

Abstract
Sigma (σ) receptors have been shown to regulate multiple ion channel types in intracardiac ganglion neurons, including voltage-gated calcium and potassium channels. However, the inhibition of these channels alone cannot fully account for σ receptor-induced changes in neuronal excitability previously reported. Whole-cell patch clamp experiments were conducted under current-clamp mode in isolated intracardiac neurons from neonatal rats to assess the effects of σ receptor activation on the active membrane properties of these cells. Bath application of the pan-selective σ receptor agonist, 1,3-Di-o-tolylguanidine (DTG), and the σ-1-selective agonist, (+)-pentazocine, significantly increased the action potential latency and decreased action potential overshoot in response to depolarizing current ramps, which suggests inhibition of voltage-gated sodium channels. Whole-cell voltage clamp experiments showed that these σ agonists reversibly decrease depolarization-activated Na+ currents in these cells at all potentials tested. The peak currents generated by membrane depolarizations were decreased in a dose dependent manner with IC50 values for DTG and (+)-pentazocine of 32 μM and 49 μM, respectively. The σ-1 receptor-selective antagonist, BD 1063 (100 nM), inhibited DTG (30 μM) block of Na+ currents by ∼ 50%, suggesting that the effects are mediated by activation of σ-1 receptors. DTG also shifted the steady-state inactivation curve of Na+ channels to more negative potentials, with the membrane potential of half-activation shifting from -49 mV to -63 mV in the absence and presence of 30 μM DTG, respectively. Taken together, these results suggest that σ-1 receptor activation decreases intracardiac ganglion neuron excitability by modulating voltage-gated Na+ channels.
Keywords: Sigma-1 receptor, sodium channel, intracardiac neurons, action potential, parasympathetic
Introduction
Mammalian intracardiac ganglia mediate all parasympathetic input to the heart, and serve as the common final pathway for autonomic regulation of cardiac function. Neurons from the mammalian intracardiac ganglion project onto pacemaker cells, myocytes, and blood vessels [1, 2]. Heart rate, atrioventricular conduction, cardiac contractility and coronary vascular tone are all modulated by the parasympathetic nervous system. Evidence suggests that these ganglia regulate cardiac function by integrating information from efferent and afferent parasympathetic, as well as sympathetic pathways [1, 2], and likely exert local control of the heart [3]. Thus, there is considerable need to understand the mechanisms by which the electrical properties of these neurons are regulated. Experiments in our laboratory have demonstrated that sigma receptor activation has a significant impact on the membrane properties of intracardiac neurons [4, 5], but the function of σ receptors in the neurons remains to be fully understood.
Sigma receptors are classified as non-opioid, non-PCP [6] receptors and are ubiquitous proteins expressed throughout the central and peripheral nervous systems. Two subtypes of the receptor have been pharmacologically identified; σ-1, which preferentially binds (+)-benzomorphans such as (+)-pentazocine and (+)-SKF-10,047 [7] and σ-2, which binds ibogaine with high affinity [8]. To date only the σ-1 subtype has been cloned [9]. These receptors have been implicated in learning and memory [10], movement disorders [11], drug dependence [12, 13] and psychological disorders [14]. In addition, sigma receptors have been shown to have significant effects on the cardiovascular system. For example, activation of sigma receptors produces positive inotropic effects in isolated rat cardiomyocytes [15], and the sigma receptor ligand, DuP 737, has been shown to decrease the threshold for ventricular fibrillation in a rat myocardial infarction model [16]. Recently, it has been shown that σ-1 receptor levels in the heart are upregulated by strong stress stimuli, and it was suggested that σ-1 receptors may be involved in the altered contractility observed under specific pathophysiological conditions, such as hypoxia [17].
Both σ-1 and σ-2 receptors are expressed in intracardiac ganglion neurons and inhibit voltage-gated K+ and Ca2+ channels, respectively [4, 5]. Inhibition of these channels is associated with depressed excitability of intracardiac neurons, but neither the changes in action potential configuration nor the complete block of action potential firing that can be produced upon σ receptor activation can be explained by inhibition of these channel types alone. Voltage-gated Na+ channels, which have not been previously shown to couple to σ receptors in intracardiac neurons, are also responsible for action potential kinetics and configuration in both nerve and muscle cells [18]. Modulation of Na+ channels influences the threshold of action potential generation, the action potential overshoot, as well as the frequency of action potential firing. Previous work has shown that sigma receptor activation not only inhibits action potential firing but also decreases the amplitude of action potential overshoot [5], suggesting that σ receptors may also affect voltage-gated Na+ channels. A recent study reported that σ-1 receptors couple to the Nav1.5 expressed in cardiomyocytes [19]. However, it has not been determined if the sodium channel subtypes expressed in intracardiac neurons (Nav1.1-3 and Nav1.7 [20]) are also modulated by σ-1 receptors. Moreover, neither the underlying biophysical changes in Na+ channel properties nor the effects of these changes on action potential firing have been characterized.
Experiments were undertaken to determine the effects of sigma receptor activation on the excitability of intrinsic cardiac neurons from neonatal rats and whether this regulation was due to the modulation of voltage-gated Na+ channels. Activation of sigma receptors increased the latency of action potential generation produced by depolarizing current ramps in cultured intracardiac neurons. Sigma agonists were shown to have inhibitory effects on the current mediated by voltage-gated Na+ channels (INa), and the pharmacological properties of the receptors were consistent with the effects being mediated specifically by σ-1 receptors. Furthermore, activation of σ receptors shifted the steady-state inactivation of Na+ channels to more negative potentials. Such a shift would effectively reduce the number of voltage-gated Na+ channels available for activation at the normal resting membrane potential of these cells (ca. -50 mV). These observations indicate that sigma receptors are capable of decreasing the parasympathetic input to the heart by inhibiting voltage-gated Na+ channels in intrinsic cardiac neurons.
Materials and methods
Preparation of cell culture
The isolation and culturing of neurons from neonatal rat intracardiac ganglia has been described previously [21]. Dissociated neurons were plated onto poly-L-lysine coated glass coverslips and incubated at 37°C for 36-72 h under a 95% air, 5% CO2 environment prior to the experiments.
Electrical recordings
Electrophysiological recording methods used were similar to those previously described [22]. Voltage-activated Na+ currents were studied in intrinsic cardiac neurons using the whole cell patch-clamp technique under voltage-clamp mode. The conventional (dialyzing) whole cell recording technique was used in experiments studying current-voltage relationships, while the amphotericin B, perforated-patch method was used in current clamp experiments. A stock solution of amphotericin B (60mg/ml) in dimethyl sulfoxide (DMSO) was prepared and diluted in pipette solution immediately prior to use to yield a final concen-tration of 198 μg/ml amphotericin B in 0.33% DMSO. Final patch pipette resistances were 1.0-1.3 MΩ to permit maximal electrical access. Junction potentials generated by the ions in the pipette and bath solutions were compensated for via the Pipette Offset control of the Axopatch 200B. Series resistance (Rs) was monitored throughout the experiment and only cells in which Rs was consistently ≤ 3 MΩ following 50% Rs compensation were used to minimize voltage error. Also, to minimize space-clamp artifact, only cells with no large visible processes were selected for the experiments. Currents through depolarization-activated Na+ channels were elicited by step depolarizations from –90 mV to more positive potentials (-50 to +90 mV for I-Vs, -10 mV for peak currents). For experiments studying the effects of sigma receptor agonists on steady-state inactivation of Na+ channels, cells were stepped to conditioning potentials for 50 msec (-120 to 0 mV) from a holding potential of -90 mV. Test currents were then recorded as the cells were depolarized to 0 mV. Action potentials were evoked by injecting current ramps (0 to 200 nA, 400 ms) into current clamped cells. The latency of action potential generation was determined as previously reported for these cells [23, 24] and was defined as the time interval between the start of the current ramp to the point at which the rising phase of the action potential depolarized to 0 mV. Membrane voltages and currents were amplified using an Axopatch 200B patch-clamp amplifier (Axon Instruments, Union City, CA), filtered at 5 kHz (-3 dB; 4-pole Bessel filter), and digitized at 20 kHz (Digidata 1200 B).
Solutions and reagents
The bath solution used when measuring Na+ currents was a physiological saline solution (INa-PSS) composed of (in mM): 70 NaCl, 70 tetraethylammonium chloride (TEA-Cl), 5 BaCl2, 1.2 MgCl2, 7.7 glucose, 0.1 cadmium chloride (CdCl2), and 10 HEPES (adjusted to pH 7.2 with NaOH). Barium and TEA were used to block voltage-gated K+ channels and cadmium was used to block voltage-gated Ca2+ channels. The pipette solution used in these experiments contained (in mM): 130 CsCl, 10 NaCl, and 10 HEPES (adjusted to pH 7.2 with CsOH). The bath solution used when measuring membrane potentials in current-clamp experiments was a physiological saline solution (AP-PSS) containing (in mM): 140 NaCl, 1.2 MgCl2, 3 KCl, 2.5 CaCl2, 7.7 glucose and 10 HEPES (adjusted to pH 7.2 with NaOH). The pipette solution used for these experiments contained (in mM): 75 K2SO4, 55 KCl, 5 MgSO4, and 10 HEPES (adjusted to pH 7.2 with N-methyl-d-glucamine) and 198 μg/ml amphotericin B. All chemical reagents used were of analytical grade. (+)-pentazocine, (1S,9S,13S)-1,13-dimethyl-10-(3-methylbut-2-en-1-yl)-10 azatricyclo [7.3.1.02,7] trideca-2, 4,6-trien-4-ol (PTZ); 1,3-Di-o-tolylguanidine (DTG); BD 1063, 1-[2-(3,4-dichlorophenyl)ethyl] -4- methylpiperazine dihydrochloride, and tetraethylammonium chloride (TEA) were purchased from Sigma Chemical (St. Louis, MO). Stock solutions of PTZ and DTG made using DMSO as the solvent and control experiments throughout the study represent the appropriate vehicle control.
Data analysis
Analyses of these data were conducted using the SigmaPlot 9 and SigmaStat 3.1 programs (Systat Software, Inc, San Jose, CA). Data points represent means ± SEM. Statistical differences were determined using paired t-tests or two-way ANOVA followed by a post-hoc Tu-key Test, as appropriate, and were considered significant if p < 0.05.
Results
Effects of sigma receptors on the latency of action potential generation
Previous studies have shown that sigma receptor modulation of K+ channels alters intracardiac ganglion neuron resting membrane potential, the number of evoked action potentials, action potential afterhyperpolarization amplitude and action potential duration [5]. These studies also suggested that sigma agonists may modulate action potential initiation. Effects of sigma ligands on the latency of action potential generation were assessed in intracardiac ganglion neurons using perforated-patch whole cell recording techniques under current clamp mode. Figure 1A shows representative membrane responses to depolarizing current ramps (0 to 200 nA, 400 ms) recorded from a single intracardiac neuron in the absence and presence of 30 μM of DTG. The latency of action potential onset was significantly increased by DTG from 23.6 ± 2.3 msec to 32.0 ± 2.7 msec, an increase of 35.6% (n = 4) (Figure 1B). Consistent with previous studies, DTG decreased action potential firing and altered action potential configuration [5]. The effects of DTG were mimicked by 50 μM (+)-pentazocine, which increased the latency of action potential firing from 27.5 ± 1.2 msec to 33.0 ± 1.3 msec, an increase of 20.0% (n=4)(Figure 1B). The effects of both sigma ligands were reversible after wash out of the drug (data not shown). This increase in firing latency cannot be explained by inhibition of either Ca2+ or K+ currents in these cells [24-26], and thus, must involve modulation of other channel types.
Figure 1.

Sigma receptor agonists increase the latency of action potential generation in rat intracardiac ganglion neurons. A, Action potentials generated by 400 ms depolarizing current ramps (0 to 200 nA) from a neuron in the absence (blue line) and presence of DTG (30 μM) (red line). Inset shows first action potentials generated by the ramps on an expanded time scale. Arrows in inset indicate start point of the injected current ramp and points at which latency times were measured. Dashed lines in represent 0 mV, and solid line above voltage traces represents the current ramp protocol used. B, Bar graph summarizing the action potential latency for multiple neurons (n=4) before (Control) and after application of the sigma receptor agonists DTG (30 μM), and (+)- pentazocine (PTZ, 50 μM). Asterisks indicate significant difference between conditions using a paired t-test (p < 0.05).
Sigma receptor activation inhibits voltage-gated Na+ channel currents
The depression of action po tential firing and changes in action potential configuration evoked by sigma receptor ac tivation suggests that voltagegated Na+ channels may be affected by these receptors. Thus, experiments were carried out to examine the relationship between σ receptors and voltage-gated Na+ channels. Voltage-gated Na+ currents were isolated in intra cardiac neurons by inhibiting Ca2+ currents with extracellular Cd2+, and inhibiting K+ currents with extracellular TEA and intracellular Cs+. Figure 2A shows a family of depolarization-activated Na+ currents recorded from a single intrinsic cardiac neuron in the absence (left traces) and presence of 100 μM DTG (right traces) in response to a set of depolarizing voltage steps between -50 and +100 mV. Under control conditions, INa was activated at approximately -30mV, was maximal at -10mV and reversed at approximately +70 mV (Figures 2B & C). The voltage dependence, kinetics and tetrodotoxin sensitivity (data not shown) of the currents observed are consistent with voltage-gated Na+ channel currents previously characterized in intracardiac ganglion neurons [26]. Bath application of 100 μM DTG (Figure 2B) or 100 μM (+)-pentazocine (Figure 2C) depressed the peak INa at potentials equal or positive to -20 mV. The effects of both DTG (Figure 2D) and (+)-pentazocine (data not shown) were reversible upon washout of drug.
Figure 2.

Inhibition of Na+ currents in rat intracardiac neurons by the sigma receptor agonists, DTG and (+)-pentazocine. A, Whole-cell Na+ currents evoked by depolarizing test pulses (-50 to +100 mV) from a holding potential of -90 mV in the absence (Control, left traces) or presence of 100 μM DTG (right traces). B, Whole-cell current-voltage relationships obtained in the absence (Control, blue circles) and presence of 100 μM DTG (DTG; red circles). C, Whole-cell current-voltage relationship obtained in the absence (Control, blue circles) and presence of 100 μM (+)-pentazocine (PTZ, red circles). Currents were normalized to the maximum control current of each cell, and data points represent means ± SE for 4 cells for both (B) and (C). D, Peak Na+ channel current amplitude before and during application of 300 μM DTG (line above graph) and after removal of DTG. Values are plotted as a function of time. Gap represents period during which lower concentrations of DTG were applied, with 300 μM DTG application commencing at t = 20 min. Inset, Family of depolarization-activated Na+ channel currents recorded from a single ICG neurons in the absence (Control) and presence of bath-applied 300 μM DTG (DTG), and following wash out of drug (Wash).
Concentration-dependent inhibition of INa by sigma ligands
Additional pharmacological experiments were undertaken to confirm that sigma receptor activation mediated the inhibitory effects on INa by sigma ligands. Selective agonists were used to identify the sigma receptor subtype mediating the observed effects. Figure 3A shows representative currents recorded from a single intracardiac ganglion neuron in the absence (Control) and presence of the pan-selective σ receptor agonist DTG at the indicated concentrations. Increasing the concentration of DTG resulted in a concomitant decrease in INa. Similarly, Figure 3B shows currents from a different neuron in the absence (Control) and presence of the σ-1 agonist, (+)-pentazocine (PTZ), at the indicated concentrations, and demonstrates that (+)-pentazocine also elicits a concentration-dependent reduction in voltage-activated Na channel currents. Plots of the mean peak INa following depolarizing voltage steps to -10 mV, as a function of drug concentration, for DTG and (+)-pentazocine are shown in Figure 3C. All of the sigma ligands tested depressed peak INa in a concentration-dependent manner. Fits of the data using the Hill equation gave half-maximal inhibitory concentration (IC50) values for DTG and (+)-pentazocine of 32 ± 3 μM and 49 ± 3 μM, respectively, with Hill coefficients of ∼1.0 for both drugs. Maximal inhibition of INa was >90% for DTG and (+)-pentazocine.
Figure 3.

Dose-dependent inhibition of depolarization-activated Na+ currents by sigma receptor agonists DTG and PTZ in rat intracardiac neurons. A, Whole-cell Na+ currents evoked from an intrinsic cardiac neuron by step depolarizations to -10 mV from a holding potential of -90 mV in the absence (Control) and presence of DTG at the indicated concentrations. B, Whole-cell Na+ currents evoked from an intracardiac neuron by step depolarizations to -10 mV from a holding potential of -90 mV in the absence (Control) and presence of (+)-pentazocine at the indicated concentrations. C, Normalized peak whole-cell INa (I/Imax), evoked by depolarizing to -10 mV from -90 mV, plotted as a function of concentration of sigma receptor agonists, DTG and (+)-pentazocine (PTZ). Data points represent mean ± SE for 4-5 neurons for each concentration. The curves represent best fits to the data using the Langmuir-Hill equation. The values for half-maximal inhibition were 32 ± 3 μM for DTG and 48 ± 3 μM for (+)-pentazocine, and the Hill coefficients were 1.0 ± 0.1 for both compounds.
Relief of DTG inhibition of voltage-gated Na+ current by the sigma antagonist, BD 1063
To confirm that σ-1 receptors modulate Na+ currents, experiments were conducted using the σ-1-selective antagonist, BD 1063, in conjunction with DTG. Figure 4A shows Na+ currents elicited by voltage steps from -90 to -10 mV from a single neuron, in the absence and presence of 30 μM DTG, without (left panel) and with 100 nM BD 1063 (right panel). Figure 4B tabulates the results from six cells depolarized in INa-PSS alone (Control), with 30 μM DTG and 100 nM BD 1063 (BD) alone and with both agents present (DTG+BD). Only the currents activated in DTG alone were significantly different from control (p < 0.05, 2-way ANOVA). Figure 4C shows the percent inhibition of INa produced by 30 μM DTG in the absence (Control) and presence of 100 nM BD 1063 (BD 1063). Where as DTG inhibited INa by over 50% when applied alone, DTG only decreased INa by less than 25% in the presence of 100 nM BD 1063. Thus, BD 1063 decreased the effects of DTG on INa by >53%, and this relief of DTG-induced inhibition was statistically significant.
Figure 4.

The σ-1 antagonist, BD 1063, blocks the effects of DTG on peak Na+ currents. A, Na+ currents evoked by voltage steps from -90 to -10 mV in a single neuron in the absence (Control; left traces) and presence of 30 μM DTG (DTG; left traces), in the presence of 100 nM BD 1063 alone (BD 1063; right traces) and following coapplication of both drugs (DTG+BD 1063; right traces). B, Tabulation of results from measurements made on six cells using the voltage protocol in (A). Bars represent mean ± SEM for normalized INa. For Control and BD 1063 (BD) values were normalized by dividing INa for each cell by the mean INa recorded from all cells under the respective conditions (i.e. Control or BD), whereas DTG and DTG + BD 1063 were normalized by dividing the INa obtained from each cell by the INa obtained from the same cell under Control conditions or in the presence of DB 1063 alone, respectively. Asterisk indicates significant difference from Control (p<0.05). C, Calculated percent inhibition of INa by 30 μM DTG in the absence (DTG) and presence of 100 nM BD 1063 (DTG + BD) (n = 6). Asterisk denotes significant difference from Control (p < 0.05).
Effects of sigma receptor activation on voltage-gated Na+ channel inactivation
Sigma receptor activation has been shown to both depress ion channel function and alter the kinetics of ion channel activation [4, 5]. Thus, a decrease in peak INa current amplitude by sigma agonists may be due to channel block or to alteration of channel kinetics. The effects of DTG (30 μM) on steady-state inactivation of Na+ currents in rat intracardiac neurons were studied using a double pulse protocol (Figure 5A, upper traces). Neurons, initially held at -90 mV, were stepped to conditioning potentials from -120 to 0 mV for 50 msec prior to a voltage step to 0 mV (50 ms) to activate the available Na+ channels. Figure 5A shows a family of current traces recorded from a voltage-clamped neuron depolarized to 0 mV following the set of conditioning pulses in the absence (Control, lower left traces) and presence (DTG, lower right traces). In the presence of DTG, the test currents obtained at more depolarized conditioning potentials were depressed to a greater extent. Inset in Figure 5A shows the currents obtained at -50 mV for both conditions scaled to each other. In contrast to steady-state inactivation kinetics, neither the rate of activation nor the time-dependent inactivation of voltage-gated Na+ channels appears to be affected by DTG. A plot of normalized Na+ peak current amplitudes (I/I(max)) as a function of conditioning potential is shown in Figure 5B. The steady-state inactivation of INa exhibited a sigmoidal dependence on the conditioning potential (V) and was best fit with a single Boltzmann function according to the equation
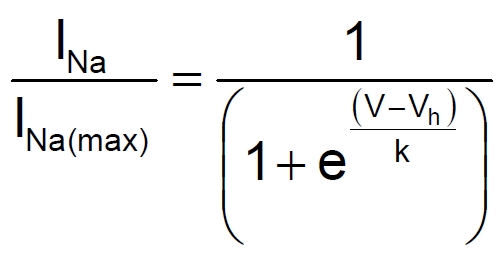 |
where Vh is the potential at which half-maximal steady-state inactivation occurs and k is the slope factor. In the absence of DTG (Control), Vh was calculated to be -49.7 ± 0.6 mV with a slope factor of 7.5. In the presence of DTG, Vh shifted to -64.3 ± 1.1 mV and k increased to 14.9. The shift in steady-state inactivation evoked by σ receptor activation was statistically significant (p < 0.001), and DTG produced a statistically significant depression in the average number of channels available for activation from -100 mV to -50 mV (p < 0.05 for all).
Figure 5.

Sigma receptor modulation of steady-state inactivation of Na+ channels in rat intracardiac ganglion neurons. A, Voltage protocol used to measure steady-state inactivation in the neurons (upper traces). Cells were held at -90 mV, stepped to a preconditioning potential (-120 to 0 mV) for 50 msec, then stepped to 0 mV and test currents measured. Family of current traces recorded from a voltage clamped neuron after stepping to 0 mV from different conditioning potentials in the absence (Control, lower left traces) or presence of 30 μM DTG (DTG, lower right traces). Time scale is expanded in reference to protocol traces, and arrows indicate the traces corresponding to the indicated conditioning potentials. Inset shows test currents obtained following a -50 mV conditioning pulse when the currents were scaled to each other. B, Relative Na+ current amplitudes as a function of conditioning potential in the absence (Control; blue circle, blue line) and presence of 30 μM DTG (DTG; red circle, red line). Data points represent means ± SE for 7 neurons. Lines represent best fit to the data using a single component Boltzmann distribution (Eq. 1), with Vh = -49.7 ± 0.6 mV and k = 7.5 ± 0.5 for Control and Vh = -64.3 ± 1.1 mV and k = 14.9 ± 1.0 in the presence of DTG. Asterisks denote significant difference from DTG group at the same preconditioning potentials (p < 0.05 for all).
Discussion
The major finding reported here is that activation of σ receptors results in suppression of voltage-gated Na+ channel function in intrinsic cardiac neurons. The inhibition of these channels contributes to changes in action potential configuration, increases the latency of action potential generation and depression of action potential firing. Moreover, sigma receptor activation shifts the voltage of half-maximal inactivation towards negative potentials, resulting in a decrease in the number of Na+ channels available for activation at the normal resting membrane potential of these cells (ca. -50 mV). The pharmacological properties of the σ receptor mediating the inhibition of INa suggest that σ-1 receptors are specifically involved.
Our laboratory has previously shown that activation of σ-1 and σ-2 receptors in intracardiac neurons results in the inhibition of voltage-gated K+ and Ca2+ channels, respectively [4, 5]. Inhibition of these channels by sigma receptor activation accounted for many of the observed changes in action potential configuration induced, including an increase in action potential duration and a decrease in action potential afterhyperpolarization [5]. Inhibition of voltage-gated K+ channels is also likely responsible for the conversion of tonic firing intracardiac neurons to phasic firing cells [5]. However, action potential firing in these neurons is abolished at high concentrations of sigma receptor agonists (mM) [5]. Inhibition of voltage-gated K+ or Ca2+ channels alone by σ receptors is unlikely to produce this effect, since previous studies have shown that blocking these channels with TEA and Cd2+, respectively, does not prevent the genesis of action potentials in intracardiac neurons [27]. The initiation of neuronal action potentials is the result of a complex interaction of the voltage- and time-dependent properties of Na+ and K+ selective ion channels [28]. In guinea-pig sympathetic neurons [23] and mudpuppy intracardiac neurons [24, 29], an outward K+ current activated by Ca2+ -induced calcium release (CICR) regulates the threshold for action potential generation. Inhibition of CICR by thapsigargin, ryanodine or Cd2+ decreases the latency of action potential generation in these neurons. Parsons et al (2002) proposed that in intracardiac neurons when membrane depolarization approaches the threshold for action potential generation, small Ca2+ influxes through voltage-dependent Ca2+ channels were sufficient to initiate CICR. The Ca2+ released from internal stores raises the intracellular Ca2+ concentrations in domains between the ER and the plasma membrane to levels high enough (≥40 μM) to activate outward K+ currents that repolarized the cell [24]. In these neurons, large conductance Ca2+- activated K+ channels (BK), which are at least in part activated by CICR, regulate the generation of action potentials. Blocking the BK current with iberiotoxin or 500 μM TEA decreases the latency of action potential generation [23, 24]. However, in mammalian intracardiac neurons, CICR inhibition did not alter the latency of action potential generation [30]. In rat intracardiac neurons, the initiation of action potential firing is facilitated by voltage-dependent Na+ channels and depressed by the M-current [22, 26]. Since blocking M-currents would decrease the latency of action potential firing [22], the increased latency of action potential firing observed following activation of σ-1 receptors must be due to the inhibition of voltage-gated Na+ channels and not due to the effects of σ-1 receptors on the M-current.
Voltage-gated Na+ channels are known to be important for the generation and conduction of action potentials in neurons. The voltage-gated Na+ channels expressed in rat intracardiac neurons are TTX sensitive, with complete blockade at 300 nM [26]. These channels undergo fast and slow inactivation, where τfast is in the millisecond range and τslow is on the order of seconds [26]. In neonatal rat intracardiac neurons, the fast inactivation process determines the maximum amplitude of the current as well as the rate of repolarization and the duration of action potentials. While σ-1 receptor activation did not appear to alter the activation rate or time-dependent inactivation of Na+ channels in these cells (see inset Figure 5A), our study shows that DTG shifts the steady-state inactivation to more negative potentials. Such a shift in the steady-state inactivation of Na+ channels to more negative potentials would lead to a greater number of Na+ channels being in the inactive state at resting membrane potentials, resulting in a decrease in INa in response to membrane depolarization. This increase in the number of inactivated Na+ channels would increase the threshold for action potential firing and consequently increase the latency of action potential firing. However, the shift in steady-state inactivation alone cannot account for the depression of peak whole-cell Na+ current at more negative membrane potentials. Thus, sigma receptor activation must be decreasing the unitary current amplitude or single channel open probability to reduce the macroscopic currents. The other sigma agonist used in this study, (+)-pentazocine, produced similar shifts in steady-state inactivation (data not shown), suggesting this is a sigma receptor dependent modulation and not particular to DTG. Interestingly, high concentrations of TTX fail to block action potentials in intracardiac neurons because these cells are capable of firing Ca2+ action potentials [26]. Thus, the complete block of action potential firing evoked by DTG likely involves inhibition of both voltage-gated Na+ and Ca2+ channels.
The pharmacological properties of the receptor subtype mediating the inhibition of INa in intracardiac neurons are consistent with σ-1 receptors. Here we show that (+)-pentazocine blocks INa with an IC50 of 48 ± 3 μM. Similarly, (+)-pentazocine has been shown to block voltage-gated K+ channels via σ-1 receptors in frog pituitary melanotrophs and rat intracardiac neurons with IC50 values of 37 μM and 76 μM, respectively [5, 31]. Further evidence that the σ-1 receptor subtype is responsible for the effects observed here is provided by the results obtained with BD 1063. BD 1063 is an inhibitor of σ receptors, which has been shown to have 50-fold higher affinity for σ-1 receptors relative to σ-2 receptors [32]. Application of 100 nM BD 1063, a concentration which would significantly affect σ-1 receptors while having negligible effects on σ-2 receptors [32], depressed the inhibitory effects of DTG by >50%. Taken together, our results suggest that σ-1 receptors couple to voltage-gated Na+ channels in intracardiac neurons.
Results from the present study demonstrate that sigma agonists inhibit voltage-gated Na+ currents in rat intracardiac neurons, which results in an increasing action potential threshold and a decrease action potential overshoot. In parasympathetic neurons, the inhibition of the action potential firing in response to depolarizing stimuli may be expected to reduce the bradycardia associated with vagal nerve stimulation in vivo, leading to tachycardia. A common side effect of general anesthetics, such as ketamine, is an increased heart rate [33]. It has been shown that ketamine inhibits Na+ channels in brainstem parasympathetic cardiac neurons [34], and some of the effects of ketamine have been recently attributed to activation of σ-1 receptors [35]. Thus, the ketamine-evoked decrease in parasympathetic input to the heart and concomitant increase the heart rate seen in surgical patients [34] may be the result of INa inhibition via the activation of σ-1 receptors in central and/or peripheral neurons. Our data suggest that the tachycardia produced by sigma ligands such as (+)-SKF-10047, DTG and (+)-3-[3-hydroxyphenyl]-N-(1-propyl) piperidine [36, 37] may be mediated in part by the inhibition of voltage-gated Na+ channels by sigma receptors in rat intracardiac neurons and the resulting decrease in parasympathetic input to the heart.
Acknowledgments
We thank Yelenis Herrera, Ph.D. and Nivia Cuevas, R.Ph. for comments on a draft of this manuscript. Grant support for this work was provided by AHA Greater Southeastern Affiliate Award Grant-In-Aid to J.C.
References
- 1.Moravec J, Moravec M. Intrinsic nerve plexus of mammalian heart: morphological basis of cardiac rhythmical activity? Int Rev Cytol. 1987;106:89–148. doi: 10.1016/s0074-7696(08)61711-8. [DOI] [PubMed] [Google Scholar]
- 2.Gagliardi M, Randall WC, Bieger D, Wurster RD, Hopkins DA, Armour JA. Activity of in vivo canine cardiac plexus neurons. Am J Physiol. 1988;255:H789–800. doi: 10.1152/ajpheart.1988.255.4.H789. [DOI] [PubMed] [Google Scholar]
- 3.Ardell JL. Structure and function of mammalian intrinsic cardiac neurons. In: Armour JAA JL, editor. In Neurocardiology. Oxford: Oxford University Press; 1994. pp. 95–114. [Google Scholar]
- 4.Zhang H, Cuevas J. Sigma receptors inhibit high-voltage-activated calcium channels in rat sympathetic and parasympathetic neurons. J Neurophysiol. 2002;87:2867–2879. doi: 10.1152/jn.2002.87.6.2867. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Cuevas J. Sigma receptor activation blocks potassium channels and depresses neuroexcitability in rat intracardiac neurons. J Pharmacol Exp Ther. 2005;313:1387–1396. doi: 10.1124/jpet.105.084152. [DOI] [PubMed] [Google Scholar]
- 6.Quirion R, Bowen WD, Itzhak Y, Junien JL, Musacchio JM, Rothman RB, Su TP, Tam SW, Taylor DP. A proposal for the classification of sigma binding sites. Trends Pharmacol Sci. 1992;13:85–86. doi: 10.1016/0165-6147(92)90030-a. [DOI] [PubMed] [Google Scholar]
- 7.Vilner BJ, Bowen WD. Modulation of cellular calcium by sigma-2 receptors: release from intracellular stores in human SK-N-SH neuroblastoma cells. J Pharmacol Exp Ther. 2000;292:900–911. [PubMed] [Google Scholar]
- 8.Bowen WD, Vilner BJ, Williams W, Bertha CM, Kuehne ME, Jacobson AE. Ibogaine and its congeners are sigma 2 receptor-selective ligands with moderate affinity. Eur J Pharmacol. 1995;279:R1–3. doi: 10.1016/0014-2999(95)00247-i. [DOI] [PubMed] [Google Scholar]
- 9.Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E, Glossmann H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci U S A. 1996;93:8072–8077. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Senda T, Matsuno K, Okamoto K, Kobayashi T, Nakata K, Mita S. Ameliorating effect of SA4503, a novel sigma 1 receptor agonist, on memory impairments induced by cholinergic dysfunction in rats. Eur J Pharmacol. 1996;315:1–10. doi: 10.1016/s0014-2999(96)00572-9. [DOI] [PubMed] [Google Scholar]
- 11.Matsumoto RR, Hemstreet MK, Lai NL, Thurkauf A, De Costa BR, Rice KC, Hellewell SB, Bowen WD, Walker JM. Drug specificity of pharmacological dystonia. Pharmacol Biochem Behav. 1990;36:151–155. doi: 10.1016/0091-3057(90)90141-4. [DOI] [PubMed] [Google Scholar]
- 12.McCracken KA, Bowen WD, Matsumoto RR. Novel sigma receptor ligands attenuate the locomotor stimulatory effects of cocaine. Eur J Pharmacol. 1999;365:35–38. doi: 10.1016/s0014-2999(98)00876-0. [DOI] [PubMed] [Google Scholar]
- 13.Ujike H, Kuroda S, Otsuki S. Sigma receptor antagonists block the development of sensitization to cocaine. Eur J Pharmacol. 1996;296:123–128. doi: 10.1016/0014-2999(95)00693-1. [DOI] [PubMed] [Google Scholar]
- 14.Snyder SH, Largent BL. Receptor mechanisms in antipsychotic drug action: focus on sigma receptors. J Neuropsychiatry Clin Neurosci. 1989;1:7–15. doi: 10.1176/jnp.1.1.7. [DOI] [PubMed] [Google Scholar]
- 15.Novakova M, Ela C, Barg J, Vogel Z, Hasin Y, Eilam Y. Inotropic action of sigma receptor ligands in isolated cardiac myocytes from adult rats. Eur J Pharmacol. 1995;286:19–30. doi: 10.1016/0014-2999(95)00424-j. [DOI] [PubMed] [Google Scholar]
- 16.Lishmanov Yu B, Maslov LN, Naryzhnaya NV, Tam SW. Ligands for opioid and sigma-receptors improve cardiac electrical stability in rat models of post-infarction cardiosclerosis and stress. Life Sci. 1999;65:PL13–17. doi: 10.1016/s0024-3205(99)00226-x. [DOI] [PubMed] [Google Scholar]
- 17.Novakova M, Bruderova V, Sulova Z, Kopacek J, Lacinova L, Kvetnansky R, Vasku A, Kaplan P, Krizanova O, Jurkovicova D. Modulation of expression of the sigma receptors in the heart of rat and mouse in normal and pathological conditions. Gen Physiol Biophys. 2007;26:110–117. [PubMed] [Google Scholar]
- 18.Hille B. Sunderland. MA: Sinauer Associates, Inc.; 2001. Ion Channels of Excitable Membranes. [Google Scholar]
- 19.Johannessen M, Ramachandran S, Riemer L, Ramos-Serrano A, Ruoho AE, Jackson MB. Voltage-gated sodium channel modulation by sigma-receptors in cardiac myocytes and heterologous systems. Am J Physiol Cell Physiol. 2009;296:C1049–1057. doi: 10.1152/ajpcell.00431.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams DJaC J. Electrophysiological properties of cardiac neurons-the foundation of neurocardiology. In: Ardell JAAa JL, editor. Basic and Clinical Neurocardiology. Oxford University Press; 2004. pp. 1–60. [Google Scholar]
- 21.Cuevas J, Adams DJ. Local anaesthetic blockade of neuronal nicotinic ACh receptor-channels in rat parasympathetic ganglion cells. Br J Pharmacol. 1994;111:663–672. doi: 10.1111/j.1476-5381.1994.tb14789.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cuevas J, Harper AA, Trequattrini C, Adams DJ. Passive and active membrane properties of isolated rat intracardiac neurons: regulation by H- and M-currents. J Neurophysiol. 1997;78:1890–1902. doi: 10.1152/jn.1997.78.4.1890. [DOI] [PubMed] [Google Scholar]
- 23.Locknar SA, Barstow KL, Tompkins JD, Merriam LA, Parsons RL. Calcium-induced calcium release regulates action potential generation in guinea-pig sympathetic neurones. J Physiol. 2004;555:627–635. doi: 10.1113/jphysiol.2003.059485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parsons RL, Barstow KL, Scornik FS. Spontaneous miniature hyperpolarizations affect threshold for action potential generation in mudpuppy cardiac neurons. J Neurophysiol. 2002;88:1119–1127. doi: 10.1152/jn.2002.88.3.1119. [DOI] [PubMed] [Google Scholar]
- 25.Hogg RC, Trequattrini C, Catacuzzeno L, Petris A, Franciolini F, Adams DJ. Mechanisms of verapamil inhibition of action potential firing in rat intracardiac ganglion neurons. J Pharmacol Exp Ther. 1999;289:1502–1508. [PubMed] [Google Scholar]
- 26.Xu ZJ, Adams DJ. Voltage-dependent sodium and calcium currents in cultured parasympathetic neurones from rat intracardiac ganglia. J Physiol. 1992;456:425–441. doi: 10.1113/jphysiol.1992.sp019344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franciolini F, Hogg R, Catacuzzeno L, Petris A, Trequattrini C, Adams DJ. Large-conductance calcium-activated potassium channels in neonatal rat intracardiac ganglion neurons. Pflugers Arch. 2001;441:629–638. doi: 10.1007/s004240000471. [DOI] [PubMed] [Google Scholar]
- 28.Horowitz LF, Hirdes W, Suh BC, Hilgemann DW, Mackie K, Hille B. Phospholipase C in living cells: activation, inhibition, Ca2+ requirement, and regulation of M current. J Gen Physiol. 2005;126:243–262. doi: 10.1085/jgp.200509309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barstow KL, Locknar SA, Merriam LA, Parsons RL. The modulation of action potential generation by calcium-induced calcium release is enhanced by mitochondrial inhibitors in mudpuppy parasympathetic neurons. Neuroscience. 2004;124:327–339. doi: 10.1016/j.neuroscience.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 30.DeHaven WI, Cuevas J. VPAC receptor modulation of neuroexcitability in intracardiac neurons: dependence on intracellular calcium mobilization and synergistic enhancement by PAC1 receptor activation. J Biol Chem. 2004;279:40609–40621. doi: 10.1074/jbc.M404743200. [DOI] [PubMed] [Google Scholar]
- 31.Soriani O, Vaudry H, Mei YA, Roman F, Cazin L. Sigma ligands stimulate the electrical activity of frog pituitary melanotrope cells through a G-protein-dependent inhibition of potassium conductances. J Pharmacol Exp Ther. 1998;286:163–171. [PubMed] [Google Scholar]
- 32.Matsumoto RR, Bowen WD, Tom MA, Vo VN, Truong DD, De Costa BR. Characterization of two novel sigma receptor ligands: antidystonic effects in rats suggest sigma receptor antagonism. Eur J Pharmacol. 1995;280:301–310. doi: 10.1016/0014-2999(95)00208-3. [DOI] [PubMed] [Google Scholar]
- 33.Hornchen U, Tauberger G. Investigations on the mechanism of the effects of ketamine (Ketanest) on circulation and respiration (au-thor's transl)] Anaesthesist. 1980;29:547–551. [PubMed] [Google Scholar]
- 34.Irnaten M, Wang J, Venkatesan P, Evans CK, Chang KS, Andresen MC, Mendelowitz D. Ketamine inhibits presynaptic and postsynaptic nicotinic excitation of identified cardiac parasympathetic neurons in nucleus ambiguus. Anesthesiology. 2002;96:667–674. doi: 10.1097/00000542-200203000-00024. [DOI] [PubMed] [Google Scholar]
- 35.Narita M, Yoshizawa K, Aoki K, Takagi M, Miyatake M, Suzuki T. A putative sigma1 receptor antagonist NE-100 attenuates the discriminative stimulus effects of ketamine in rats. Addict Biol. 2001;6:373–376. doi: 10.1080/13556210020077091. [DOI] [PubMed] [Google Scholar]
- 36.Wu KM, Martin WR. Cardiovascular and respiratory effects of an opioid kappa agonist ethylketazocine and sigma agonist Nallylnormetazocine in acutely decerebrated dogs. Pharmacol Biochem Behav. 1989;34:405–411. doi: 10.1016/0091-3057(89)90334-1. [DOI] [PubMed] [Google Scholar]
- 37.Lishmanov Iu B, Maslov LN, Krylatov AV, Ugdyzhekova DS, Gilligan P, Tam SW. The anti-arrhythmia and pro-arrhythmia properties of sigma-receptor ligands] Eksp Klin Farmakol. 2000;63:39–43. [PubMed] [Google Scholar]