

Abstract
Bone marrow-derived cells contribute to repair of injured tissue and to the maintenance of tissue homeostasis, but the extent to which perturbations of systemic homeostasis modulate this contribution is unknown. Accordingly, hematopoietic chimeras were used to determine contributions of bone marrow-derived cells to hepatocytes, skeletal muscle myocytes, and cardiomyocytes in healthy young, healthy old, and young obese diabetic mice. Mice with multiple genomic copies of a non-expressed β-globin/pBR322 sequence served as bone marrow donors. Because detection of the integrated sequence does not involve gene expression and many copies of the sequence are present, the sensitivity of detection is high and is not influenced by the state of cell differentiation. Our data indicate that bone marrow contributes a significant fraction of hepatocytes in old and diabetic mice, but half as many in young mice. They also show that bone marrow is a significant source of new cardiomyocytes at all ages and that this contribution is unaffected by diabetes. Additionally we found that bone marrow makes a substantial contribution to skeletal myocyte replacement that decreases with age. In summary, bone marrow-derived cells contribute significantly to normal non-hematopoietic cell replacement, a contribution that is altered by overall homeostatic state in a tissue specific manner. These data are significant if we are to understand if, and if so how, bone marrow-derived cell dysfunction contributes to tissue damage and senescence.
Keywords: Adult bone marrow stem cells, diabetes mellitus, aging, homeostasis, bone marrow cells, mice
Introduction
Numerous studies indicate that bone marrow (BM)-derived cells can differentiate into or fuse with non-hematopoietic cell types. Much of this work, in adult animals, has centered on BM-derived cell contributions to tissue repair. Less attention has been paid to their role in tissue homeostasis, with almost no consideration of how the systemic milieu might influence their behavior. Considering that the US population is rapidly aging, and that diabetes is epidemic, the influence of age and diabetes on BM-derived cell contributions to hepatocytes, skeletal myocytes, and cardiomyocytes are of particular interest. The liver plays a crucial role in maintaining homeostasis. It is among the tissues most compromised by aging [1, 2]. Furthermore, liver dysfunction, Type 2 diabetes and obesity are strongly linked [3]. In skeletal muscle, both aging and diabetes lead to changes in cellular metabolism that result in overproduction of reactive oxygen species [4, 5]. Aging also results in muscle loss and atrophy even when high activity levels are maintained [6, 7]. Heart function deteriorates in both aging and diabetes, and though many of these changes are due to extrinsic factors such as hypertension and atherosclerosis, myocardial dysfunction per se is also present [5, 8-10].
Mice whose bone marrow (BM) has been replaced with BM cells whose fate can be tracked, have made it possible to study the integration of BM-derived cells into non-hematopoietic tissues. Using such chimeras, we have demonstrated that ∼10% of endothelial cells in neovessels that developed in response to an injury were derived from BM [11]. Others have reported the contribution of BM to the endothelium in injured tissues ranging from less than 1% of endothelial cells to as high as 26% of vessels containing BM-derived cells [11-15]. In our earlier study, we also examined the role of BM cells in maintaining homeostasis in the pre-existing vasculature and found that ∼1% of endothelial cells in blood vessels were derived from the BM during a 4-month period following BM transplantation.
Since our earlier study, other reports on the role of BM cells in maintaining normal tissue homeostasis, including the liver, heart, and skeletal muscle have appeared. One examined the contribution of BM cells to the liver, and concluded that approximately 0.1% of hepatocytes were BM-derived 2-3 months after BM transplant in the absence of tissue injury, and that there was no evidence of cell fusion in the uninjured liver [16]. Two other reports emphasized the lack of a significant contribution of BM cells to the oval cell compartment, but are nevertheless consistent with a low level of BM cell contribution to hepatocytes and/or oval cells. In the first study, after many months of treatment to induce hepatocyte and oval cell turnover, it was found that hepatocytes were predominantly not of BM origin, and analysis of mice after secondary transplant indicated that <1% of liver repopulating cells were of BM origin [17]. The second study reported that 0.2%, and 0.1-0.7% of oval cells were of BM origin in uninjured and injured liver, respectively [18]. Overall estimates of BM contributions to the injured or transplanted liver range from <1% to 62%, but rejection status profoundly affects this number [19-23].
In skeletal and cardiac muscle, low levels of BM cell contribution to the healthy muscle have been reported, with no evidence of cell fusion in uninjured cardiomyocytes, skeletal myocytes, or hepatocytes, though it was observed in regenerating skeletal muscle [16]. A report by La Barge and Blau indicated that BM cells make a negligible contribution to uninjured skeletal muscle, but can contribute to injured muscle, and do so by first moving into the stem (satellite) cell niche and subsequently contributing nuclei to myofibers [24]. Estimates of BM-derived cardiomyocytes range from 0% to 20% [15, 25-30]. Laflamme et al reported an average of 0.04% host-derived cardiomyocytes in transplanted hearts, but in one patient with acute rejection, as many as 29% of myocytes in local “hot spots” were host-derived [31].
Some investigators suggest that apparent large contributions of BM cells to non-hematopoietic tissues are due to artifact. It is possible, however, that the failure to detect BM-derived cells in tissues is due to poor sensitivity. Down-regulation of reporter trans-genes or selective removal of transgenic cells may influence data, since even strain differences alter the expression of GFP transgenes [32]. In addition to technical factors, physiological differences probably contribute to the apparently contradictory results. Age, proliferative state, and the source of the donor cells as well as the methodology used to isolate and transplant them can impact results [33]. Moreover, within a strain significant differences in physiological characteristics may affect results. For example, we find that it is relatively simple to induce hindlimb ischemia in nu/nu mice from one vendor, but not in mice from a different vendor, and even mice from the same colony housed in two different locations exhibit different physiologic characteristics within weeks of separation. Thus, variations in the contribution of BM cells to tissues could represent real physiological differences and suggest that non-hematopoietic differentiation of BM-derived cells is exquisitely sensitive to perturbations of homeostasis.
In light of this, we investigated how the overall physiologic state of an animal can influence the ability of BM-derived cells to contribute to tissue homeostasis by examining the extent of BM cell differentiation and integration into tissues of young adult, old, and young adult obese diabetic mice maintained under identical conditions. Mice that carry multiple copies of β-globin/pBR322 sequences integrated into their genome [34] served as BM donors [11]. BM-derived cells can be identified after non-isotopic in situ hybridization to the integrated pBR322 sequences by the presence of an unambiguous dark nuclear dot. Because detection does not involve gene expression, possible effects of differentiation on marker expression are avoided.
Materials and methods
Mouse strains and chimeras
All animal procedures were approved by the University of Iowa Institutional Animal Care and Use Committee. Mice were all housed and bred in the same room. C57BL/6-Tg(ACTbEGFP)10sb/J mice heterozygous for ubiquitous expression of enhanced green fluorescent protein (EGFP) [35] were crossed with C57BL/6.Tg(β-globin/pBR322) mice carrying multiple copies of the β-globin/pBR322 gene integrated into their genome at a single locus [34]. F1 mice were backcrossed into C57BL/6.Tg(β-globin/pBR322) mice to obtain β-globin/pBR322+/+EGFP+/- mice. These mice, which served as BM donors, carry the Ly 5.2 antigen and are congenic with B6.SJL-PtrcaPep3b/Boy recipient mice who have the Ly 5.1 allotype [36]. The presence of Ly 5.2 made it possible to distinguish donor from recipient cells in the circulation, which is difficult to do by in situ hybridization due to cell detachment when probing blood smears. B6.Cg-m +/+ Leprdb recipients are obese and affected mice become hyperglycemic at 1- 2 months and progress to polyphagia, polydipsia, polyuria and overt diabetes over time. The mice are congenic with the donors and have the Ly 5.2 allotype, so assessment of blood chimerism was performed using EGFP analysis only.
Young (2-3 mo) and old (20-24 mo) B6.SJL-PtrcaPep3b/Boy (Ly 5.1+) mice or B6.Cg-m +/+ Leprdb (2-3 mo) mice were given 100mg/L neomycin (Sigma) and 10mg/L polymyxin-B sulfate (Sigma) in acidified water ad libitum for one week, then whole-body irradiated at 6 Gy and 4 hr later at 5 Gy to destroy their hematopoietic system. The BM of the irradiated mice was reconstituted via tail vein injection of 1×107 freshly harvested whole BM cells. Cells from young mice were injected into young mice (n=5), from old mice into old mice (n=5), and from young mice into diabetic mice (n=7). Twelve weeks later mice were anesthetized and an aliquot of blood removed for blood smears for Ly 5.2 histocompatibility antigen immunostaining (BD Pharmingen), or subjected to FACS sorting for EGFP expressing cells, or both procedures to determine percent chimerism. Next, mice were perfused with PBS and hindlimb hamstring muscles (semitendonosus, biceps femoris, and semimembranosus muscles), liver, and heart were harvested and fixed with methyl Carnoys overnight and then paraffin embedded. Tissue from C57BL/6.Tg(β-globin/pBR322) and B6.SJL-PtrcaPep3b/Boy mice were also collected as positive and negative β-globin/pBR322 controls, respectively.
Immunolabeling, in situ hybridization, and morphometry
Sections were immunolabeled with biotinylated antibodies to laminin (rabbit polyclonal, Sigma, St Louis) or desmin (mouse monoclonal, DAKO, Carpinteria, CA) for skeletal myocytes, CD45 (mouse monoclonal, clone 30F11, Pharmingen) for blood cells, desmin for cardiomyocytes, and hepatocyte specific antigen (mouse monoclonal, Cell Marque, Hot Springs, AK) for hepatocytes. Antibodies were detected by incubation with horseradish perox-idase-conjugated streptavidin (Vector Laboratories, Burlingame, CA) followed by detection with Vector Red (Vector Laboratories). In skeletal muscle, myocytes were identified as large striated cells surrounded by anti-laminin staining. Nuclei within the laminin rings were counted as myocyte nuclei. In other sections, large striated CD45 negative cells were counted as skeletal muscle myocytes. In the heart, large cells with striations, whose nuclei were surrounded by a small region of clear cytoplasm, and that were CD45 negative or desmin positive, were counted as cardiomyocytes. Hepatocyte-specific antigen positive cells with large round nuclei surrounded by extensive cytoplasm were considered to be hepatocytes. Potential Kupffer cells were identified as CD45 negative cells with oval shaped nuclei.
Immunolabeling was followed by non-isotopic in situ hybridization with a digoxygenin-labeled (Roche, Indianapolis, IN) pBR322 (New England BioLabs, Beverly, MA) probe and detection with 2,2'-diaminobenzidine as described [11]. Only cells with visible nuclei were counted. Nuclei were examined under high power and both the total number of hepatocytes, cardiomyocytes, and skeletal muscle myocytes and the number of each cell type positive for the pBR322 probe (i.e., brown nuclear dot) was counted. Five to fifteen 4 mm sections from different regions of each tissue were analyzed and 1200-3100 nuclei were counted for each tissue in each mouse. The corrected percent of blood-derived cells (%BMcor) was computed using the formula:
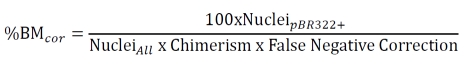 |
Where: False negative correction = Labeled nuclei / All nuclei in C57BL/6.Tg(β-globin/ pBR322); Chimerism = (Ly5.2+PBMC or EGFP+PBMC) / Total PBMCs; PBMC = peripheral blood mononuclear cell.
Y chromosome in situ hybridization
Sections (5 μm) of liver, heart, and skeletal muscle were deparaffinized, incubated in 8% sodium thiocyanate (10 min at 80°C), washed in PBS, digested with 4 mg/ml pepsin in 0.1M HCl (10 min at 37°C), then quenched in 0.2% glycine in 2X PBS. After 10 min post-fixation in 4% paraformaldehyde, tissues were washed and dehydrated through graded alcohols. Biotinylated Y chromosome paint (Open Biosystems, Huntsville, AL) was applied to sections, then denatured for 10 min at 60°C and hybridized overnight at 37°C. Sections were washed 5 min each in 50% formamide in 2X SSC at 37°C three times and then three times in 2X SSC. Finally, sections were washed in 0.05% Tween-20 in 4X SSC at 37°C for 10 min and rinsed in PBS. Sections were incubated in a 1:200 dilution of Alexa 488 streptavidin (Molecular Probes, Eugene, OR) for 1 hour at room temperature to visualize the probe. Sections were DAPI stained and coverslipped.
Sections were imaged using confocal microscopy with Z scan capture at 0.3 μm increments. Individual nuclei were examined throughout the entire nucleus to detect Alexa 488 labeled chromosomes. The number of positive signals per nucleus in at least 1000 nuclei per tissue was determined. Only labeled chromosomes in nuclei for which the entire nucleus could be visualized were counted.
Statistical analyses were performed for each tissue by One-way ANOVA followed by a Tukey's HSD post-hoc analysis for multiple simultaneous comparisons. P< 0.05 was considered statistically significant.
Results
Chimera characteristics
Percent hematopoietic chimerism was determined for all mice by either FACS analysis for EGFP fluorescence or Ly5.2 staining. Both EGFP and Ly5.2 staining were done for some mice, and results were similar for each approach. Overall, blood chimerism ranged from 46 to 89% (mean 67.2 + 5.3%). The final calculation of percent hematopoietic origin of different cell types was corrected for percent hematopoietic chimerism. A second correction factor was used to correct for the fact that histological sections do not always include the entire nucleus, so that the presence of the β-globin/pBR322 marker can be missed. False-negative rates of 2.4%, 3.2%, and 5.9% were found in liver hepatocytes, myocardium, and skeletal muscle, respectively, using tissue from non-chimeric mice homozygous for β-globin/pBR322. One thousand nuclei in each of the three cell types were examined in B6.SJL-PtrcaPep3b/Boy mice. No false positives were observed.
Liver
Sections of liver were immunolabeled with anti-hepatocyte specific antigen or anti-CD45 antibodies to delineate hepatocytes or blood cells, respectively, prior to pBR322 in situ hybridization. Because hepatocytes are morphologically quite distinct from other liver cells, neither of these labels provided additional accuracy in identification of hepatocytes but did show that BM-derived cells expressed he-patocyte-specific antigen. Nuclei with one or two clearly visible brown dots were identified as BM-derived cells. (Figure 1 A-B) Integration of BM-derived cells into hepatocytes was relatively low, but the percentage of BM-derived hepatocytes in old and diabetic mice was more than double that of young mice. (P<0.01) (Figure 2)
Figure 1.

Bone marrow-derived cells in quiescent tissue. Brightfield images of tissue sections after in situ hybridization with digoxygenin labeled pBR322 probe to detect bone marrow-derived cells in chimeric mice. A & B) Liver section of young (A) and diabetic (B) mouse methyl green stained to delineate nuclei. Arrows point to small black dots indicating hybridization to pBR322 probe to hepatocyte nuclei. Arrowhead in (B) shows labeled cell, possibly a Kupffer cell. C & D) Desmin labeled sections of skeletal muscle with labeled nuclei (arrows) in young mice. E & F) Desmin labeled sections of myocardium with labeled nuclei (arrows) in an old (E) and young (F) heart. Bar = 10 μm.
Figure 2.

Quantitation of BM-derived nuclei in quiescent hepatocytes, skeletal muscle, and cardiomyocytes in chimeric mice generated by lethally irradiating mice and replacing their BM with cells carrying multiple copies of pBR322 sequences in their genome. Young BM cells were transplanted into young mice (Y to Y), old cells into old mice (O to O), and young cells into obese diabetic mice (Y to D). The mean percentage and SEM of pBr322+ nuclei detected by in situ hybridization among the indicated cell types 12 weeks after bone marrow replacement is shown. Data were compared by One way ANOVA with Tukey's HSD post-hoc analysis.
*P<0.05 relative to all other groups.
Skeletal muscle
Sections of hamstring muscles (semitendonosus, biceps femoris, and semimembranosus muscles) were immunolabeled with antilaminin to delineate the endomysium of skeletal muscle fibers prior to in situ labeling to identify pBR322+ (i.e., BM-derived) cells. However, due to the close proximity of laminin to the satellite cell and myofiber nuclei, we were unable to unambiguously identify pBR322+ nuclei. Sections were instead labeled with anti-desmin to identify skeletal muscle followed by pBR322 in situ hybridization, which allowed us to identify pBR322+ nuclei. Only nuclei that were clearly associated with desmin positive cytoplasm were counted as (either satellite or differentiated) skeletal muscle cells. (Figure 1 C-D)
Although many thousands of nuclei were examined for each animal, only nuclei that could be unambiguously identified as skeletal muscle cells were counted. Nuclei with one or two clearly visible brown dots were identified as BM-derived. The mean percentage of BM-derived nuclei was greater than 3% in all groups, but the percentage in old mice was only 57% that of young mice (P<0.02) (Figure 2).
Cardiomyocytes
Sections of ventricular myocardium were immunolabeled with anti-desmin and anti-CD45 antibodies to delineate myocardial cells and blood cells, respectively, prior to pBR322 in situ hybridization to identify BM-derived cells. Anti-desmin positive cells with striations in the cytoplasm were considered cardiomyocytes. Difficulties in unambiguously identifying cells arise because occasionally blood cells with little cytoplasm intercalate between cardiomyocytes. If the cardiomyocyte nucleus is not in that particular section, these blood cell nuclei might be mistaken for a cardiomyocyte nucleus. Thus, anti-CD45 labeling was used so that blood cells were not inadvertently counted as cardiomyocytes. In eosin-stained and desmin-labeled sections, the cytoplasm around the nucleus of cardiomyocytes tends to be more lightly stained because myofibrils are thicker and more closely packed at the periphery of the fibers. This morphological feature was also used to assist in identifying cardiomyocytes. (Figure 1 E-F) Nuclei with one or two clearly visible brown dots were identified as BM-derived. As with skeletal myocytes, the percentage of BM-derived nuclei was surprisingly high, but was similar among all groups. (Figure 2).
Fusion and differentiation
BM-derived cells could have differentiated or fused with tissue resident cells. However, no bi-nucleate cardiomyocytes with pBR322+ nuclei were observed in any conditions. We also saw no pBR322+ nuclei in bi-nucleate hepatocytes. Skeletal muscle is multinucleate.
To determine if BM cell nuclei fused with tissue resident nuclei, the nuclei of male mice that received male bone marrow transplants were examined for the presence of two Y-chromosomes. Sections of liver, heart, and skeletal muscle were subjected to fluorescence in situ hybridization for Y-chromosomes and stained with DAPI to determine if a single or two Y-chromosomes were present. No nuclei containing two Y-chromosomes were detected in skeletal or cardiac muscle of chimeric or control male non-chimeric mice. We performed a similar analysis on hepatocytes, which are commonly polyploid. Many nuclei (3.4 - 26.6%) in both non-chimeric and chimeric mice contained multiple Y-chromosomes, but there was no relationship between the percentage of polyploid cells and whether or not a mouse was chimeric.
Discussion
Our data indicate that BM cells may play a significant role in maintaining tissue homeostasis, at least in some genetic backgrounds. BM cells infiltrate the heart, skeletal muscle, and liver, and once there, some assume a morphology and express antigens similar to those of cells in the local environment (i.e., cardiomyocytes, skeletal muscle myocytes, and hepatocytes, respectively). The contribution of BM cells to various tissues is modulated by systemic physiological changes brought about by both aging and diabetes. These changes are, however, tissue specific for each of these altered physiological states.
The BM cell contributions reported here are higher than in some other reports, probably in part because there is no reliance on gene expression to detect BM-derived cells. Detection of the male Y chromosome in sex-mismatched transplants has this advantage as well, but relies on detection of a single chromosome and has a false negative rate typically of 60%. In contrast, two chromosomes are targets in the β-globin/pBr322 BM cells, and our false negative rate ranged from 2.4-5.9% in this study [31]. Still, as noted above, the higher rates are more likely due to the biological characteristics of this system.
Liver
The liver is an organ capable of self-regeneration. Even under basal conditions, there is a constant balance between cell death and cell proliferation. Basal proliferation has been reported to be 12-25 fold lower in old than in young rats, while rates of hepatocyte cell death appear to be similar [2, 37-43]. Thus, homeostatic regulation of hepatocyte number through replication and cell death may become less competent with age, perhaps leading to a greater reliance on BM cells to maintain hepatocyte homeostasis. Our data indicate that BM-derived cells contribute a readily detectable fraction of hepatocytes superimposed upon regulation of hepatocyte number by regulation of hepatocyte proliferation and death. The physiological significance of the BM contribution to hepatocyte homeostasis awaits future studies examining the functional consequences of ablating the BM contribution to hepatocyte replacement. However, the increased fractional contribution of BM-derived hepatocytes in older animals and in young diabetic mice suggests that this contribution may have functional consequences.
Another potential explanation for the relative increase in BM-derived cells in the older and possibly in the diabetic animals is that postradiation liver damage is greater in these mice than in young mice after radiation. However, exogenous BM cells have been found to have very poor direct engraftment potential into non-hematopoietic tissue, so it is unlikely that the cells engrafted directly into the tissue. It is also improbable that the cells engrafted into the BM and moved back into the blood stream before repair of the radiation-induced liver damage was completed.
Skeletal muscle
We found that 5.1% of myonuclei were BM-derived after 12 weeks in young mice, suggesting that unlike the liver, BM-derived myocytes may significantly supplement myocytes derived from local proliferation even in young animals in skeletal muscle. Reported rates of BM-derived cell integration into skeletal muscle have varied from negligible to as many as 4.4% of myonuclei 2 months after BM transplantation and 5.5% after 3 months in young sarcoglycan deficient mice in another [24, 44]. Also, it was found that 1.7% of satellite cells were BM-derived at 3 months, and 1.3-2.6% of myonuclei were BM-derived at 6 months with GFP tracing [45]. Our results are in general agreement with the published reports that detected BM-derived cells in the muscle, and the higher contribution levels that we observe may result from using a more reliable and readily detected tracking system.
Satellite cell replicative capacity does not appear to be lost with age, but the fraction of satellite cell nuclei in muscle decreases by approximately 60% in old relative to young muscle [46-49]. If BM cells enter the muscle as satellite cells and replicate in the same way as endogenous satellite cells, one would expect a 60% loss in BM-derived cells in the muscle. Interestingly, we observed a 43% decrease in BM-derived myonuclei in the older mice. Of course, other mechanisms may account for the reduction in recruitment of BM cells to the muscle compartment.
Cardiomyocytes
Though recent evidence suggests that cardiomyocytes may proliferate, such proliferation in a non-ischemic undamaged heart is rare [25, 50]. Our findings that 5% of young and 6% of old cardiomyocytes are BM-derived indicate that BM cells might be the primary means of replenishing cardiomyocytes. We cannot distinguish whether they might do so by first intercalating into the myocardium as cardiomyocyte progenitors or via direct cardiopoiesis.
Myocyte loss in a healthy human heart is low but potentially significant, with about 0.025% of heart muscle being necrotic or apoptotic at any given time [51]. In diabetes, this number is increased approximately 4 fold to 0.1% [51]. Thus, though diabetic hearts may experience greater cell death, we find that they do not recruit BM cells at an increased rate. Unless cardiomyocytes are supplied by another source, this imbalance could have a significant impact on the myocardium, ultimately leading to hypertrophic myocytes in the diabetic heart.
Conclusion
BM cell contribution to non-hematopoietic lineages is readily detectable and the fractional extent of this contribution depends on the context and physiological status. We cannot conclude that BM cells are major players in maintaining tissue homeostasis in all species or individuals within a species. Nevertheless, our data indicate that they do play a significant role in normal tissue maintenance in mice, and that their contribution is affected by the overall physiological status of the organism.
Acknowledgments
Confocal imaging was performed at the University of Iowa Central Microscopy Facility, and mouse irradiation at the University of Iowa Radiation Biology Core.
References
- 1.Hall DM, Xu L, Drake VJ, Oberley LW, Oberley TD, Moseley PL, Kregel KC. Aging reduces adaptive capacity and stress protein expression in the liver after heat stress. J. Appl. Physiol. 2000;89:749–759. doi: 10.1152/jappl.2000.89.2.749. [DOI] [PubMed] [Google Scholar]
- 2.Zhang HJ, LX, VJ D, Xie L, Oberley LW, Kregel KC. Heat-induced liver injury in old rats is associated with exaggerated oxidative stress and altered transcription factor activation. Faseb J. 2003;17:226–250. doi: 10.1096/fj.03-0139fje. [DOI] [PubMed] [Google Scholar]
- 3.den Boer M, Voshol PJ, Kuipers F, Havekes LM, Romijn JA. Hepatic steatosis: a mediator of the metabolic syndrome. Lessons from animal models. Arterioscler Thromb Vasc Biol. 2004;24:644–649. doi: 10.1161/01.ATV.0000116217.57583.6e. [DOI] [PubMed] [Google Scholar]
- 4.Barazzoni R. Skeletal muscle mitochondrial protein metabolism and function in ageing and type 2 diabetes. Curr Opin Clin Nutr Metab Care. 2004;7:97–102. doi: 10.1097/00075197-200401000-00015. [DOI] [PubMed] [Google Scholar]
- 5.Rebrin I, Kamzalov S, Sohal RS. Effects of age and caloric restriction on glutathione redox state in mice. Free Radic Biol Med. 2003;35:626–635. doi: 10.1016/s0891-5849(03)00388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yarasheski KE. Exercise, aging, and muscle protein metabolism. J Gerontol A Biol Sci Med Sci. 2003;58:M918–922. doi: 10.1093/gerona/58.10.m918. [DOI] [PubMed] [Google Scholar]
- 7.Hunter GR, McCarthy JP, Bamman MM. Effects of resistance training on older adults. Sports Med. 2004;34:329–348. doi: 10.2165/00007256-200434050-00005. [DOI] [PubMed] [Google Scholar]
- 8.Young LH. Insulin resistance and the effects of thiazolidinediones on cardiac metabolism. Am J Med. 2003;115(Suppl 8A):75S–80S. doi: 10.1016/j.amjmed.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 9.Goldspink DF, Burniston JG, Tan LB. Cardiomyocyte death and the ageing and failing heart. Exp Physiol. 2003;88:447–458. doi: 10.1113/eph8802549. [DOI] [PubMed] [Google Scholar]
- 10.Takeda N, Nakamura I, Hatanaka T, Ohkubo T, Nagano M. Myocardial mechanical and myosin isoenzyme alterations in streptozotocin-diabetic rats. Jpn Heart J. 1988;29:455–463. doi: 10.1536/ihj.29.455. [DOI] [PubMed] [Google Scholar]
- 11.Crosby JR, Kaminski WE, Schatteman GC, Martin JC, Raines EW, Seifert RA, Bowen-Pope DF. Endothelial cells of hematopoietic origin make a significant contribution to adult blood vessel formation. Circ Res. 2000;87:728–730. doi: 10.1161/01.res.87.9.728. [DOI] [PubMed] [Google Scholar]
- 12.Jackson KA, Majka SM, Wang H, Pocius J, Hartley CJ, Majesky MW, Entman ML, Michael LH, Hirschi KK, Goodell MA. Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. J Clin Invest. 2001;107:1395–1402. doi: 10.1172/JCI12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, Wang J, Homma S, Edwards NM, Itescu S. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med. 2001;7:430–436. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- 14.Lagaaij EL, Cramer-Knijnenburg GF, van Kemenade FJ, van Es LA, Bruijn JA, van Krieken JH. Endothelial cell chimerism after renal transplantation and vascular rejection. Lancet. 2001;357:33–37. doi: 10.1016/S0140-6736(00)03569-8. [DOI] [PubMed] [Google Scholar]
- 15.Quaini F, Urbanek K, Beltrami AP, Finato N, Beltrami CA, Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Chimerism of the transplanted heart. N Engl J Med. 2002;346:5–15. doi: 10.1056/NEJMoa012081. [DOI] [PubMed] [Google Scholar]
- 16.Harris RG, Herzog EL, Bruscia EM, Grove JE, Van Arnam JS, Krause DS. Lack of a fusion requirement for development of bone marrow-derived epithelia. Science. 2004;305:90–93. doi: 10.1126/science.1098925. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Foster M, Al-Dhalimy M, Lagasse E, Finegold M, Grompe M. The origin and liver repopulating capacity of murine oval cells. Proc Natl Acad Sci U S A. 2003;100(Suppl 1):11881–11888. doi: 10.1073/pnas.1734199100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Menthena A, Deb N, Oertel M, Grozdanov PN, Sandhu J, Shah S, Guha C, Shafritz DA, Dabeva MD. Bone marrow progenitors are not the source of expanding oval cells in injured liver. Stem Cells. 2004;22:1049–1061. doi: 10.1634/stemcells.22-6-1049. [DOI] [PubMed] [Google Scholar]
- 19.Fujii H, Hirose T, Oe S, Yasuchika K, Azuma H, Fujikawa T, Nagao M, Yamaoka Y. Contribution of bone marrow cells to liver regeneration after partial hepatectomy in mice.[comment] Journal of Hepatology. 2002;36:653–659. doi: 10.1016/s0168-8278(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 20.Avital I, Feraresso C, Aoki T, Hui T, Rozga J, Demetriou AA, Muraca M. Bone marrow-derived liver stem cell and mature hepatocyte engraftment in livers undergoing rejection. Surgery. 2002;132:384–390. doi: 10.1067/msy.2002.125785. [DOI] [PubMed] [Google Scholar]
- 21.Wang X, Ge S, McNamara G, Hao QL, Crooks GM, Nolta JA. Albumin-expressing hepatocyte-like cells develop in the livers of immune-deficient mice that received transplants of highly purified human hematopoietic stem cells. Blood. 2003;101:4201–4208. doi: 10.1182/blood-2002-05-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Theise ND, Nimmakayalu M, Gardner R, Illei PB, Morgan G, Teperman L, Henegariu O, Krause DS. Liver from bone marrow in humans. Hepatology. 2000;32:11–16. doi: 10.1053/jhep.2000.9124. [DOI] [PubMed] [Google Scholar]
- 23.Petersen BE, Bowen WC, Patrene KD, Mars WM, Sullivan AK, Murase N, Boggs SS, Greenberger JS, Goff JP. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284:1168–1170. doi: 10.1126/science.284.5417.1168. [DOI] [PubMed] [Google Scholar]
- 24.LaBarge MA, Blau HM. Biological progression from adult bone marrow to mononucleate muscle stem cell to multinucleate muscle fiber in response to injury. Cell. 2002;111:589–601. doi: 10.1016/s0092-8674(02)01078-4. [DOI] [PubMed] [Google Scholar]
- 25.Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, Entman ML, Schneider MD. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12313–12318. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deb A, Wang S, Skelding KA, Miller D, Simper D, Caplice NM. Bone marrow-derived cardiomyocytes are present in adult human heart: A study of gender-mismatched bone marrow transplantation patients. Circulation. 2003;107:1247–1249. doi: 10.1161/01.cir.0000061910.39145.f0. [DOI] [PubMed] [Google Scholar]
- 27.Glaser R, Lu MM, Narula N, Epstein JA. Smooth muscle cells, but not myocytes, of host origin in transplanted human hearts. Circulation. 2002;106:17–19. doi: 10.1161/01.cir.0000021923.58307.8f. [DOI] [PubMed] [Google Scholar]
- 28.Muller P, Pfeiffer P, Koglin J, Schafers HJ, Seeland U, Janzen I, Urbschat S, Bohm M. Cardiomyocytes of noncardiac origin in myocardial biopsies of human transplanted hearts. Circulation. 2002;106:31–35. doi: 10.1161/01.cir.0000022405.68464.ca. [DOI] [PubMed] [Google Scholar]
- 29.Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, Rubart M, Pasumarthi KB, Virag JI, Bartelmez SH, Poppa V, Bradford G, Dowell JD, Williams DA, Field LJ. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature. 2004;428:664–668. doi: 10.1038/nature02446. [DOI] [PubMed] [Google Scholar]
- 30.Balsam LB, Wagers AJ, Christensen JL, Kofidis T, Weissman IL, Robbins RC. Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature. 2004;428:668–673. doi: 10.1038/nature02460. [DOI] [PubMed] [Google Scholar]
- 31.Laflamme MA, Myerson D, Saffitz JE, Murry CE. Evidence for cardiomyocyte repopulation by extracardiac progenitors in transplanted human hearts. Circ Res. 2002;90:634–640. doi: 10.1161/01.res.0000014822.62629.eb. [DOI] [PubMed] [Google Scholar]
- 32.Mothe AJ, Kulbatski I, van Bendegem RL, Lee L, Kobayashi E, Keating A, Tator CH. Analysis of green fluorescent protein expression in transgenic rats for tracking transplanted neural stem/progenitor cells. J Histochem Cytochem. 2005;53:1215–1226. doi: 10.1369/jhc.5A6639.2005. [DOI] [PubMed] [Google Scholar]
- 33.Theise ND. On experimental design and discourse in plasticity research. Stem Cell Rev. 2005;1:9–13. doi: 10.1385/SCR:1:1:009. [DOI] [PubMed] [Google Scholar]
- 34.Lo C. Localization of low abundance DNA sequences in tissue sections by in situ hybridization. Cell Science. 1986;81:143–162. doi: 10.1242/jcs.81.1.143. [DOI] [PubMed] [Google Scholar]
- 35.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 36.Morgan GM, McKenzie IF. Cell membrane alloantigenic determinants of several unusual mouse strains. J Immunogenet. 1980;7:393–399. doi: 10.1111/j.1744-313x.1980.tb00733.x. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Chong E, Herman B. Age-associated increases in the activity of multiple caspases in Fisher 344 rat organs. Exp Gerontol. 2002;37:777–789. doi: 10.1016/s0531-5565(02)00013-x. [DOI] [PubMed] [Google Scholar]
- 38.Ikeyama S, Wang XT, Li J, Podlutsky A, Martindale JL, Kokkonen G, van Huizen R, Gorospe M, Holbrook NJ. Expression of the proapoptotic gene gadd153/chop is elevated in liver with aging and sensitizes cells to oxidant injury. J Biol Chem. 2003;278:16726–16731. doi: 10.1074/jbc.M300677200. [DOI] [PubMed] [Google Scholar]
- 39.Higami Y, Shimokawa I, Okimoto T, Tomita M, Yuo T, Ikeda T. Effect of aging and dietary restriction on hepatocyte proliferation and death in male F344 rats. Cell Tissue Res. 1997;288:69–77. doi: 10.1007/s004410050793. [DOI] [PubMed] [Google Scholar]
- 40.Bralet MP, Branchereau S, Brechot C, Ferry N. Cell lineage study in the liver using retroviral mediated gene transfer. Evidence against the streaming of hepatocytes in normal liver. Am J Pathol. 1994;144:896–905. [PMC free article] [PubMed] [Google Scholar]
- 41.Magami Y, Azuma T, Inokuchi H, Kokuno S, Moriyasu F, Kawai K, Hattori T. Cell proliferation and renewal of normal hepatocytes and bile duct cells in adult mouse liver. Liver. 2002;22:419–425. doi: 10.1034/j.1600-0676.2002.01702.x. [DOI] [PubMed] [Google Scholar]
- 42.Sawada N, Ishikawa T. Reduction of potential for replicative but not unscheduled DNA synthesis in hepatocytes isolated from aged as compared to young rats. Cancer Res. 1988;48:1618–1622. [PubMed] [Google Scholar]
- 43.Roth SM, Martel GF, Ivey FM, Lemmer JT, Metter EJ, Hurley BF, Rogers MA. Skeletal muscle satellite cell populations in healthy young and older men and women. Anat Rec. 2000;260:351–358. doi: 10.1002/1097-0185(200012)260:4<350::AID-AR30>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 44.Lapidos KA, Chen YE, Earley JU, Heydemann A, Huber JM, Chien M, Ma A, McNally EM. Transplanted hematopoietic stem cells demonstrate impaired sarcoglycan expression after engraftment into cardiac and skeletal muscle. J Clin Invest. 2004;114:1577–1585. doi: 10.1172/JCI23071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dreyfus PA, Chretien F, Chazaud B, Kirova Y, Caramelle P, Garcia L, Butler-Browne G, Gherardi RK. Adult bone marrow-derived stem cells in muscle connective tissue and satellite cell niches. American Journal of Pathology. 2004;164:773–779. doi: 10.1016/S0002-9440(10)63165-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmalbruch H, Hellhammer U. The number of satellite cells in normal human muscle. Anat Rec. 1976;185:279–287. doi: 10.1002/ar.1091850303. [DOI] [PubMed] [Google Scholar]
- 47.Tome FM, Fardeau M. Nuclear changes in muscle disorders. Methods Achiev Exp Pathol. 1986;12:261–296. [PubMed] [Google Scholar]
- 48.Ishimoto S, Goto I, Ohta M, Kuroiwa Y. A quantitative study of the muscle satellite cells in various neuromuscular disorders. J Neurol Sci. 1983;62:303–314. doi: 10.1016/0022-510x(83)90207-1. [DOI] [PubMed] [Google Scholar]
- 49.Renault V, Thornell LE, Eriksson PO, Butler-Browne G, Mouly V, Thorne LE. Regenerative potential of human skeletal muscle during aging. Aging Cell. 2002;1:132–139. doi: 10.1046/j.1474-9728.2002.00017.x. [DOI] [PubMed] [Google Scholar]
- 50.Beltrami AP, Barlucchi L, Torella AR, Baker MB, Limana F, Chimenti S, Kasahara T, Rota M, Musso E, Kajstura J, Nadal-Ginard B, Anversa P. Adult Cardiac Stem Cells Are Multipotent and Support Myocardial Regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 51.Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B, Anversa P. Myocardial cell death in human diabetes. Circ Res. 2000;87:1123–1132. doi: 10.1161/01.res.87.12.1123. [DOI] [PubMed] [Google Scholar]