

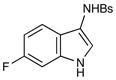
The pyrroloindoline natural products have attracted considerable interest both because of their biological activity and their unique structures (Figure 1). i Nature presumably produces these architecturally fascinating compounds via electrophilic cyclization cascades involving appropriately functionalized tryptamine and tryptophan precursors. In the laboratory, the use of chemical oxidants (e.g., m-CPBA, dioxirane, and NBS) to trigger similar cascades has been a common strategy for the total synthesis of these target molecules.ii Recently, several research groups have become interested in the synthesis of the subclass of pyrroloindoline alkaloids such as psychotrimine (1)iii and chaetomin (2)iv that feature a carbon-nitrogen bond at the C3 position of the oxidized indole.v However, only a limited number of methods for the direct electrophilic introduction of nitrogen at this position have been developed,vb,d,g,vi and thus the synthesis of aminated indole alkaloids remains a formidable synthetic challenge, especially in an enantioselective fashion.
Figure 1.

3-Aminopyrroloindoline natural products.
For the past several years, we have been investigating the oxidative functionalization of alkenes with N-sulfonyl oxaziridinesvii (“Davis’ oxaziridines,” e.g., 3) in the presence of copper(II) catalysts, which we have shown results in highly regioselective olefin oxyamination.viii Much of our effort has been focused on elucidating the mechanism for the formation of the 1,3-oxazolidine products. In a recent paper,ix we summarized our accumulated evidence that suggests that the cationic mechanism we originally proposed is not operational, and that the reaction instead involves a radical intermediate. As a further test of this conclusion, we elected to study the reactivity of indoles under the oxaziridine-mediated oxyamination conditions we have developed (Scheme 1). The regiochemistry of electrophilic additions to indoles is well established and is known to result in preferential attack at the C3 position due to the stability of the intermediate iminium cation (e.g., 5). This analysis suggests that a cationic mechanism would result in the formation of regioisomer 6 featuring oxygen at C3. Indeed, Dmitrienko observed the formation of this regioisomer in the uncatalyzed reaction between oxaziridine 3 and electron-rich indoles and suggested a similar cationic intermediate to account for its formation.x On the other hand, radical additions to indoles have also been the subject of extensive investigation, and the regiochemistry of these reactions prefers initial attack at C2 due to the stability of the intermediate benzylic radical (e.g., 7).xi A radical mechanism for the copper-catalyzed oxyamination would therefore result in the formation of regioisomer 8 complementary to that expected from a cationic mechanism.
Scheme 1.

Oxyamination of indoles as a regiochemical probe of mechanism. Bs = benzenesulfonyl.
Our investigation began with an examination of the reaction between oxaziridine 3 and indoles in the presence of copper(II) catalysts. Under these strongly oxidizing conditions, we found that electron-rich N-alkyl and N-unsubstituted indoles underwent rapid and exothermic decomposition to an intractable mixture of products. On the other hand, the reaction of N-acylindole 4 proceeded smoothly within 1.5 h in 85% yield and 2:1 diastereoselectivity. Importantly, both diastereomers of the oxyaminated product (8) corresponded to the benzylic amine regioisomer that would be predicted from a radical mechanism, xii which provides further evidence against the cationic mechanism we originally proposed.
Similarly, exclusive formation of the benzylic amine regioisomer is observed in the oxyamination of all indole substrates we have examined to date. As Figure 2 indicates, a range of structurally varied N-acylindoles are suitable substrates for oxyamination. These include substrates bearing electron-donating and –withdrawing moieties at various positions on the indole (e.g., 10–13) or substituted with more complex N-acyl groups (14). Unfortunately, tryptamines and other indoles substituted at C3 afforded somewhat lower yields, which we speculate may be attributable to unproductive side oxidations of these more electron-rich substrates. (15). After some optimization, we found that the yield could be improved by performing the reaction in acetone with higher loadings of the copper(II) catalyst and using less reactive 3,3-dimethyl oxaziridine 9 as the terminal oxidant (16). This modification enables the oxyamination of a variety of more electron-rich indoles bearing alkyl substituents on the indole olefin (17–21). The functional group compatibility of this process is quite good; aryl halides, ethers, esters, amides, and carbamates are all easily tolerated under both sets of reaction conditions.
Figure 2.

Scope of indole oxyamination. Yields reflect the averaged isolated yields of two reproducible experiments. [a] Conditions: 2 mol% CuCl2, 2 mol% Bu4N+Cl−, 2 equiv oxaziridine, CH2Cl2, 23 °C [b] Conditions: 10 mol% CuCl2, 10 mol% Bu4N+Cl−, 2 equiv oxaziridine, acetone, 23 °C.
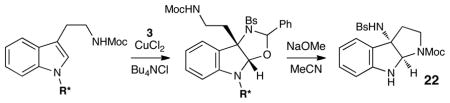
Although the initial impetus for this study was to provide additional evidence for a radical mechanism, we quickly recognized that the aminal products could serve as synthetically valuable precursors to a variety of heterocyclic systems found in bioactive compounds, including pyrroloindoline natural products such as 1 and 2. Indeed, treatment of tryptamine-derived aminal 18 with NaOH results in the facile cleavage of the N-acetyl group with concomitant elimination of acetone; under the basic conditions of this hydrolysis, the N-Moc carbamate undergoes spontaneous cyclization upon the imine intermediate to produce pyrroloindoline 22 in 85% yield (Table 1, entry 1). Cyclization of the homotryptamine-derived aminal 19 was also successful, affording α-carboline 23 in good yield (entry 2). This cyclization cascade can also be terminated with oxygen nucleophiles; the latent alcohol of O-acyl tryptophol-derived aminal 20 is revealed under the basic conditions of the reaction and affords furoindoline 24 in excellent yields (entry 3). Substrates without internal nucleophiles were also subjected to these conditions. The imine intermediate arising from hydrolysis of 8 presumably undergoes rearomatization to afford 3-aminoindole 25 (entry 4).xiii This rearomatization is also successful for various substituted indolines (entries 5 and 6).
Table 1.
Base-promoted transformations of aminal products.[a]
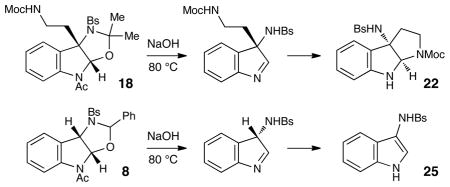 | ||||
|---|---|---|---|---|
| Entry | Animal | Product | Time | Yield[b] |
| 1 |
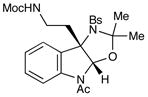 18 |
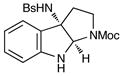 22 |
7 h | 85% |
| 2 |
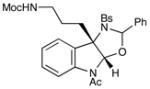 19 |
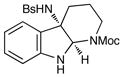 23 |
7 h | 75% |
| 3 |
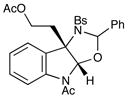 20 |
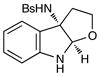 24 |
14 h | 95% |
| 4 |
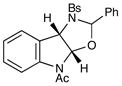 8 |
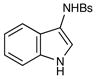 25 |
14 h | 88% |
| 5 |
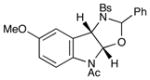 11 |
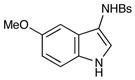 26 |
26 h | 87% |
| 6 |
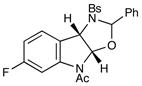 12 |
 27 |
14 h | 93% |
Conditions: 6 M NaOH, dioxane, 80 °C.
Averaged isolated yields of two reproducible experiments.
The challenge of effecting the enantioselective synthesis of pyrroloindolines has been addressed by several methods, including asymmetric Heck cyclizations, xiv organocatalytic cascade cyclizations of tryptamines, xv and electrophilic cyclizations of enantiopure tryptophans.xvi We wondered if an asymmetric version of this copper-catalyzed oxyamination might also serve as a platform for the enantioselective synthesis of C3-amine-substituted pyrroloindolines exemplified by the core of psychotrimine (1). Our strategy took advantage of two key observations. First, we found that a variety of acyl groups can be tolerated on the indole nitrogen, including amino acid derivatives (e.g., 14). Second, based on our mechanistic hypothesis, the initial steteodetermining step of the oxyamination process should occur on the C2 position of the indole, proximal to the N-acyl moiety. Taking these ideas together, we wondered if the use of a chiral amino acid auxiliary on indole nitrogen could influence the stereochemical outcome of the initial C–O bond formation.
Thus, we prepared a variety of tryptamines modified at N1 with N-Boc-aminoacyl groups, which could be installed in one step with excellent yields (see Supporting Information for details), and examined these substrates in a two-step oxyamination–cyclization sequence (Table 2).xvii Surprisingly, we found that the enantiopurity of the pyrroloindoline product increased as the steric demand of the amino acid auxiliary decreased (entries 1–5). Of the auxiliaries screened, N-Boc proline provided the highest enantioselectivity and afforded 22 in 73% ee (entry 6). The identity of the N-protecting group on the prolyl moiety proved to be critical; smaller carbamates such as Cbz provided significantly lower enantioselectivities (entry 7). Further optimization showed that the performing the reaction in chloroform provided a marked increase in the enantioselectivity of the process (entry 8). Finally, the enantioselectivity of the process could be further improved to 91% ee by conducting the reaction at –30 °C, although the reaction time was somewhat increased (entry 9). In an initial exploration of the generality of this two-step sequence, we found that a variety of enantioenriched indole derivatives could be prepared in 86–91% ee (Figure 3).xviii
Table 2.
Asymmetric synthesis of 22.[a]
 | |||||
|---|---|---|---|---|---|
| Entry | R*[b] | solvent | T | Time | ee[c] |
| 1 | L-Boc-t-Leu | acetone | 23 °C | 6 h | <5% |
| 2 | L-Boc-Phg | acetone | 23 °C | 6 h | <5% |
| 3 | L-Boc-Val | acetone | 23 °C | 6 h | 20% |
| 4 | L-Boc-Phe | acetone | 23 °C | 6 h | 36% |
| 5 | L-Boc-Ala | acetone | 23 °C | 6 h | 57% |
| 6 | L-Boc-Pro | acetone | 23 °C | 1 h | 73% |
| 7 | L-Cbz-Pro | acetone | 23 °C | 2 h | 48% |
| 8 | L-Boc-Pro | CHCl3 | 23 °C | 2 h | 86% |
| 9 | L-Boc-Pro | CHCl3 | −30 °C | 24 h | 91% |
Oxyamination conditions: 10 mol% CuCl2, 10 mol% Bu4N+Cl−, 2 equiv of 3. Cyclization conditions: NaOMe, MeCN, 23 °C.
t-Leu = tert-leucine, Phg = phenylglycine.
Determined by chiral SFC analysis.
Figure 3.

Enantioenriched oxidized indole derivatives. See Supporting Information for experimental details.
The ground state conformation of N-prolylindole 28, as predicted by a molecular mechanics geometry minimization (Figure 4), suggests a possible rationale for the absolute sense of stereoinduction in this process. This model is consistent with the known propensity of N-acylindoles to strongly prefer a Z conformation about the N–acyl bond.xix In addition, the prolyl moiety adopts a conformation that minimizes the pseudo-A(1,3) strain between its α stereogenic center and the C-2 position of the indole. This places the bulky N-Boc group in an orientation that shields the Re face of the indole and leaves the Si face open to attack by the oxaziridine. This model is thus consistent with the absolute sense of stereoinduction observed in this transformation. It is also consistent with the observations that the enantioselectivity increases with decreasing steric bulk of the aminoacyl side chain (Table 3, entries 1–6) and that smaller protecting groups on the prolyl nitrogen are less effective at differentiating the prochiral faces of the indole (entry 7).
Figure 4.

Ground state conformation of 28 predicted by computational minimization (Sybyl force field, Spartan).
In summary, we have demonstrated that the copper(II)-catalyzed oxyamination of indoles affords aminals in high yields and excellent regioselectivity. The regiochemical outcome of this process is consistent with a radical intermediate and is thus further corroboration of our recently revised mechanistic model. The aminal products arising from these studies can be easily transformed into 3-aminoindoles, α-carbolines, pyrroloindolines, and other synthetically valuable heterocyclic structures. Finally, we have used our understanding of this process to design a chiral auxiliary-controlled asymmetric oxyamination of tryptamines, which enables the enantioselective construction of C3-aminopyrroloindolines that may be useful in the synthesis of oxidized indole natural products. Future studies in our lab will focus on utilizing this method for the total synthesis of psychotrimine.
Supplementary Material
Acknowledgments
We thank Kevin Williamson for obtaining X-ray quality crystals of 8, Lara Spencer for determining crystal structures, and Dr. Prakash Raman for insightful discussions. Financial support was provided by the NSF (CHE-0645447) and NIH (GM084022). The NMR spectroscopy facility at UW-Madison is funded by the NIH (S10 RR04981-01) and NSF (CHE-9629688).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- i.For recent reviews, see: Steven A, Overman LE. Angew Chem. 2007;119:5584–5605.Angew Chem Int Ed. 2007;46:5488–5508. doi: 10.1002/anie.200700612.Kim J, Movassaghi M. Chem Soc Rev. 2009;38:3035–3050. doi: 10.1039/b819925f.
- ii.a) Ohno M, Spade TF, Witkop B. J Am Chem Soc. 1970;92:343–348. doi: 10.1021/ja00705a643. [DOI] [PubMed] [Google Scholar]; b) Savige WE. Aust J Chem. 1975;28:2275–2287. [Google Scholar]; c) Nakagawa M, Kato S, Kataoka S, Hino T. J Am Chem Soc. 1979;101:3136–3137. [Google Scholar]; d) Nakagawa M, Taniguchi M, Sodeoka M, Ito M, Yamaguchi K, Hino T. J Am Chem Soc. 1983;105:3709–3710. [Google Scholar]; e) Baran PS, Guerrero CA, Corey EJ. J Am Chem Soc. 2003;125:5628–5629. doi: 10.1021/ja034491+. [DOI] [PubMed] [Google Scholar]; f) Hewitt PR, Cleator E, Ley SV. Org Biomol Chem. 2004;2:2415–2417. doi: 10.1039/B410180D. [DOI] [PubMed] [Google Scholar]
- iii.Takayama H, Mori I, Kitajima M, Aimi N, Lajis NH. Org Lett. 2004;6:2945–2948. doi: 10.1021/ol048971x. [DOI] [PubMed] [Google Scholar]
- iv.a) Waksman SA, Bugie E. J Bacteriol. 1944;48:527–530. doi: 10.1128/jb.48.5.527-530.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Geiger WB, Conn JE, Waksman SA. J Bacteriol. 1944;48:531–536. doi: 10.1128/jb.48.5.531-536.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- v.a) Matsuda Y, Kitajima M, Takayama H. Org Lett. 2008;10:125–128. doi: 10.1021/ol702637r. [DOI] [PubMed] [Google Scholar]; b) Newhouse T, Baran PS. J Am Chem Soc. 2008;130:10886–10887. doi: 10.1021/ja8042307. [DOI] [PubMed] [Google Scholar]; c) Espejo VR, Rainier JD. J Am Chem Soc. 2008;130:12894–12895. doi: 10.1021/ja8061908. [DOI] [PubMed] [Google Scholar]; d) Newhouse T, Lewis CA, Baran PS. J Am Chem Soc. 2009;131:6360–6361. doi: 10.1021/ja901573x. [DOI] [PubMed] [Google Scholar]; e) Takahashi N, Ito T, Matsuda Y, Kogure N, Kitajima M, Takayama H. Chem Comm. 2010;46:2501–2503. doi: 10.1039/b923918a. [DOI] [PubMed] [Google Scholar]; f) Espejo VR, Rainier JD. Org Lett. 2010;12:2154–2157. doi: 10.1021/ol100672z. [DOI] [PubMed] [Google Scholar]; g) Newhouse T, Lewis CA, Eastman KJ, Baran PS. J Am Chem Soc. 2010;132:7119–7137. doi: 10.1021/ja1009458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vi.For other key examples, see: Campbell N, Cooper RC. J Chem Soc. 1935:1208–1211.Bahadur GA, Bailey AS, Scott PW, Vandrevala MH. J Chem Soc Perkin Trans 1. 1980:2870–2877.Sanchis-Llorca RT, Sepulveda-Arques J, Zaballos-Garcia E, Jones RA. Heterocycles. 1987;26:401–404.Kumar PR. Heterocycles. 1987;26:1257–1262.Hashimoto Y, Ishizaki T, Shudo K. Tetrahedron. 1991;47:1837–1860.Pelkey ET, Gribble GW. Synthesis. 1999;7:1117–1122.Beaumont S, Pons V, Retailleau P, Dodd RH, Dauban P. Angew Chem. 2010;122:1678–1681. doi: 10.1002/anie.200906650.Angew Chem Int Ed. 2010;49:1634–1637.
- vii.a) Davis FA, Nadir UK, Kluger EW. J Chem Soc, Chem Commun. 1977:25–26. [Google Scholar]; b) Davis FA, Jenkins R, Jr, Yocklovich SG. Tetrahedron Lett. 1978;19:5171–5174. [Google Scholar]
- viii.a) Michaelis DJ, Shaffer CJ, Yoon TP. J Am Chem Soc. 2007;129:1866–1867. doi: 10.1021/ja067894t. [DOI] [PubMed] [Google Scholar]; b) Michaelis DJ, Ischay MA, Yoon TP. J Am Chem Soc. 2008;130:6610–6615. doi: 10.1021/ja800495r. [DOI] [PubMed] [Google Scholar]; (c) Michaelis DJ, Williamson KS, Yoon TP. Tetrahedron. 2009;65:5118–5124. doi: 10.1016/j.tet.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ix.Benkovics T, Du J, Guzei IA, Yoon TP. J Org Chem. 2009;74:5545–5552. doi: 10.1021/jo900902k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- x.Mithani S, Drew DM, Rydberg EH, Taylor NJ, Mooibroek S, Dmitrienko GI. J Am Chem Soc. 1997;119:1159–1160. [Google Scholar]
- xi.a) Artis DR, Cho IS, Jaime-Figueroa S, Muchowski JM. J Org Chem. 1994;59:2456–2466. [Google Scholar]; b) Byers JH, Campbell JE, Knapp FH, Thissell JG. Tetrahedron Lett. 1999;40:2677–2680. [Google Scholar]; c) Furst L, Matsuura BS, Narayanam JMR, Tucker JW, Stephenson CRJ. Org Lett. 2010 doi: 10.1021/ol101146f. in press. [DOI] [PubMed] [Google Scholar]
- xii.The regiochemistry of both diastereomers was unabiguously determined by single-crystal X-ray diffraction. CCDC-785147 and CCDC-785148 contain the supplementary crystallographic data for each diastereomer of 8. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- xiii.For studies on the biological activity of 3-aminoindoles, see: Bahekar RH, Jain MR, Goel A, Patel DN, Prajapati VM, Gupta AA, Jadav PA, Patel PR. Bioorg Med Chem. 2007;15:3248–3265. doi: 10.1016/j.bmc.2007.02.029.Pews-Davtyan A, Tillack A, Schmöle AC, Ortinau S, Frech MJ, Rolfs A, Beller M. Org Biomol Chem. 2010;8:1149–1153. doi: 10.1039/b920861e.
- xiv.a) Ashimori A, Matsuura T, Overman LE, Poon DJ. J Org Chem. 1993;58:6949–6951. [Google Scholar]; b) Matsuura T, Overman LE, Poon DJ. J Am Chem Soc. 1998;120:6500–6503. [Google Scholar]; c) Overman LE, Paone DV, Stearns BA. J Am Chem Soc. 1999;121:7702–7703. [Google Scholar]
- xv.a) Austin JF, Kim SG, Sinz CJ, Xiao WJ, MacMillan DWC. Proc Nat Assoc Sci. 2004;101:5482–5487. doi: 10.1073/pnas.0308177101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jones SB, Simmons B, MacMillan DWC. J Am Chem Soc. 2009;131:13606–13607. doi: 10.1021/ja906472m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xvi.a) Marsden SP, Depew KM, Danishefsky SJ. J Am Chem Soc. 1994;116:11143–11144. [Google Scholar]; b) Kamenecka TM, Danishefsky SJ. Angew Chem, Int Ed. 1998;37:2995–2998. doi: 10.1002/(SICI)1521-3773(19981116)37:21<2995::AID-ANIE2995>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]; c) Depew KM, Marsden SP, Zatorska D, Zatorski A, Bornmann WG, Danishefsky SJ. J Am Chem Soc. 1999;121:11953–11963. [Google Scholar]; d) Schkeryantz JM, Woo JCG, Siliphaivanh P, Depew KM, Danishefsky SJ. J Am Chem Soc. 1999;121:11964–11975. [Google Scholar]; e) Crich D, Huang X. J Org Chem. 1999;64:7218–7223. doi: 10.1021/jo982385y. [DOI] [PubMed] [Google Scholar]
- xvii.To the best of our knowledge, this constitutes the first example of an N-aminoacyl auxiliary for asymmetric reactions of indoles. Proline-derived groups, however, have been used to direct nucleophilic additions to β-carbolines. See: Itoh T, Matsuya Y, Enomoto Y, Nagata K, Miyazaki M, Ohsawa A. Synlett. 1999:1799–1801.
- xviii.The absolute configuration of 23 was determined by single-crystal X-ray diffraction; the configurations of 22 and 24 were assigned by analogy. CCDC-785149 contains the supplementary crystallographic data for 23. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- xix.Elguero J, Marzin C, Peek ME. Org Magn Resonance. 1975;7:445–450. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.