1999;276:R152–R161. doi: 10.1152/ajpregu.1999.276.1.R152. [DOI] [PubMed] [Google Scholar]
- 14.Glowinski I, Iversen LL. Regional studies of catecholamine metabolism in the rat brain. I. The disposition of (3H) dopamine and (3H) dopa in various regions of the brain. J Neurochem. 1966;13:655–669. doi: 10.1111/j.1471-4159.1966.tb09873.x. [DOI] [PubMed] [Google Scholar]
- 15.Gunawan S, Walton N, Treiman D. High performance liquid chromatography determination of selected amino acids in rat brain by precolumn derivatization with phenylisothiocyanate. J Chroma. 1990;503(1):177–187. doi: 10.1016/s0021-9673(01)81499-0. [DOI] [PubMed] [Google Scholar]
- 16.Halliwell B, Chirico S. Lipid peroxidation: its mechanism, measurement, and significance. Am J Clin Nutr. 1993;57:715S–725S. doi: 10.1093/ajcn/57.5.715S. [DOI] [PubMed] [Google Scholar]
- 17.Benjamin JA, Iwata R, Hazalett J. Kinetics of entry of proteins into the myelin membrane. J Neurochem. 1978;31:1077–1085. doi: 10.1111/j.1471-4159.1978.tb00150.x. [DOI] [PubMed] [Google Scholar]
- 18.Gutteridge JM, Quinlan GJ. Malondialdhyde formation from lipid peroxides in the thiobarbituric acid test: the role of lipid radicals, iron salt and metal chelators. J Appl Biochem. 1983;5(4-5):293–299. [PubMed] [Google Scholar]
- 19.Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121–126. doi: 10.1016/s0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- 20.Bradford M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 21.Ng F, Berk M, Dean 0, Bush A. Oxidative stress in psychiatric disorders: evidence base and therapeutic implications. Int J Neuropsychopharmacol. 2008 doi: 10.1017/S1461145707008401. Published online. [DOI] [PubMed] [Google Scholar]
- 22.Grippo AJ, Johnson AK. Biological mechanisms in the relationship between depression and heart disease. Neurosci Biobehav Rev. 2002;26:941–962. doi: 10.1016/s0149-7634(03)00003-4. [DOI] [PubMed] [Google Scholar]
- 23.Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction, Toxicol Lett. 1995;82-83:969–974. doi: 10.1016/0378-4274(95)03532-x. [DOI] [PubMed] [Google Scholar]
- 24.Greenlund US, Deckwerth TL, Johnson EM. Super oxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed cell death. Neuron. 1995;14:303–316. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 25.Koenen KC, Driver KL, Oscar-Berman M, Wolfe J, Folsom S, Huang MT, Schlesinger L. Measures of prefrontal system dysfunction in posttraumatic stress disorder. Brain Cogn. 2001;45:64–78. doi: 10.1006/brcg.2000.1256. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Chlan-Fourney AV, Juorio VL, Bennetts S, Shrikhande M, Bowen RC. Antidepressants up regulate messenger RNA levels of the neuroprotective enzyme super oxide dismutase (S0D1) J Psychiatry Neurosci. 2000;25:43–47. [PMC free article] [PubMed] [Google Scholar]
- 27.Duman RS, Nakagawa S, Malberg J. Regulation of adult neurogenesis by antidepressant treatment. Neuropsychopharmacology. 2001;25(6):836–844. doi: 10.1016/S0893-133X(01)00358-X. [DOI] [PubMed] [Google Scholar]
- 28.Kim E, Shirvalkar P, Herrera DG. Regulation of neurogenesis in the aging vertebrate brain: role of oxidative stress and neuropsychiatric factors. Clin Neurosci Res. 2003;2:285–293. [Google Scholar]
- 29.Mattson MP. Modification of ion homeostasis by lipid per oxidation: roles in neuronal degeneration and adaptive plasticity, Trends Neurosci. 1998;20:53–57. doi: 10.1016/s0166-2236(97)01188-0. [DOI] [PubMed] [Google Scholar]
- 30.Van-der-Vliet A, Bast A. Effect of oxidative stress on receptors and signal transmission. Chem Biol Interact. 1992;85:95–116. doi: 10.1016/0009-2797(92)90055-p. [DOI] [PubMed] [Google Scholar]
- 31.Britt SG, Chiu VW, Redpath GT, VandenBerg SR. Elimination of ascorbic acid-induced membrane lipid per oxidation and serotonin receptor loss by Trolox-C, a water-soluble analogue of vitamin E. J Recept Res. 1992;12:181–200. doi: 10.3109/10799899209074791. Depressive Disorder. Massachusetts General. [DOI] [PubMed] [Google Scholar]
- 32. Massachusetts General Hospital. Brain GABA Hospital Depression Clinical and Research Levels and Treatment Response in Major Program Website 2008.