

Abstract
Wide-field temporal focusing is a novel technique that provides optical sectioning for imaging without the need for beam scanning. However, illuminating over large areas greatly reduces the photon density which limits the technique applicability to small regions, precluding functional imaging of cellular networks. Here we present a strategy that combines beam shaping and temporal focusing of amplified pulses (>1 µJ/pulse) for fast imaging of cells from the central nervous system in acute slices. Multiphoton video-rate imaging over total areas as wide as 4800 µm2 with an optical sectioning under 10 µm at 800 nm is achieved with our setup, leading to imaging of calcium dynamics of multiple cells simultaneously in thick tissue.
OCIS codes: (180.4315) Nonlinear microscopy, (070.6120) Spatial light modulators, (320.5540) Pulse shaping, (170.2655) Functional monitoring and imaging
1. Introduction
A major challenge toward understanding brain function is to succeed in monitoring the activity of individual cells, such as neurons, astrocytes, oligodendrocytes and microglia, which constitute the basic functional elements of the nervous system. As these cells operate in complex networks, it is also necessary to monitor their activity simultaneously over large areas. Furthermore, brain cells are highly ramified and can cover hundreds of micrometers. In the case of neurons, their complex dendritic arbor is thought to integrate numerous excitatory or inhibitory synaptic inputs in order to propagate information throughout the network. However, the rules that govern such integration are poorly understood because of the lack of approaches available to measure with high spatial and temporal resolution the responses of the entire dendritic arbor of a neuron to complex synaptic inputs. Understanding how individual neurons communicate and integrate information is important because such processes are believed to be misregulated in several brain disorders, making their study critical for developing therapeutic strategies.
Functional imaging of cellular activity requires both high temporal resolution and good spatial discrimination. The benefits of wide-field illumination and camera-based imaging for measuring network activity are important: multiple intercellular connections can be covered while the high number of CCD pixels makes efficient use of the illumination. Although standard wide-field illumination works well in cell cultures, for acute slices and in vivo preparations it demands optical sectioning to reject fluorescence from out-of-focus tissue. Several techniques exist to provide optical sectioning with wide-field illumination, but it usually comes at the price of temporal resolution. Post-processing techniques such as structured illumination [1] and HiLo microscopy [2] require multiple images to be acquired for each imaging plane. Light sheet illumination [3], either in one or- two-photon excitation, requires right-angle detection and agarfilled recording chambers that are not applicable to functional neuronal imaging. Fast scanning techniques such as multi-faceted polygonal mirrors, spinning disks and multi-foci illumination are not compatible with random-access. Acousto-optic deflectors add significant amount of complexity when working with femtosecond laser pulses [4]. Temporal focusing (TF) on the other hand allows optical sectioning with wide-field illumination making use of the intrinsic low efficiency of multiphoton absorption [5]. The principle of TF was first introduced in 2005 to provide optical sectioning to multiphoton excitation without the need of a scanning unit [5–7]. Briefly, a femtosecond pulse is temporally stretched prior to the objective and it is only at the focal plane that it is short enough to produce multiphoton excitation. However, as with other multiphoton widefield imaging schemes [8, 9], illuminating over large areas decreases considerably the photon density at the focal plane such that wide-field TF is often used for low intensity applications. Recently, TF was used as illumination source for multiphoton PALM microscopy [10] and for two-photon-activation of ChR2 [11, 12].
We overcome this limitation by using a regenerative laser amplifier as an illumination source. The extra intensity of the amplified pulses provides enough peak irradiance to extend the imaging area without affecting the temporal resolution. To get random-access over wide field-of-views, TF can readily be coupled with a spatial light modulator (SLM) at the microsope Fourier plane [13–16]. The combination of high intensity pulses TF with digital holography led to a truly wide-field and scanless multiphoton microscope exhibiting high optical sectioning at high imaging frame rate.
Here, we report the first use of TF combined with spatial light modulation to create a fast imaging modality for functional neuronal activity measurements. It is shown that out-of-focus fluorescence background rejection by TF of amplified pulses allows for neuronal kinetics measurements in thick samples. This is demonstrated with an improved signal-to-background ratio when monitoring the spontaneous variations in free calcium ion concentration in cells from neuronal acute slices.
2. Materials and methods
2.1. Microscopy setup for fluorescence imaging
The microscopy setup is shown in Fig. 1(a). The laser source is a regenerative laser amplifier (Coherent, Inc. RegA 9000, California) pumped by a Titanium:sapphire laser (Coherent, Inc. Mira 900-F, California). The RegA (800 nm, >1 µJ, <200 fs, 250 kHz) beam diameter is expanded to 1 cm to match the SLM active surface (Hamamatsu LCOS-SLM X10468-02, Japan). Because the device is a phase-only modulator, it has to be placed at a Fourier plane of the system. The incoming wavefront is phase-modulated by the SLM using a custom-made software that calculates the appropriate phase mask from a user-defined target intensity image. This calculation is done by means of an iterative Fourier transform algorithm described elsewhere [17]. Briefly, a reference image is acquired using a mercury arc lamp or white light. Regions-of-interest (ROIs) are user-selected and used in the algorithm to calculate the phase mask. The beam is Fourier-transformed by a 300 mm focal length achromatic lens (FF) onto a diffractive grating (DG) (1200 lines/mm, Edmund Optics NT43-210, New Jersey) placed at the conjugate plane of the objective. A block is placed little before the grating to remove the undiffracted beam (not shown). Because the zeroth order beam is at the center of the field-of-view, the beam block generates a dark area of 20 µm × 24 µm at the sample plane when imaged with a 40X magnification. The spatially dispersed wavelengths are then recombined by a telescope comprised of another 300 mm focal length achromatic lens (FG) and a microscope objective (Olympus LUMPlanFI/IR 40X/0.8W). Because the SLM and DG are diffractive elements, the laser intensity at the sample is 20% of the incident power. The RegA output is attenuated to 600 mW at most. The fluorescence from the sample is collected in epi-detection configuration and imaged with a commercial inverted microscope (Olympus IX-71, Japan) and an EMCCD camera (Princeton Instruments ProEM 512, New Jersey). The images are analyzed with FIJI software (http://pacific.mpi-cbg.de/).
Fig. 1.
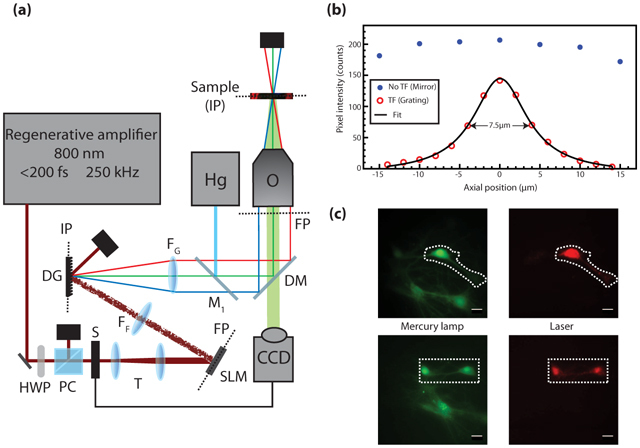
(a) Microscopy Setup. HWP: half-wave plate, PC: polarizer cube, S: camera-controlled shutter, T: telescope comprised of two achromatic lenses (f = −75 mm and +125 mm), SLM: spatial light modulator, FP: Fourier plane, FF: Fourier achromatic lens (f = 300 mm), DG: diffractive grating, IP: image plane, FG: achromatic lens for collimating the diffracted beam (f = 300 mm), Hg: mercury arc lamp, M1: Flip mirror, DM: excitation-detection dichroic filters, O: microscope objective, CCD: EMCCD camera. (b) Fluorescence intensity of a thin layer of fluorescein scanned through the focal plane with and without TF by replacing the DG by a mirror. (c) Fluorescence of live cultured cells loaded with Fluo-4 AM calcium indicator. (Green) Wide-field mercury arc lamp excitation (λexc = 450 nm, λem = 525 nm longpass), (Dashed line) targeted illumination, (Red) TF patterned illumination (λexc = 800 nm, λem = 535/40 nm). All scale bars are 20 µm.
A mercury arc lamp (100W, Olympus) is used for wide-field one-photon illumination experiments (Hg). The switch between modalities is done by a flip mirror (M1). The IX-71 microscope is readily coupled with a halogen lamp for white-light transmission microscopy (not shown on Fig. 1).
2.2. Slice preparation, immunohistochemistry and fluorophore loading
For immunohistochemistry, adult Sprague-Dawley rats were deeply anesthetized with a mixture of ketamine/xylazine (i.p.) and perfused transcardially with 4% paraformaldehyde in 0.1M phosphate buffer (PB), pH 7.4. Spinal cord was removed and postfixed in 4% paraformaldehyde in PB for 2 hours, rinsed and cut in 80 µm-thick parasagittal sections in a Vibratome (Leica). Immunohistochemistry was carried out by incubating spinal sections with rabbit anti-GFAP antibodies (1:1000, Dako), overnight at 4°C. Primary antibodies were visualized using goat anti-rabbit AlexaFluor 594 secondary antibodies (1:200, Molecular Probes) incubated for 2h at room temperature.
For calcium imaging, acute hippocampal slices (300 µm-thick) were prepared from 6–8 days-old Sprague-Dawley rats. After being anesthetized with isoflurane, animals were decapitated and their brains were quickly removed and placed into an ice-cold solution of oxygenated sucrose-rich oxygenated artificial cerebro-spinal fluid (aCSF) and sliced as described elsewhere [18]. Slices were allowed to rest at room temperature and recover for at least 1 hour before further handling.
Prior to calcium imaging experiments, a subregion to be observed within an hippocampal slice was loaded with the calcium indicator Fluo-4 AM by pressure ejection from a pipetter (concentration 0.9 µM, Invitrogen) (adapted from [19]). Fluo-4 fluorescence signal increases with the chelation of calcium ions. The slice was maintained, protected from light, in 3 ml of oxygenated aCSF imaging solution containing (in mM): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 MgCl2, 26 NaHCO3, 10 glucose and 2 CaCl2 (pH=7.4, 300 mosmol/l).
2.3. Ethics statement
All experiments were done in accordance with the Université Laval animal welfare guidelines.
3. Results
3.1. Optical sectioning capability and patterned illumination
In order to obtain images exhibiting high signal-to-background ratio when imaging thick sample, one needs to ensure that the fluorescence from out-of-focus planes is minimized. This is achieved here by means of temporal focusing. Optical sectioning characterization is done by scanning a thin layer (~1 µm) of fluorescein isothiocyanate (FITC) through the focal plane. The fluorescence intensity is then fitted by a Lorentzian function and the system optical sectioning is defined as the full-width at half-maximum of that function. With our TF parameters, the measured optical sectioning is 7.5 µm. As a comparison, canceling the TF by replacing the DG by a mirror shows little to no optical sectioning over the same scanning range, as shown in Fig. 1(b). Furthermore, because the maximum illumination area is limited by the available laser irradiance, the SLM is used to efficiently redistribute the light irradiance over the sample (Fig. 1(c)). Although the flatness of the intensity distribution in a ROI depends on the ROI size and lateral location [11, 20], the intensity variations inside the used ROI are not limiting for quantitative measurements since the fluorescence intensity is integrated over the ROI. Also, digital holography generates intensity inhomogeneities (speckles) that can correspond to light fluctuations on the order of 50%. As discussed in Sec. 4, those variations are fixed for a given phase mask and are thus not a problem for functional imaging.
3.2. Cellular imaging in thick rat spinal cord slice
To demonstrate the applicability of the technique to thick sample imaging, we measured a z-stack (15 optical planes separated by 5 µm) from astrocytes immunostained against glial fibrillary acidic protein (GFAP) in a fixed 80 µm thick rat spinal cord slice (described in Section 2.2). The immunostained astrocytes are visualized with an AlexaFluor 594 (Invitrogen) secondary antibody (Fig. 2). Standard one-photon fluorescence from a single optical plane is used as reference for phase mask calculation (Fig. 2(a), left). To appreciate the optical sectioning obtained with our setup, Fig. 2(b) shows the orthogonal view along the dashed line in Fig. 2(a) for one-photon imaging (left panel) and TF patterned illumination (right panel). In the one-photon experiment, fluorescence from the cell body is always present even if the astrocyte is not at the imaging plane (Fig. 2(b), left panel). Conversely, the same axial projection with the TF setup over an ROI defined by the SLM (4200 µm2) shows that two-photon excitation volume is confined to the image plane, effectively reducing out-of-focus background (Fig. 2(b), right panel). The average laser irradiance at the sample is 0.07 mW/µm2, corresponding to 140 GW/cm2 for a single 200 fs pulse. Furthermore, five different phase masks are averaged for each imaging plane in order to reduce the speckling effect of the SLM.
Fig. 2.
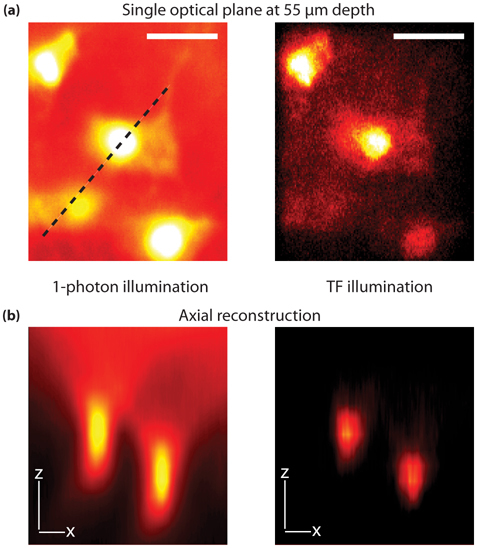
Comparison between one-photon and TF imaging of astrocytes in 80 µm spinal cord slice immunolabeled for glial fibrillary acidic protein (GFAP), revealed with AlexaFluo 594 secondary antibody. (a) Image of a single optical plane at 55 µm depth by one-photon (left, λexc = 450 nm, λem = 650/50 nm) and TF (right, λexc = 800 nm, λem = 650/50 nm) illumination. Total field-of-view is 4200 µm2. (b) Orthogonal view along the dashed line in (a) from a z-stack comprised of 15 images separated by 5 µm for one-photon (left) and TF (right) illumination. The axial reconstruction is done by FIJI software Volume Viewer plugin and with the Volume II interpolation to ensure continuity between imaging planes. The use of TF allows one to reduce the out-of-focus background. All scale bars are 20 µm.
3.3. Imaging of intracellular calcium dynamics in rat acute hippocampal slice
The major advantage of this approach is that it should allow high speed imaging of a neuronal network with high signal-to-background ratio in thick tissue. To test this, we compared the dynamics of Fluo-4 fluorescence from the same field-of-view measured either by standard one-photon or TF patterned illumination (Fig. 3). Spontaneous elevations in free calcium reflect electrical activity from the neurons. Prior to the imaging, Fluo-4 AM is loaded as described in Sec. 2.2 and then the cells are superfused with an oxygenated aCSF. A fluorescence image is then taken to identify neurons and draw an ROI around each soma. Under basal neuronal activity, the fluorescence is low, indicating low levels of intracellular free calcium from healthy neurons (Fig. 3(a)). The TF imaging experiment is done using three square illumination areas of 40×40 µm2 (total area of 4800 µm2) where healthy cells are identified (Fig. 3(b)). Large rectangular regions are used to image all cellular structures (cell bodies, axons, dendrites) as well as the surrounding background to provide context for the measurement. We used an average irradiance of 0.02 mW/µm2 (peak irradiance of 40 GW/cm2). The imaging workflow is as follows: an image is taken every 10 seconds with an exposure time of 30 ms for 15 minutes. Fig. 3(c) shows spontaneous calcium transients revealed by fluorescence variations (ΔF/F) from the same region and in one case from the same cell (ROI #2 and 6) using either one-photon (left) or TF illumination (right). The individual traces and averaged responses from 4 cells (Fig. 3(d)) show a ~3 fold larger ΔF/F when the measurements are made with TF illumination. These results indicate a significant benefit of reducing out-of-focus fluorescence with the optical sectioning of the TF-based illumination for functional measurements of neuronal activity in a brain slice.
Fig. 3.
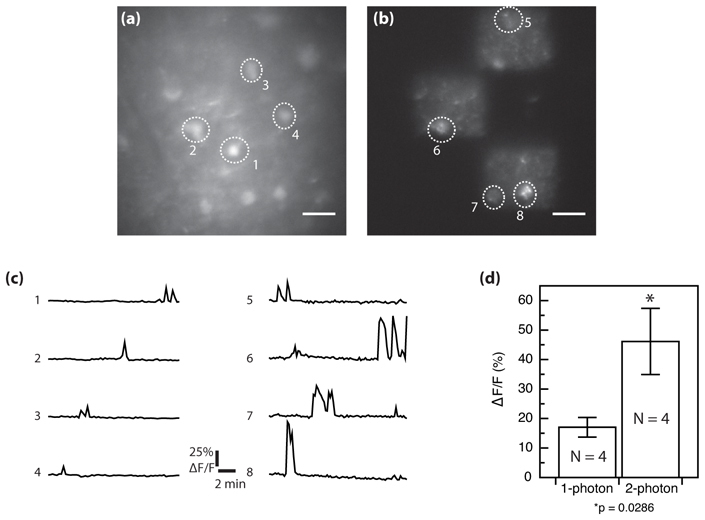
Comparison between one-photon and TF functional imaging in brain slice from two independent spontaneous activity measurements. Fluorescence from Fluo-4 AM calcium indicator loaded in a P8 300 µm rat hippocampal slice. (a) one-photon wide-field imaging. (b) Wide-field TF imaging of the same region with patterned illumination. Three square subregions were selected for illumination using the SLM. (c) Fluorescence fluctuations (ΔF/F) corresponding to intracellular free calcium elevation revealed by one-photon wide-field (left) and by TF patterned illumination (right). (d) Mean (± SEM) (ΔF/F) amplitudes from 4 cells for each imaging condition. Statistical test is Mann-Whitney and the difference between means is significant (p = 0.0286). Scale bars in (a) and (b) are 20 µm.
4. Discussion
We successfully overcome the low photon density in wide-field TF by using a regenerative laser amplifier producing >1 µJ pulses. Illumination with such high laser intensity allows for efficient two-photon absorption over large areas with commonly used biological fluorophores. In this case, with a maximum incident average power on the SLM of 600 mW, the largest illumination area obtainable with enough signal for 30 ms exposure time is 8100 µm2 at 40X magnification with a peak irradiance of 40 GW/cm2. In Fig. 2(b), the average laser irradiance at the sample was 0.07 mW/µm2 over the 4200 µm2 area. Compared to other custom-made scanning video-rate microscopes (1 mW/pixel, 16 pixels/µm2, 80 MHz, 200 fs) [21], the average irradiance is approximately 200 times lower, while the instantaneous irradiance is comparable (approximately 100 GW/cm2). Because the two-photon absorption depends on the square of the peak intensity, there is a real gain by diminishing the average irradiance at the sample and increasing the instantaneous intensity. The low repetition rate of the amplified system (250 kHz) also provides more dead time between pulses (1 pulse every 4 µs compared to 1 pulse every 12.5 ns for a 80 MHz Ti:Sapph) to let the photochemical system return to its fundamental state before the next pulse, hence reducing the photo- and thermal damage [22].
The coupling of an SLM with TF is essential to obtain a truly scanless microscope. Even with amplified pulses, the excitation field is limited to 90×90 µm2 for 30 frames-per-second (fps) imaging. Patterned illumination allows efficient random-access to the entire field-of-view defined by the maximum lateral size accessible by the SLM (180×180 µm2 with a 40X microscope objective) (equation 5 in [20]) without any moving parts.
The axial resolution of our setup (Fig. 1(b)) is slightly lower but of the same order as previously reported values [15, 16]. In order to decrease the out-of-focus excitation, one needs to increase the DG groove density or the objective NA. Nevertheless, Fig. 2 shows that much of the out-of-focus background present in standard wide-field imaging is absent with TF. Rejecting non-contributing noise is important when imaging changes of fluorescence as a reporter of cellular dynamics. Also, conversely to 3D structural imaging, functional imaging does not require thin optical section. The contributing signal occurs from all planes in cells that often span more than 10 µm. Imaging the whole cell body while rejecting the major out-of-focus background leads to better signal (Fig. 3(c) and 3(d)). Nevertheless, TF is essential and gives the flexibility to image large or small ROIs with sectioning.
Similarly, the transverse resolution is decreased by the laser speckles induced by the SLM. The pixelized phase-only modulator cannot perfectly shape the incident beam and random interference zones occur at the sample plane, leading to illumination hotspots. On the other hand, high frame rate transfer with CCD cameras required pixel binning, reducing the lateral resolution. Speckles are thus not a problem for high speed dynamics imaging, considering they are fixed for a given phase mask. If temporal resolution is not imperative, uniform illumination can easily be achieved by averaging speckle variations over time (rotating diffuser [15], phase mask shift-averaging [23]). Another technique is the generalized phase contrast method (GPC). TFGPC has recently been implemented for photostimulation patterns generation, where uniform illumination is essential for uniform stimulation [11].
The temporal resolution of the microscope is limited by the readout time of the camera. Figure 3 shows that sufficient fluorescence photons are available with 30 ms exposure time. Smaller ROI on the camera (50×50 µm2) can lead to imaging speed as high as 10 ms/frame (100 fps), which is more than enough to monitor fast cellular dynamics. Indeed, transient calcium signals in dendrites and spines are on the order of hundred milliseconds. Because these structures are spatially extended, network spatiotemporal dynamics are challenging to measure properly. By enabling multi-region imaging, our setup makes possible the measurements of the dynamics at multiple places simultaneously, providing contextual network information (Fig. 3(b), 3(c)).
5. Conclusion
Temporal focusing coupled with patterned illumination of high intensity pulses is a premium tool to assess fast functional biological questions in thick samples. Our optically-sectioned wide-field and random-access illumination setup provides the necessary temporal resolution for hundred milliseconds kinetics measurements in addition to providing contextual information. The technique is already used for calcium imaging and will be extended to voltage imaging. With proper stimulation protocols, this technique is very well adapted to assess dendritic integration. Whereas imaging individual action potential requires millisecond temporal resolution, the envelope of action potential bursts evoked in network communication can be followed with video-rate imaging [24]. Even for slower cellular dynamics imaging, less illumination time is beneficial. Fluorescence imaging can lead to phototoxicity so keeping excitation light as low as possible is crucial. Another use of the SLM is the holographic photolysis of caged neurotransmitters [17, 25]. However, it is important to note that the wavelength of the system is fixed at 800 nm and combined imaging and stimulation requires particular care and specific caged compounds. Nevertheless, by properly coupling contextual and high frame rate imaging with simultaneous multispine stimulations, it is expected that much will be learned about the dendritic role of synaptic activity integration.
Acknowledgments
We would like to thank Yves De Koninck for providing the amplified laser setup and the inverted microscope, Francine Nault for the preparation of rat hippocampal cultures, Hugues Dufour for helping in the development of the setup electronics and Steve Begin for critical reading of the manuscript. The research was supported by the Canadian Foundation for Innovation infrastructure fund, Discovery grants from the Natural Sciences and Engineering Research Council of Canada (NSERC, P.D.K. and D.C.). O. D.-T. and B.A. are supported by an NSERC training Program in Biophotonics. S.P. is supported by a Canadian Institute of Health Research (CIHR) training program in Neurophysics. P.D.K. is the holder of a CIHR Career Award and D.C. is the holder of a Canada Research Chair in Biophotonics.
References and links
- 1.Neil M. A., Juskaitis R., Wilson T., “Method of obtaining optical sectioning by using structured light in a conventional microscope,” Opt. Lett. 22(24), 1905–1907 (1997). 10.1364/OL.22.001905 [DOI] [PubMed] [Google Scholar]
- 2.Lim D., Chu K. K., Mertz J., “Wide-field fluorescence sectioning with hybrid speckle and uniform-illumination microscopy,” Opt. Lett. 33(16), 1819–1821 (2008). 10.1364/OL.33.001819 [DOI] [PubMed] [Google Scholar]
- 3.Palero J., Santos S. I. C. O., Artigas D., Loza-Alvarez P., “A simple scanless two-photon fluorescence microscope using selective plane illumination,” Opt. Express 18(8), 8491–8498 (2010). 10.1364/OE.18.008491 [DOI] [PubMed] [Google Scholar]
- 4.Saggau P., “New methods and uses for fast optical scanning,” Curr. Opin. Neurobiol. 16(5), 543–550 (2006). 10.1016/j.conb.2006.08.011 [DOI] [PubMed] [Google Scholar]
- 5.Oron D., Tal E., Silberberg Y., “Scanningless depth-resolved microscopy,” Opt. Express 13(5), 1468–1476 (2005). 10.1364/OPEX.13.001468 [DOI] [PubMed] [Google Scholar]
- 6.Zhu G. H., van Howe J., Durst M., Zipfel W., Xu C., “Simultaneous spatial and temporal focusing of femtosecond pulses,” Opt. Express 13(6), 2153–2159 (2005). 10.1364/OPEX.13.002153 [DOI] [PubMed] [Google Scholar]
- 7.Oron D., Silberberg Y., “Harmonic generation with temporally focused ultrashort pulses,” J. Opt. Soc. Am. B 22(12), 2660–2663 (2005). 10.1364/JOSAB.22.002660 [DOI] [Google Scholar]
- 8.Hwang J. Y., Moffatt-Blue C., Equils O., Fujita M., Jeong J., Khazenzon N. M., Lindsley E., Ljubimova J., Nowatzyk A. G., Farkas D. L., Wachsmann-Hogiu S., “Multimode optical imaging of small animals: Development and applications,” Proc. SPIE 6441, 644105–, 644105-10. (2007). 10.1117/12.711582 [DOI] [Google Scholar]
- 9.Hwang J. Y., Wachsmann-Hogiu S., Ramanujan V. K., Nowatzyk A. G., Koronyo Y., Medina-Kauwe L. K., Gross Z., Gray H. B., Farkas D. L., “Multimodal wide-field two-photon excitation imaging: characterization of the technique for in vivo applications,” Biomed. Opt. Express 2(2), 356–364 (2011). 10.1364/BOE.2.000356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaziri A., Tang J., Shroff H., Shank C. V., “Multilayer three-dimensional super resolution imaging of thick biological samples,” Proc. Natl. Acad. Sci. U.S.A. 105(51), 20221–20226 (2008). 10.1073/pnas.0810636105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.E. Papagiakoumou, F. Anselmi, A. B`egue, V. deSars, J. Gl¨uckstad, E. Y. Isacoff, and V. Emiliani, “Scanless two-photon excitation of channelrhodopsin-2,” Nat. Methods (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrasfalvy B. K., Zemelman B. V., Tang J., Vaziri A., “Two-photon single-cell optogenetic control of neuronal activity by sculpted light,” Proc. Natl. Acad. Sci. U.S.A. 107(26), 11981–11986 (2010). 10.1073/pnas.1006620107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oron D., Silberberg Y., “Spatiotemporal coherent control using shaped, temporally focused pulses,” Opt. Express 13(24), 9903–9908 (2005). 10.1364/OPEX.13.009903 [DOI] [PubMed] [Google Scholar]
- 14.Suchowski H., Oron D., Silberberg Y., “Generation of a dark nonlinear focus by spatio-temporal coherent control,” Opt. Commun. 264(2), 482–487 (2006). 10.1016/j.optcom.2006.02.064 [DOI] [Google Scholar]
- 15.Papagiakoumou E., de Sars V., Oron D., Emiliani V., “Patterned two-photon illumination by spatiotemporal shaping of ultrashort pulses,” Opt. Express 16(26), 22039–22047 (2008). 10.1364/OE.16.022039 [DOI] [PubMed] [Google Scholar]
- 16.Papagiakoumou E., de Sars V., Emiliani V., Oron D., “Temporal focusing with spatially modulated excitation,” Opt. Express 17(7), 5391–5401 (2009). 10.1364/OE.17.005391 [DOI] [PubMed] [Google Scholar]
- 17.Lutz C., Otis T. S., DeSars V., Charpak S., DiGregorio D. A., Emiliani V., “Holographic photolysis of caged neurotransmitters,” Nat. Methods 5(9), 821–827 (2008). 10.1038/nmeth.1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Topolnik L., Azzi M., Morin F., Kougioumoutzakis A., Lacaille J. C., “mGluR1/5 subtype-specific calcium signalling and induction of long-term potentiation in rat hippocampal oriens/alveus interneurones,” J. Physiol. 575(1), 115–131 (2006). 10.1113/jphysiol.2006.112896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.R. Yuste, and A. Konnerth, Imaging in Neuroscience and Development (CSHL Press, 2005). [Google Scholar]
- 20.Golan L., Reutsky I., Farah N., Shoham S., “Design and characteristics of holographic neural photo-stimulation systems,” J. Neural Eng. 6(6), 066004 (2009). 10.1088/1741-2560/6/6/066004 [DOI] [PubMed] [Google Scholar]
- 21.Veilleux I., Spencer J. A., Biss D. P., Cote D., Lin C. P., “In vivo cell tracking with video rate multimodality laser scanning microscopy,” IEEE J. Sel. Top. Quantum Electron. 14(1), 10–18 (2008). 10.1109/JSTQE.2007.912751 [DOI] [Google Scholar]
- 22.J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 3rd ed. (Springer, 2006). [Google Scholar]
- 23.Golan L., Shoham S., “Speckle elimination using shift-averaging in high-rate holographic projection,” Opt. Express 17(3), 1330–1339 (2009). 10.1364/OE.17.001330 [DOI] [PubMed] [Google Scholar]
- 24.Akemann W., Mutoh H., Perron A., Rossier J., Knöpfel T., “Imaging brain electric signals with genetically targeted voltage-sensitive fluorescent proteins,” Nat. Methods 7(8), 643–649 (2010). 10.1038/nmeth.1479 [DOI] [PubMed] [Google Scholar]
- 25.Nikolenko V., Watson B. O., Araya R., Woodruff A., Peterka D. S., Yuste R., “SLM microscopy: scanless two-photon imaging and photostimulation with spatial light modulators,” Front. Neural Circuits 2, 5 (2008). 10.3389/neuro.04.005.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]