

Abstract
A highly general, predictably selective C—H oxidation method for the direct, catalytic synthesis of complex allylic esters is introduced. This Pd(II)/sulfoxide-catalyzed method allows a wide range of complex aryl and alkyl carboxylic acids to couple directly with terminal olefins to furnish (E)-allylic esters in synthetically useful yields and selectivities (16 examples, E:Z ≥ 9:1) and without the use of stoichiometric coupling reagents or unstable intermediates. Strategic advantages of constructing allylic esters via C—H oxidation vs. C—C bond forming methods are evaluated and discussed in four “case studies”.
Introduction
Well-accepted bond forming strategies exist for the construction of heteroatom rich complex compounds through the coupling of simple pre-oxidized molecules. Inherently, oxygenated functional groups often require oxidation state changes and protection/de-protection sequences both to install and be compatible with further manipulations on the molecule. Selective C-H oxidation of pre-assembled hydrocarbon frameworks represents an alternative strategy for the rapid assembly of complex oxygen1,2 and nitrogen 1,3 rich structures at late stages of synthesis. When these reactions are predictably selective, mild, and incorporate the desired functionality without the need for further manipulation, unnecessary functional group manipulations (FGMs) can be bypassed, reducing synthetic steps and increasing overall yield.4
Complex Allylic Ester Synthesis
Esterification, one of the most important reactions in organic synthesis, involves coupling pre-oxidized carboxylic acid and alcohol fragments.5 Significant “synthetic overhead” is required to install these oxidized moieties in the correct oxidation state. Moreover, coupling generally involves stoichiometric amounts of a condensation reagent, or the generation of an activated, and often unstable, acid derivative. Although catalytic esterification methods exist, they suffer from limited scope and often require one coupling partner to be used in large excess.5 A catalytic, general esterification method that oxidatively couples a hydrocarbon with a carboxylic acid would be a significant advance.
Common approaches to linear (E)-allylic esters are shown in Figure 1. A Horner-Wadsworth-Emmons (HWE) or stabilized Wittig olefination approach generally involves taking a pre-oxidized starting material through a four step route: (1) aldehyde formation via oxidation [O] or reduction [H-], (2) olefination to form the α,β-unsaturated ester, (3) reduction to the allylic alcohol, and (4) acylation to obtain the target ester. If other functionality on the molecule is incompatible with this diverse set of conditions (i.e. oxidation, base, reduction), protecting group manipulations are also required. An alternative olefination strategy involves cross-metathesis of a terminal olefin with a pre-formed allylic ester.6 Although highly efficient, challenges associated with this route are predictable control of olefin geometry and the requirement for an excess of one of the olefin coupling partners to achieve high yields. Additionally, as in the HWE route, esterification often requires extensive screening of stoichiometric reagents, many of which generate waste that is difficult to remove from the product. We anticipated that the direct, catalytic coupling of terminal olefins with carboxylic acids via a predictably selective C—H esterification reaction would streamline the synthesis of certain (E)-allylic esters by minimizing the need for oxygenated intermediates.
Figure 1.

Common strategies for generating complex allylic esters.
Allylic C—H Esterification
In 2004, we first described a DMSO-promoted, Pd(OAc)2-catalyzed allylic C—H oxidation of α-olefins with solvent quantities of acetic acid (AcOH) to furnish linear (E)-allylic acetates with high regio- and stereoselectivities and outstanding functional group tolerance (Figure 2A).2a As synthetic intermediates acetates often serve as precursors to more complex esters via the intermediacy of alcohols. Ideally, any carboxylic acid could be directly incorporated into the hydrocarbon framework via C—H oxidation to furnish complex esters. Although this allylic C—H acetoxylation method has since been explored extensively by other researchers with respect to ligands, oxidants, and activators, 2f, 2h–j no general linear allylic esterification method has emerged. In a preliminary study directed towards streamlining polyol synthesis, we developed specific conditions to couple p-anisic acid and a chiral homoallylic ether to directly furnish a hexose precursor (Figure 2B).4b Unfortunately, these conditions did not prove to be general (vide infra). In this article, we describe a general reaction manifold for the linear allylic C—H oxidation reaction (LAO) that enables coupling of a wide range of carboxylic acids and α-olefins to furnish complex linear (E)-allylic ester products (Figure 2C).
Figure 2.

Progress toward a general allylic C—H esterification protocol
Results and Discussion
Design Principles
Our working mechanistic hypothesis for the Pd(OAc)2/DMSO catalyzed LAO reaction suggests that a carboxylate counterion on the palladium is needed to effect C—H cleavage to form a π-allylPd intermediate and that high concentrations of DMSO and AcOH are optimal for effecting functionalization.7 Within this original reaction manifold, challenges encountered in expanding the scope of the LAO to a general esterification method included: (1) formation of allylic acetate byproducts from the Pd(OAc)2 catalyst, (2) requirement for high equivalents of carboxylic acid, and (3) poor solubilities of many carboxylic acids in DMSO. Switching to a Pd(CH3CN)4(BF4)2 catalyst that can undergo counterion exchange with the carboxylic acids eliminated acetate by-products. The introduction of N,N-diisopropylethylamine (DIPEA), believed to promote functionalization through deprotonation of the acid, enabled lowering the amount of carboxylic acid from solvent quantities to 1.5–3 equiv.3,4b Increasing the amount of DIPEA from 50 to 70 mol% and lowering the amount of DMSO to 1.4 equiv. made possible using more CH2Cl2 as a solvent to improve solubility of both the α-olefin and the carboxylic acid. Collectively, these changes significantly widened the range of complex allylic esters that could be generated via C—H oxidation.
Reaction Scope
The expansive scope and streamlining potential of this methodology is represented in the construction of known allylic ester intermediates (Table 1). Oxidative coupling of unsaturated aryl acids and α-bromo-carboxylic acids with allyl arenes provides a direct and modular route to compounds 18 and 29 (entries 1–5), some of which have been shown to exhibit antibacterial and antifungal activities8 (entry 1). It is notable that 1.5 equivalents of acid can be used with only a moderate reduction in isolated yield (entry 2). This feature of the reaction is particularly significant when using complex carboxylic acids that require lengthy synthetic sequences to prepare (vide infra). A decrease in the reaction time (72h → 24h) and catalyst loadings (10 mol% → 5 mol%) is possible while maintaining synthetically useful yields (entries 3 and 4, respectively). It is interesting to note that under these modified conditions even fatty acids, insoluble at high concentrations of DMSO, are useful functionalization reagents (entry 6).10
Table 1.
Linear allylic oxidation (LAO) for the construction of known allylic ester intermediates.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Isolated Yielda | E:Zb | L:Bb (crude) | steps | ||
| C-H | C-C | |||||
| 1 |
1 |
70 % | >20:1 | >20:1 | 1 | 3 ref. 8 |
| 2 | 61 %c | >20:1 | >20:1 | |||
| 3 | 53 %d | >20:1 | >20:1 | |||
| 4 | 65 %e | >20:1 | >20:1 | |||
| 5 |
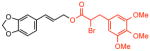 2 |
68 % | 16:1 | >20:1 | 2 | 5 ref. 9 |
| 6 |
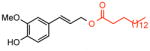 3 |
64 % | >20:1 | >20:1 | 1 | 3 ref. 10 |
| 7 |
4 |
60 % | 11:1 | 6:1 | 2 | 4 ref. 11 |
| 8 | 31 % | 10:1 | 9:1 | |||
| 9 |
5 |
50 % | 11:1 | 9:1 | 2 | 4 ref. 12 |
| 10 | 23 % | 7:1 | 11:1 | |||
| 11 |
(+)-6 |
54 %c | 17:1h | >20:1i | 2 | 2 ref. 14 |
| 12 |
(+)-7 |
72 % | 18:1h | >20:1 | 2 | 4 ref. 15 |
Isolated yields of >20:1 LB. Unless otherwise noted, E:Z does not change after purification.
crude values by 1H NMR.
1.5 equiv. acid.
24 h reaction time.
5 mol % Pd[CH3CN]4(BF4)2.
using previously published conditions, ref. 4b.
THP protected version of this compound was made.
determined after methanolysis and acetylation by 1H NMR.
determined by 1H NMR of purified material.
Bi-functional allylic esters 411 and 5 12 have been synthesized via N,N-dicyclohexyl carbodiimide/4-dimethylaminopyridine (DCC/DMAP) coupling of the respective carboxylic acids and mono-protected (E)-pent-2-en-1,5-diol (synthesized via a HWE route, Table 1, entries 7 and 9). Although DCC is one of the most widely used coupling reagents to form esters in organic synthesis, it is a potent skin irritant and generates by-products (dicyclohexylurea) that are relatively insoluble and difficult to remove.13 In contrast, direct, oxidative coupling of the same carboxylic acids with silyl-protected pentenol afforded 4 and 5 in good yields, half the number of steps, and without the use of any stoichiometric condensing reagents (entries 7 and 9). We have found that the reduced form of the quinone oxidant is easily removed via aqueous, basic extraction during the workup procedure (see Experimental). Significantly, reactions run under the previously reported conditions for benzoylation (Fig. 2B) resulted in significantly lower yields. (Table 1, entries 8 and 10).
Both α- and β-amino acids can be used as coupling partners in the oxidative esterification reaction without any epimerization. It is significant to note that t-butyl ester (+)-6 can be synthesized via a C—C bond-forming route with equal efficiency to the C—H oxidation route by alkylating symmetric dibromo-2-butene with the enolate of acetic acid t-butyl ester (Table 1, entry 11).14 However, when a more complex ester is required, for example to afford orthogonally protected aspartate (+)-7,15 the C—H esterification reaction enables a significant streamlining of the route (entry 12). It is notable that in all cases examined, the E:Z selectivity of this C—H esterification reaction does not drop below 10:1.
Streamlining Synthesis
A series of case studies were undertaken to evaluate the strategic advantages of constructing complex allylic esters via C—H oxidation routes versus C—C bond forming routes. A total synthesis of (−)-lepadiformine features an (E)-allylic ester intermediate (−)-8 that undergoes diastereoselective amino acid enolate Claisen rearrangement followed by a ring closing metathesis to forge the complex tricyclic backbone (Scheme 1).16 Starting from a fully oxidized starting material 10 (1,4-butane-diol), allylic ester (−)-8 was generated through a classic series of reactions: monoprotection, oxidation, HWE olefination, reduction, and DCC-mediated esterification with cyclic amino acid (−)-9. This route is reliable and generally high-yielding; however, it requires 5 steps, 2 of which are oxidation state manipulations. Direct installation of the (E)-allylic ester from the catalytic coupling of a terminal olefin and carboxylic acid affords a dramatic streamlining effect on this route. Using 1.5 equivalents of the same cyclic amino acid (−)-9, oxidative C—H esterification of TBDPS-protected 5-hexen-1-ol 13 provided (−)-8 in only 2 steps and 50% overall yield. The importance of using fragment coupling quantities of reagents is underscored here, as an 8-step sequence is required to synthesize carboxylic acid (−)-9.
Scheme 1.

C—H oxidation vs C—C bond forming strategies for the synthesis of key linear allylic ester intermediate (−)-8 in the synthesis of (−)-lepadiformine.
Synthetic routes for chiral molecules are often driven by practical considerations of availability of chiral starting material. In addition to providing more expedient routes to (E)-allylic esters, a C—H oxidation approach expands the options for using simpler chiral starting materials by minimizing total oxygenation. Chiral (E)-allylic ester (−)-14, an intermediate en route to (−)-laulimalide, was previously obtained from a highly oxygenated chiral pool material (S)-β-hydroxy-γ-butyrolactone (−)-15.17 Manipulation of the pre-installed oxygen functionality to arrive at the desired structure required a six-step HWE route comprised of 2 protections, serial reductions, and esterification (Scheme 2). In contrast, C—H esterification is a simplifying transform that enables targeting less oxygenated intermediates. For example, precursors for LAO, optically enriched bis-homoallylic alcohols, can be directly accessed via allylation of chiral terminal epoxides that are now readily available via hydrolytic kinetic resolution (HKR) methodology.18 Thus, an alternative approach to (−)-14 starts with allylation and protection of commercially available tert-butyldimethylsilyl (S)-(+)-glycidyl ether to afford enantiomerically enriched bis-homoallylic ether (−)-18 in just 2 steps. Benzoic acid was used as the coupling partner to afford the desired (E)-allylic ester (−)-14 in half the number of steps (3 steps) and comparable overall yield to the olefination route (Scheme 2).
Scheme 2.

C—H oxidation vs C—C bond formation routes in the synthesis of linear E-allylic ester intermediate (−)-14 en route to (−)-laulimalide.
While terminal olefins may also be used as intermediates in olefination sequences, FGMs are generally required. To illustrate this point, we compare a C—H oxidation strategy to an olefination strategy to allylic ester (±)-19, which serves as a precursor to trans-fused polycyclic ethers in brevetoxins (Scheme 3).19 Both routes start with alkylation and protection of cyclohexene oxide to rapidly afford homologous terminal olefin intermediates 21 and 24. The established HWE sequence to allylic alcohol 22 requires that the terminal olefin be oxidatively cleaved to afford the aldehyde precursor for olefination followed by reduction of the allylic ester to the desired oxidation state. The resulting (E)-allylic alcohol 22 was subsequently coupled to an acylchloride to form the desired (E)-allylic ester (±)-19 in a total of 6 steps and 19% overall yield (Scheme 3). In the C—H esterification route, olefin 24 can be coupled directly to 2,4-dichlorobenzoic acid to furnish desired product (±)-19 in a total of only 3 steps and 47% overall yield (Scheme 3).
Scheme 3.

C—H oxidation vs C—C bond forming routes proceeding via analogous terminal olefin intermediates.
An olefination strategy that is analogous to a C—H oxidation strategy for the construction of (E)-allylic esters is cross metathesis. These C—C bond forming routes are expedient because they also utilize terminal olefins and install the desired oxygen moiety directly without further manipulation (Figure 1). Challenges associated with this method center around the ability to control and predict E:Z selectivity of the newly formed internal olefin. In contrast, linear C-H oxidation methodology under these mild conditions generates E-allylic oxygenates with selectivities that are 10:1 or higher. Both the olefination and C—H oxidation routes to macrocyclic lactam (+)-25, a peptidomimetic, began with alkylation of macrocyclic amide 26 to furnish homologous compounds 27 and 30 (Scheme 4).20 Allylation of the amino acid Boc-L-phenylalanine via DIC-mediated esterification was required to furnish metathesis coupling partner (−)-29. Cross-metathesis coupling of allylated compounds 27 and (−)-29 (2 equiv.) provided phenylalanine derived macrocycle (+)-25 in 28% overall yield with an E:Z selectivity of 1.2:1. Using a linear C—H esterification strategy, direct C—H esterification of 30 with 1.5 equiv. of commercial Boc-L-phenylalanine (+)-28 furnished (+)-25 in 40% overall yield with an E:Z selectivity of 17:1 (Scheme 4).
Scheme 4.

C—H oxidation vs cross-metathesis route for the formation of complex allylic esters.
Allylic C—H Acetoxylation Revisited
Although our previously reported allylic C—H acetoxylation reaction showed broad functional group tolerance, acid sensitive substrates were not well tolerated under those conditions which employed solvent quantities of AcOH. We hypothesized that these new conditions employing only 1.5–3.0 equiv. of carboxylic acid and 70 mol% base may further expand the scope of this powerful transformation (Table 2).
Table 2.
Linear allylic C—H acetoxylation (LAO) with acid sensitive substrates.
 | |||||
|---|---|---|---|---|---|
| Entry | Product | Conditions | Isolated Yieldc | E:Zd | L:B (crude)d |
| 1 |
31 |
old | 51 % | 8:1 | 12:1 |
| 2 | new | 75 % | 9:1 | >20:1 | |
| 3 |
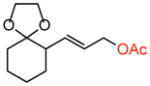 32 |
old | 50 %e | >20:1 | >20:1 |
| 4 | new | 64 % | >20:1 | >20:1 | |
| 5 |
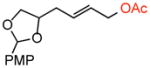 33 |
old | <5 % | --- | --- |
| 6 | new | 53 % | 12:1 | 12:1 | |
| 7 |
34 |
old | 17 % | 8:1 | 4:1 |
| 8 | new | 58 % | 12:1 | 8:1 | |
| 9 | oldf, g | 7 % | 5:1 | 3:1 | |
| 10 |
35 |
old | 32 % | 8:1 | 4:1 |
| 11 | new | 69 % | 11:1 | 10:1 | |
Pd(OAc)2 (10 mol %), DMSO: AcOH (1:1; 0.17M), 4Å MS, BQ (2 equiv.), air, 41°C, 72h.
Pd[CH3CN]4(BF4)2 (10 mol%), DMSO:CH2CI2(1:5; 2.0M), AcOH (3 equiv.), 4Å MS, PhBQ (2 equiv.), air, 41°C, 72h.
isolated yields of >20:1 L:B. Unless otherwise noted, E:Z does not change after purification.
crude values by 1H NMR
Previously reported, see ref. 4a.
Pd(OAc)2 (10 mol%), DMSO (0.17M), AcOH (3 equiv.), 4Å MS, BQ (2 equiv.), air, 41 °C, 72h.
When the reaction was run under the “old” conditions at 0.33M DMSO, 3 equiv. AcOH only trace reactivity was observed: 10% yield.
Terminal olefin substrates containing moderately acid-sensitive moieties such as primary tert-butyl N-tosyl carbamates and ketals showed improvements in yield (51% → 86%; 50% → 63%, respectively) with no erosion of selectivities under the new conditions (Table 2, entries 1 and 2). However, substrates containing highly acid-sensitive functionality, i.e. primary TBS ethers, triphenylmethyl (Tr) ethers, and p-methoxybenzyl (PMB)-acetals, all showed significant improvements in isolated yields (Table 2, entries 5–8, 10–11). Significantly, when AcOH loadings were reduced to 3 equiv. under the original reaction conditions, only trace reactivity was observed (Table 2, entry 9).
Conclusion
In summary, this study introduces the first general, predictably selective C—H oxidation method for the direct synthesis of complex allylic esters. The ability to forge esters using a catalytic method that couples two highly stable compounds, carboxylic acids and terminal olefins, provides an attractive alternative to methods that use stoichiometric amounts of coupling reagents or require reactive, unstable intermediates. The milder conditions that employ low loadings of carboxylic acid and catalytic base also enable broadening the substrate scope of the allylic C—H acetoxylation reaction to include acid-sensitive moieties. Strategic as well as practical advantages emerge when comparing C—H oxidation versus C—C bond forming routes for the synthesis of complex allylic esters. Introduction of oxygen functionality late in a sequence, without the need for further manipulation, provides a significant streamlining of the route by eliminating FGMs such as oxidation state changes, protection/deprotection sequences, and functional group transformations. Moreover, the ability to utilize simpler, less oxygenated intermediates expands the options with respect to chiral starting materials, often leading to more efficient routes. Based on the generality and predictable selectivity of this C—H oxidation method along with the strategic advantages it enables, we anticipate that it will find widespread use in complex molecule syntheses.
Experimental Procedures
A typical procedure for the Pd mediated oxidation of terminal olefins to linear E-allylic esters is described. To a 4 mL borosilicate vial was first added Pd(CH3CN)4(BF4)2 (44.4 mg, 0.1 mmol, 10 mol %) under argon atmosphere. The following reagents were then added in one portion under ambient atmosphere: phenyl benzoquinone (368 mg, 2.0 mmol, 2 equiv.), carboxylic acid (3.0 mmol, 3.0 equiv.), two 4Å molecular beads (50 mg). Finally, DMSO (100 μL, 1.4 mmol, 1.4 equiv.), CH2Cl2 (500 μL), and DIPEA (121.0 μL, 0.7 mmol, 0.7 equiv.) were added sequentially via glass syringe followed by a Teflon© stir bar. This solution was stirred at 41 °C for 5 minutes before starting material (1.0 mmol, 1.0 equiv.) was added neat. The vial was then capped and stirred at 41 °C for 72 hours. Upon completion as determined by NMR, the reaction was transferred to a separatory funnel using minimal methylene chloride (~2 mL) [Note 1]. The solution was diluted with diethyl ether (50 mL) and washed with 5% K-2CO3 (aq.) solution twice [Note 2]. The organic layer was dried with MgSO4, filtered, and reduced in vacuo. Purification was achieved via flash silica gel chromatography. Notes: (1) The excess quinone may be reduced by addition of solid Na2SO3 (2 g) to a reaction mixture diluted with 50 mL of EtOAc and 50 mL of a 5% K2CO3 (aq.) solution. The resulting biphasic mixture is then stirred rapidly for 30 minutes before continuing with the extraction. (2) If an inseparable emulsion forms, filter the solution through a pressed pad of celite with 20–30 mL diethyl ether.
Full experimental details and characterization data are given in the Supporting Information.
Supplementary Material
Acknowledgments
Financial support was provided by NIH/NIGMS (grant no. GM076153) and kind gifts from Pfizer, Eli Lilly, Bristol-Myers Squibb, and Amgen. We thank Johnson Matthey for a generous gift of Pd(OAc)2. We thank P. Gormisky for checking our experimental procedure.
Footnotes
Supporting Information Available: Experimental procedures, full characterization, and additional experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.General C—H oxidation, aminations, alkylations: Muller P, Fruit C. Chem Rev. 2003;103:2905. doi: 10.1021/cr020043t.Espino CG, DuBois J. In: Modern Rhodium-Catalyzed Organic Reactions. Evans PA, editor. Wiley-VCH; Weinheim: 2005. p. 379.Dick AR, Sanford MS. Tetrahedron. 2006;62:2439. references therein.Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:174. doi: 10.1021/cr0509760.Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597.Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485.Lewis JC, Bergman RG, Ellman JA. Acc Chem Res. 2008;41:1013. doi: 10.1021/ar800042p.Young AJ, White MC. J Am Chem Soc. 2008;130:14090. doi: 10.1021/ja806867p.Lafrance M, Lapointe D, Fagnou K. Tetrahedron. 2008;64:6015.Chen X, Engle KM, Wang D–H, Yu J-Q. Angew Chem Int Ed. 2009;48:5094. doi: 10.1002/anie.200806273.Phipps RJ, Gaunt MJ. Science. 2009;323:1593. doi: 10.1126/science.1169975.Chen MS, White MC. Science. 2010;327:566. doi: 10.1126/science.1183602.Jazzar R, Hitce J, Renaudat A, Sofack-Kreutzer J, Baudoin O. Chem Eur J. 2010;16:2654. doi: 10.1002/chem.200902374.Das S, Incarvito CD, Crabtree RH, Brudvig GW. Science. 2006;312:1941. doi: 10.1126/science.1127899.
- 2.Pd-catalyzed allylic C—H oxidations: Chen MS, White MC. J Am Chem Soc. 2004;126:1346. doi: 10.1021/ja039107n.Chen MS, Prabagaran N, Labenz NA, White MC. J Am Chem Soc. 2005;127:6970. doi: 10.1021/ja0500198.Fraunhoffer KJ, Prabagaran N, Sirois LE, White MC. J Am Chem Soc. 2006;128:9032. doi: 10.1021/ja063096r.Delcamp JH, White MC. J Am Chem Soc. 2006;128:15076. doi: 10.1021/ja066563d.Covell DJ, White MC. Angew Chem Int Ed. 2008;47:6448. doi: 10.1002/anie.200802106.Mitsudome T, Umetani T, Nosaka N, Mori K, Mizugaki T, Ebitani K, Kaneda K. Angew Chem Int Ed. 2006;45:481. doi: 10.1002/anie.200502886.Pilarski LT, Selander N, Bose D, Szabo KJ. Org Lett. 2009;11:5518. doi: 10.1021/ol9023369.Lin BL, Labinger JA, Bercaw JE. Can J Chem. 2009;87:264.Thiery E, Aouf C, Belloy J, Harakat D, Le Bras J, Muzart J. J Org Chem. 2010;75:1771. doi: 10.1021/jo902587u.Henderson WH, Check CT, Proust N, Stambuli JP. Org Lett. 2010;12:824. doi: 10.1021/ol902905w.
- 3.Pd-catalyzed allylic C—H aminations: Larock RC, Hightower TR, Hasvold LA, Peterson KP. J Org Chem. 1996;61:3584. doi: 10.1021/jo952088i.Fraunhoffer KJ, White MC. J Am Chem Soc. 2007;129:7274. doi: 10.1021/ja071905g.Reed SA, White MC. J Am Chem Soc. 2008;130:3316. doi: 10.1021/ja710206u.Liu G, Yin G, Wu L. Angew Chem Int Ed. 2008;47:4733. doi: 10.1002/anie.200801009.Rice GT, White MC. J Am Chem Soc. 2009;131:11707. doi: 10.1021/ja9054959.Reed SA, Mazzotti AR, White MC. J Am Chem Soc. 2009;131:11701. doi: 10.1021/ja903939k.Nahra F, Liron F, Prestat G, Mealli C, Messaoudi A, Poli G. Chem Eur J. 2009;15:11078. doi: 10.1002/chem.200901946.Shimizu Y, Obora Y, Ishii Y. Org Lett. 2010;12:1372. doi: 10.1021/ol100292g.
- 4.Streamlining synthesis via late-stage C—H oxidation: a) For first explicit demonstration of this concept see: Fraunhoffer KJ, Bachovchin DA, White MC. Org Lett. 2005;7:223. doi: 10.1021/ol047800p.Covell DJ, Vermeulen NA, White MC. Angew Chem Int Ed. 2006;45:8217. doi: 10.1002/anie.200603321.Stang EM, White MC. Nat Chem. 2009;1:547. doi: 10.1038/nchem.351.For excellent reviews: Hoffman RW. Synthesis. 2006;21:3531.Hoffman RW. Elements of Synthesis Planning. Springer; Heidelberg: 2009. For some elegant examples of late stage C—H hydroxylation and amination see: Wender PA, Hilinski MK, Mayweg AVW. Org Lett. 2005;7:79. doi: 10.1021/ol047859w.Hinman A, Du Bois J. J Am Chem Soc. 2003;125:11510. doi: 10.1021/ja0368305.h) ref. 1e, 1l.
- 5.a) Otera J. Esterification. Wiley-VCH; Weinheim: 2003. and references therein. [Google Scholar]; b) Ishihara K. Tetrahedron. 2009;65:1085. [Google Scholar]
- 6.Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 7.Under a C—H cleavage reaction manifold, high linear regioselectivity may be rationalized by outer sphere functionalization at the least sterically hindered position of a cationic π-allylPd(DMSO)2 intermediate. However, we cannot exclude the possibility of an alternative, albeit unprecedented, mechanism involving anti-Markovnikov oxy-palladation followed by regioselective β-hydride elimination.2a Significantly, when using catalytic amounts of a bidentate bis-sulfoxide ligand, high branched regioselectivity for ester formation is observed.2a,b Mechanistic studies suggest this regio outcome arises from inner sphere functionalization from an electronically dissymmetric π-allylPd(BQ)carboxylate species.2b,c, 4c
- 8.a) Mahajan RP, Patil UK, Patil SL. Indian Journal of Chemistry: Section B. 2007;46B:1459. [Google Scholar]; b) Mali RS, Papalkar AS. J Chem Research (S) 2001:433. [Google Scholar]
- 9.Belletire JL, Mahmoodi NO. J Nat Prod. 1992;55:194. doi: 10.1021/np50080a006. [DOI] [PubMed] [Google Scholar]
- 10.Kanjilal S, Shanker KS, Rao KS, Reddy KK, Rao BVSK, Kumar KBS, Kantam ML, Prasad RBN. Eur J Lipid Sci Technol. 2008;110:1175. [Google Scholar]
- 11.a) Castro PJL, Owen SN, Seward EM, Swain CJ, Williams BJ. 2002016343. PCT Int Appl WO. 2002; b) Hubshwerlen C, Angehrn R, Gubernator K, Page MGP, Specklin JL. J Med Chem. 1998;41:3972. doi: 10.1021/jm9800245. [DOI] [PubMed] [Google Scholar]
- 12.a) Dell C, Khan KM, Knight DW. J Chem Soc, Perkin Trans. 1994;1:341. [Google Scholar]; b) Dell C, Khan KM, Knight DW. J Chem Soc, Chem Commun. 1989:1812. [Google Scholar]; c) Ren Q, Dai L, Zhang H, Tan W, Xu Z, Ye T. Synlett. 2008;15:2379. [Google Scholar]
- 13.Isobe T, Ishikawa T. J Org Chem. 1999;64:6984. [Google Scholar]
- 14.Nakahara Y, Ando S, Itakura M, Kumabe N, Hojo H, Ito Y, Hakahara Y. Tetrahedron Lett. 2000;41:6489. [Google Scholar]
- 15.Visitin C, Aliev AE, Riddall D, Baker D, Okuyama M, Hoi PM, Hiley R, Selwood DL. Org Lett. 2005;7:1699. doi: 10.1021/ol0502578. [DOI] [PubMed] [Google Scholar]
- 16.a) Carter RG, Weldon DJ. Org Lett. 2000;2:3913. doi: 10.1021/ol006674w. [DOI] [PubMed] [Google Scholar]; b) Lee M, Lee T, Kim E, Ko H, Kim D, Kim S. Org Lett. 2006;8:745. doi: 10.1021/ol053010j. [DOI] [PubMed] [Google Scholar]
- 17.Messenger BT, Davidson BS. Tetrahedron Lett. 2001;42:801. [Google Scholar]
- 18.Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J Am Chem Soc. 2002;124:1307. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 19.Bartlett PA, Ting PC. J Org Chem. 1986;51:2230. [Google Scholar]
- 20.Enholm E, Low T. J Org Chem. 2006;71:2272. doi: 10.1021/jo052415e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.