

Abstract
There is considerable interest in the development of an inhibitor of aldo-keto reductase (AKR) 1C3 (type 5 17β-hydroxysteroid dehydrogenase and prostaglandin F synthase) as a potential therapeutic for both hormone-dependent and hormone-independent cancers. AKR1C3 catalyzes the reduction of 4-androstene-3,17-dione to testosterone and estrone to 17β-estradiol in target tissues, which will promote the proliferation of hormone dependent prostate and breast cancers, respectively. AKR1C3 also catalyzes the reduction of prostaglandin (PG) H2 to PGF2α and PGD2 to 9α,11β-PGF2, which will limit the formation of anti-proliferative prostaglandins, including 15-deoxy-Δ12,14-PGJ2, and contribute to proliferative signaling. AKR1C3 is overexpressed in a wide variety of cancers, including breast and prostate cancer. An inhibitor of AKR1C3 should not inhibit the closely related isoforms AKR1C1 and AKR1C2, as they are involved in other key steroid hormone biotransformations in target tissues. Several structural leads have been explored as inhibitors of AKR1C3, including non-steroidal anti-inflammatory drugs, steroid hormone analogues, flavonoids, cyclopentanes, and benzodiazepines. Inspection of the available crystal structures of AKR1C3 with multiple ligands bound, along with the crystal structures of the other AKR1C isoforms, provides a structural basis for the rational design of isoform specific inhibitors of AKR1C3. We find that there are subpockets involved in ligand binding that are considerably different in AKR1C3 relative to the closely related AKR1C1 or AKR1C2 isoforms. These pockets can be used to further improve the binding affinity and selectivity of the currently available AKR1C3 inhibitors.
Keywords: Aldo-keto reductase, steroid hormones, prostaglandins, nuclear receptors, structure-based inhibitor design
1. Rationale for AKR1C3 Inhibition
Aldo-keto reductase (AKR) 1C31(insert foot note) catalyzes the NADPH dependent stereospecific reduction of carbonyl moieties on substrates of importance to the pre-receptor regulation of signaling pathways involved in cell proliferation (Figure 1). The interconversion of a ketone group with a hydroxyl group on lipophilic ligands can drastically alter their affinity for their cognate receptors. In the development of hormone dependent cancers, the reduction of a ketone at the 17-position of steroid hormones to the corresponding alcohol, catalyzed by the reductive 17β-hydroxysteroid dehydrogenases (HSDs), is of particular importance. Each of the 17β-HSD isoforms that perform this reaction is a target for the development of new pharmacological agents; see other reviews in this issue for a discussion of inhibitors of these other enzymes [1-3]. This article is focused on AKR1C3 (type 5 17β-HSD), which reduces both steroids and prostaglandins.
Figure 1.

Reactions catalyzed by AKR1C3. AKR1C3 will enhance proliferative signaling in various hormone-responsive cells through the formation of androgens and estrogens with increased affinity for the androgen receptor (AR) and estrogen receptor (ER), respectively, as well as through reduction of progesterone to a metabolite with decreased affinity for the progesterone receptor (PR). AKR1C3 will also catalyze prostaglandin (PG) reduction reactions that enhance proliferative pathways by activating the F prostanoid (FP) receptor and preventing formation of anti-proliferative 15-deoxy-Δ12,14-PGJ2, which forms through spontaneous dehydration and rearrangement of PGD2.
AKR1C3 reduces the 17-position of 4-androstene-3,17-dione (a weak androgen) to form testosterone (a potent androgen) and the 17-position of estrone (a weak estrogen) to form 17β-estradiol (a potent estrogen), leading to trans-activation of the androgen and estrogen receptors, respectively[4, 5]. It can also act at the 20 position of progesterone and deoxycorticosterone, forming 20α-hydroxy metabolites with reduced affinities for the progesterone and mineralcorticoid receptors, respectively [6]. Finally, as a prostaglandin (PG) F synthase, AKR1C3 catalyzes the reduction of the endoperoxide PGH2 to yield PGF2α and the stereospecific reduction of PGD2 to 9α,11β-PGF2[4, 7-9]. This reaction occurs at the same active site, which has considerable flexibility to accommodate diverse ligands (see section 3). In the absence of AKR1C3 activity, PGD2 spontaneously dehydrates and rearranges to form the PGJ2prostanoids[4]. The PGF2 isomers are pro-inflammatory and enhance proliferation, while the PGJ2 products, particularly 15-deoxy-Δ12,14-PGJ2 (15dPGJ2), are anti-inflammatory, promote differentiation, and are anti-neoplastic via several mechanisms[10-15].
The products of reactions catalyzed by AKR1C3 promote tumor growth. AKR1C3 is therefore an important target for the prevention or treatment of both hormone-dependent and hormone-independent cancers. AKR1C3 likely contributes to the development of castrate resistant prostate cancer through the intratumoral formation of the active androgen testosterone[16]. Transcript levels and measurement of testosterone: 5α-dihydrotestosterone (5α-DHT) ratios indicate a reliance on the formation of testosterone by AKR1C3 in castrate resistant disease, as well as a decreased dependence on the activity of 5α-reductase[17, 18]. In the breast, AKR1C3 catalyzes the reduction of 4-androstene-3,17-dione to testosterone, which can undergo aromatization to form 17β-estradiol. In addition, AKR1C3 also reduces estrone to 17β-estradiol. Consistent with these activities AKR1C3 has been shown to promote proliferation of MCF-7 hormone-dependent breast cancer cells [4]. In the endometrium, AKR1C3 could increase estrogen levels and decrease progesterone levels and thus promote endometrial cancer cell proliferation [19]. By increasing proliferative PGF2 isomers and decreasing anti-proliferative PGJ2 products, the prostaglandin F synthase activities of AKR1C3 have the potential to impact both hormone-dependent and hormone-independent cancers. In particular, prostaglandin metabolism by AKR1C3 has been shown to prevent differentiation of leukemia cells and AKR1C3 inhibition is being explored as a treatment for acute myelogenous leukemia [20]. AKR1C3 is over-expressed across a wide variety of cancers, including those of the breast and prostate, and its expression increases with tumor aggressiveness [21-26].
AKR1C3 catalyzed reactions also play important roles in other physiological and pathological processes that may be targets for therapeutic intervention. Androgen production by AKR1C3 is likely involved in the development of benign prostatic hyperplasia [27]. Increased estrogen and decreased progesterone receptor signaling due to increased AKR1C3 activity could contribute to endometriosis and dysmenorrhea [28]. PGF2 isomers formed by AKR1C3stimulate smooth muscle contraction during parturition, while the AKR1C3 substrate progesterone prevents parturition, so AKR1C3 inhibitors may be useful as progestational agents [29, 30]. Interestingly, indomethacin has proven effective at stopping premature parturition [31]. This effect is thought to result from its inhibition of the prostaglandin H synthases (PGHS), but indomethacin also inhibits AKR1C3 in the same therapeutic dose range [32]. Use of indomethacin for this purpose is limited due to side-effects in the fetus that likely stem from the inhibition of PGHS activities in developing organ systems [31]. Because PGF2α is involved in preventing adipocyte differentiation and 15dPGJ2 promotes differentiation of adipocytes, inhibiting the prostaglandin F synthase activities of AKR1C3 may have beneficial effects in diabetes similar to those observed with peroxisome proliferator activated receptor γ agonists [33, 34].
A specific inhibitor of AKR1C3 would be a valuable tool to better understand AKR1C3 and its contribution to normal physiology and disease. One challenge in the development of an AKR1C3 inhibitor is that there are three other closely related enzyme isoforms, AKR1C1, AKR1C2, and AKR1C4, which share greater than 84% sequence homology with AKR1C3 and are also involved in steroid hormone metabolism [35]. AKR1C4 is liver specific, while AKR1C1 and AKR1C2 are widely expressed across tissue types. Inhibition of these three isoforms, particularly AKR1C1 and AKR1C2, is not desirable in prostate cancer as they act primarily as 3-ketosteroid reductaseson 5α-DHT. For example, AKR1C1 reduces 5α-DHT to 5α-androstane-3α,17β-diol (a pro-apoptotic ligand of estrogen receptor β) and AKR1C2 reduces 5α-DHT to 5α-androstane-3α,17β-diol (a weak androgen) [36]. Inhibition of these activities would promote proliferative signaling in the prostate. Given their similar expression patterns and substrate specificities, an inhibitor of AKR1C3 that does not inhibit AKR1C1 or AKR1C2 is needed to explore the role of AKR1C3 in normal and aberrant cell signaling.
2. Types of AKR1C3 Inhibitors
2.1 Overview of AKR1C3 Inhibitors
AKR1C3 is inhibited by several structural classes of compounds. Structures of representative compounds from each known class of inhibitors and their potency towards AKR1C isoforms are shown in Figure 2. Although there is significant structural diversity in the types of compounds that inhibit AKR1C3, they all contain one or more rings and at least one carbonyl group. Interestingly, many of the compounds that inhibit AKR1C3 have already been described as being effective in the chemoprevention of cancer. For instance, non-steroidal anti-inflammatory drugs (NSAIDs) and medroxyprogesterone acetate (MPA), which are AKR1C3 inhibitors, as well as consumption of fruits and vegetables, which contain AKR1C3 inhibitors, have all been shown to substantially reduce the risk of colon cancer [37-40]. While this may be a coincidence, it raises the question as to whether AKR1C3 inhibition has already been used as a means to reduce the risk of cancer.
Figure 2.
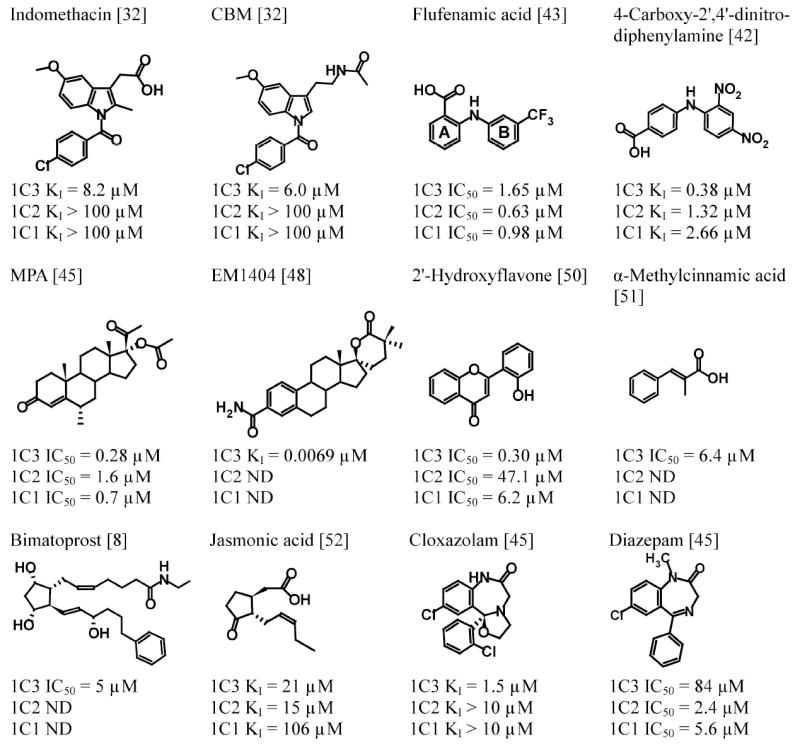
Chemical structures of representative AKR1C3 inhibitors and their inhibitory potency towards AKR1C3 and closely related isoforms AKR1C1 and AKR1C2.ND, not determined; CBM, 4-chlorobenzoyl melatonin; MPA, medroxyprogesterone acetate. The A- and B-rings of flufenamic acid are indicated.
2.2 Non-steroidal Anti-inflammatory Drugs (NSAIDs)
One class of compounds that our laboratory discovered as AKR1C inhibitors are the NSAIDs and their analogues [32, 41-43]. In general, NSAIDs inhibit the AKR1C isoforms with potencies similar to those observed for the inhibition of their putative targets the PGHS. Given that NSAIDs have been extensively used in humans, their analogues are predicted to be well tolerated. Two classes of NSAIDs particularly stand out for their potential to lead to a selective inhibitor of AKR1C3 [32]. Indomethacin exhibits a strong selectivity for AKR1C3 over AKR1C1 and AKR1C2, while the N-phenylanthranilic acids are the most potent NSAID inhibitors of AKR1C3. The availability of crystal structures of AKR1C3 with both indomethacin and the N-phenylanthranilic acid flufenamic acid bound allows predictions of structure activity relationships for the AKR1C enzymes (discussed in detail in section 3). Moreover, the structure activity relationships for the inhibition of the PGHS enzymes by NSAIDs have been well studied and can be exploited to develop inhibitors that are inactive towards PGHS. Based on these structure-activity relationships, we have rationally designed an indomethacin analogue, N-(4-chlorobenzoyl)melatonin (CBM), that inhibits AKR1C3 but does not inhibit AKR1C1, AKR1C2, or the PGHS enzymes [32]. We have also designed N-phenylanthranilic acid analogues, such as 4-carboxy-2’,4’-dinitrodiphenylamine, that inhibit all of the AKR1C enzymes but not the PGHS enzymes [42]. For a more detailed review of AKR1C inhibition by NSAIDs, see Byrns and Penning [44].
2.3 Steroids
Steroidal inhibitors that are analogs of AKR1C3 substrates, such as MPA [45] or estrogen lactones [46, 47], have been used as AKR1C3 inhibitors. Although MPA is a potent inhibitor of both recombinant AKR1C3 and AKR1C3 expressed in cells [20, 45], it does not exhibit AKR1C3 selectivity over other AKR1C isoforms and it binds to multiple steroid hormone receptors. It is therefore not a good inhibitor for probing AKR1C3 function, but it may be interesting to explore whether inhibition of AKR1C3 might contribute to the pharmacological effects of MPA. In endometriosis or endometrial cancer, where both AKR1C3 inhibition and progesterone receptor activation are desired, MPA could represent a useful therapeutic agent. Based on its inhibition of AKR1C3, MPA is currently being tested in early clinical trials for the treatment of acute myeloid leukemia in combination with bezafibrate, which stimulates lipid peroxidation-mediated PGD2 formation [20].
Estrogen lactones are the most potent inhibitors of AKR1C3 currently described, with a number of compounds exhibiting IC50 values in the low nM range [46-48]. One disadvantage of these inhibitors is that they have not been tested against the other AKR1C isoforms, although the relative bulk of the lactone ring is likely to limit inhibition of AKR1C1 and AKR1C2 (see section 3.2). Many of these compounds do inhibit other 17β-HSD enzymes, with inhibition of the oxidative type 2 17β-HSD posing a particular concern. Type 2 17β-HSD performs the oxidation of testosterone and 17β-estradiol to yield 4-androstene-3,17-dione and estrone, respectively, acting in direct opposition to AKR1C3 [46]. Recently, steroidal lactones that exhibit minimal inhibition of type 2 17β-HSD and have low nanomolar affinities for AKR1C3 have been described. For example, a 3-desoxyestradiol derivative with a dimethylatedspiro-δ-lactone at position 17 inhibited AKR1C3 with an IC50 of 2.9 nM and exhibited little inhibition of type 2 17β-HSD or affinity for steroid hormone receptors [47]. Qiu et al have described a crystal structure for AKR1C3 bound to another estrogen spiro-δ-lactone, EM1404 (KI = 6.9 nM)[48].
2.4 Flavonoids and Cinnamic Acids
Several plant compounds and their analogs have also been explored as AKR1C3 inhibitors. One promising class of phytocompounds is the flavonoids [49, 50]. While most of these compounds are not specific for AKR1C3 over the other AKR1C isoforms, 2’-hydroxyflavone exhibits an IC50 value of 300 nMfor AKR1C3 and exhibits a 20 fold and >100 fold selectivity over AKR1C1 and AKR1C2, respectively [50]. In addition, flavonoid precursors, the cinnamic acids, have also been shown to inhibit AKR1C3 with IC50 values in the micromolar range [51]. Flavonoids are phytoestrogens, and these compounds likely have a number of additional targets in vivo, so they may not represent the best route to an AKR1C3-selective drug. However, the inhibition of AKR1C3 by these compounds could contribute to their observed therapeutic benefits, which include chemoprevention of cancer [40].
2.5 Cyclopentane Derivatives
The cyclopentane ring is a key structural feature in prostaglandins, which has led to the study of several cyclopentanes as AKR1C3 inhibitors. One such compound, bimatoprost, is an anti-glaucoma drug targeting the prostamide F2 receptor to relieve ocular pressure [8]. Bimatoprost is a structural analogue of the AKR1C3 product prostaglandin F2α and yields an IC50 value against AKR1C3 in the low micromolar range [8]. It has not been screened for inhibition of the other AKR1C isoforms. Jasmonic acids are structural analogues of prostaglandins that are found in plants and act as plant hormones regulating defense and plant development[52]. Jasmonic acid inhibits AKR1C3 with a KI of 21 μM and exhibits similar inhibition of the other AKR1C isoforms. Other cyclopentane derivatives have been described by Stefane et al [53], with KI values as low as 16 μM, but have relatively low selectivity and inhibit AKR1C1. In general, the currently described cyclopentane based inhibitors do not appear to be good leads for the development of selective AKR1C3 inhibitors. However, the much lower KM of PGD2 for AKR1C3 as compared to the other AKR1C isoforms suggests that a selective inhibitor from this class should be achievable [7].
2.6 Benzodiazepines
Benzodiazepines have also been tested for AKR1C inhibition [54]. Because AKR1C enzymes are involved in the biotransformation of neurosteroids and expressed in the brain, it has been proposed that their inhibition might contribute to the anti-anxiety effects of these drugs. Most of the benzodiazepines, including diazepam (Valium), were inhibitors of all of the AKR1C enzymes, but exhibited more potent inhibition of the other three isoforms relative to AKR1C3. However, cloxazolam, a prodrug used in Brazil and parts of Europe, was a relatively potent (KI = 1.5 μM) AKR1C3 inhibitor that did not substantially inhibit the other AKR1C isoforms. A cloxazolam analogue that did not undergo metabolic activation to an anxiolytic could represent an interesting path to an AKR1C3 specific inhibitor.
2.7 Considerations when Assaying AKR1C3 Inhibition
One concern in the development of AKR1C3 inhibitors is that the inhibitory potency of compounds towards the AKR1C enzymes can vary considerably with assay conditions, making it difficult to compare results across studies. For instance, we have observed an approximately 10-fold increase in affinity of indomethacin for both AKR1C2 and AKR1C3 with a change in cosolvent from 4% ethanol to 4% acetonitrile or DMSO. We have also observed that AKR1C2 inhibition by indomethacin is enhanced when its preferred cofactor for oxidation (NADP+) is replaced by NAD+ (Byrns, Adeniji, Jin & Penning, unpublished observations). This sensitivity to assay conditions suggests that results may vary considerably between groups and makes it difficult to assess the results of studies when detailed assay conditions are not included, or when trying to compare results with purified recombinant enzyme to experiments performed in cells. Another consideration is that in order to use IC50 values to compare the relative potency of an inhibitor towards different enzymes or isoforms, or to the work of other laboratories, it is best to determine the pattern of inhibition for a representative lead compound to determine whether the mode of inhibition is competitive. If the pattern of inhibition is competitive, inhibition experiments should be performed with the concentration of the substrate set to the KM value of the respective enzyme and the IC50 value can be converted to a Ki value using the Cheng-Pruss of relationship. Indomethacin and its analogs are potent competitive inhibitors of the AKR1C3 catalyzed reduction of 4-androstene-3,17-dione, and N-phenylanthranilic acids are potent competitive inhibitors of the AKR1C3 catalyzed oxidation of 1-acenaphthenol [32, 42]. However, with different substrates the same inhibitor, e.g. indomethacin, can yield different inhibition patterns [32]. During the AKR1C3 dependent reduction of 9,10-phenanthrenequinone, indomethacin can preferentially bind to the E•NADP+ complex to prevent cofactor release, and thus demonstrates an uncompetitive inhibition pattern. This suggests that it can be problematic to compare IC50 values unless a pattern of inhibition for a lead compound for a structural series is established and unless the assay conditions are kept invariant.
3. Structural Insights into Selective AKR1C3 Inhibition from the Available Crystal Structures
3.1 Available Crystal Structures
Crystal structures of the four human AKR1C enzymes have been actively pursued by several groups. Ten crystal structures of different AKR1C3 ternary complexes have been deposited into the Protein Data Bank (Table 1)[48, 55-59]. These structures have provided a strong basis for understanding the activities of existing inhibitors and for rational design of AKR1C3 inhibitors with better selectivity and potency.
Table 1.
Available crystal structures of AKR1C3 in the Protein Data Bank.
| PDB ID | Ligands | Reference |
|---|---|---|
| 1RY0 | NADP+, PGD2 | [55] |
| 1RY8 | NADP+, rutin | [55] |
| 1S1P | NADP+, 2-methyl-2,4-pentanediol, acetate ion | [56] |
| 1S1R | NADP+, 2-methyl-2,4-pentanediol, acetate ion | [56] |
| 1S2A | NADP+, indomethacin, unknown atom or ion, DMSO | [56] |
| 1S2C | NADP+, flufenamic acid, DMSO | [56] |
| 1XF0 | NADP+, 4-androstene-3,17-dione, acetate ion | [57] |
| 2F38 | NADP+, bimatoprost | [58] |
| 1ZQ5 | NADP+, EM1404, acetate ion | [48] |
| 2FGB | NADP+, hexaethylene glycol, acetate ion | [48] |
3.2 Characteristics of the AKR1C3 Substrate Binding Site
In the ten crystal structures of AKR1C3, the enzyme is complexed with the cofactor NADP+ and a second ligand of different types, including PGD2, 4-androstene-3,17-dione, inhibitors, and solvent molecules (Table 1)[48, 55-59]. Close inspection of these structures reveals that the binding site for the second ligand is large and can be dissected into the following sub-sites: oxyanion site, steroid channel, and three sub-pockets that we have namedSP1, SP2, and SP3. One common feature of all ten AKR1C3 structures is an occupied SP1 site (albeit to different degree), while the occupancy of other sites varies with different ligands. The occupancies of sub-sites in AKR1C3 by four inhibitors EM1404, bimatoprost, flufenamic acid, and indomethacin are depicted in Figure 3 (A-D)[48, 56, 58]. The binding mode of PGD2 is similar to that of bimatoprost, while 4-androstene-3,17-dione is bound nonproductively in similar fashion to EM1404 [48, 55, 57, 58].
Figure 3.
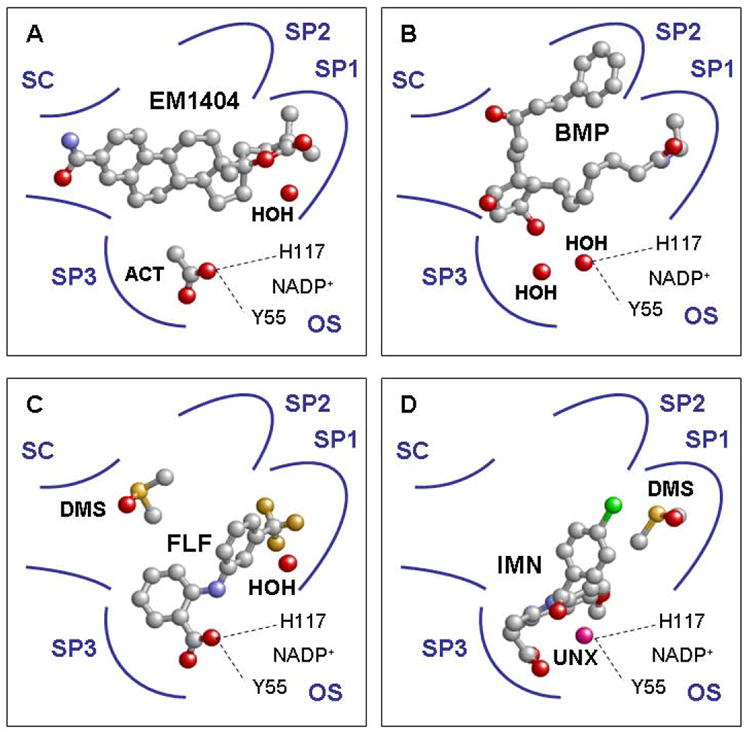
Occupancies of AKR1C3 ligand binding sub-sites by inhibitors EM1404, bimatoprost, flufenamic acid, and indomethacin (A through D). SC =steroid channel; OS = oxyanion site; ACT = acetate ion; HOH = water; BMP = bimatoprost; FLF = flufenamic acid; DMS = dimethyl sulfoxide; IMN = indomethacin; UNX = unknown atom or ion; SP1- 3 = subpockets 1-3. A, the lactone portion of EM1404 is bound in SP1 while the estrone portion extends into the steroid channel (PDB ID:1ZQ5[48]). B, the α-chain of bimatoprost is bound in SP1 while the β-chain of the inhibitor is bound in SP2 (PDB ID:2F38[58]). C, the trifluoromethyl substituted B ring of flufenamic acid is located in SP1 and the carboxylic acid moiety of the moleculeisanchored at the oxyanion site (PDB ID:1S2C[56]). D, the bridge carbonyl group of indomethacin is positioned via an unknown atom located at the oxyanion site. The p-chlorobenzoyl group of indomethacin projects into SP1, while the indole ring and carboxylic acid is bound in the well defined SP3 (PDB ID:1S2A[56]). Ligands are depicted in ball-and-stick representation and the following atomic coloring scheme: C = grey; O = red; N = blue; F = orange; S = yellow; Cl = green; unknown = pink.
The oxyanion siterefers to the conserved site that anchors the oxyanion intermediate formed during the enzyme reaction and consists of the catalytic residues Y55, H117 and NADP+in all AKR1C enzymes [59]. This position is often found occupied by the oxygen atom of a carboxylic acid, ketone, or hydroxyl group of a ligand. Strong hydrogen bonding interactions form between the occupant and Y55 and H117 (2.6 Å and 2.8 Å, respectively). The positioning of the carbonyl group of a substrate at the oxyanion site is believed to be essential for productive binding, since the anchoring brings the carbonyl group in proximity to the cofactor, allowing the reaction to proceed [59-61]. Similarly, it is believed that the binding of the carboxylate or ketone group of an NSAID at the oxyanion site explains the general inhibition of AKR1C enzymes by NSAIDs. In the AKR1C3 structure, the oxyanion sitedid not significantly contribute to the binding of 4-androstene-3,17-dione, EM1404 and bimatoprost (Figure 3A and 3B) in AKR1C3, as the site was occupied by an acetate ion in the crystal structures of the AKR1C3•NADP+•4-androstene-3,17-dione and AKR1C3•NADP+•EM1404 complexes, and by a water moleculein the crystal structure of the AKR1C3•NADP+•bimatoprost complex[48, 57, 58]. In contrast, the carboxylic acid moiety of flufenamic acid isanchored at the oxyanion site of AKR1C3, while the bridge carbonyl group of indomethacin binds via an unidentified atom to the oxyanion site of the enzyme (Figure 3C and 3D)[56].
The steroid channel refers to the elongated open channel that is also conserved in all AKR1C enzymes [59]. W227 and L/V54 are important gate keepers for the steroid channel and determine the positional and stereochemical specificities of the steroid transforming activity of AKR1C enzymes [22, 60, 61]. Interestingly, the steroid channel does not appear to be an important inhibitor binding site for AKR1C3. With the exception of EM1404, which has its steroid ring structure partially occupying the steroid channel, this channel is left empty bybimatoprost, flufenamic acid, and indomethacin (Figure 3)[48, 56, 58].
The SP1 sub-pocket is formed by residues S118, N167, F306, F311, and Y319 in AKR1C3, and is the only site that is occupied in all ten crystal structures of this enzyme. As such, it accommodates the lactone moiety of EM1404 (Figure 3A)[48], the D ring of 4-androstene-3,17-dione [57], the 12β-chain of PGD2[55], the 8α-chain of bimatoprost[58] (Figure 3B), or the –CF3substituted B-ring of flufenamic acid[56](Figure 3C). The p-chlorobenzoyl group of indomethacin also projects into this site, although in that structure a solvent molecule from the crystallization solution (DMSO) was bound in the bottom of SP1 [56] (Figure 3D).
The SP2 sub-pocket refers to a pocket formed by residues W86, S129, W227 and F311 in AKR1C3. This pocket is where the 8α-chain of PGD2 and the 12β-chain of bimatoprost were bound[55, 58]. It appears that this sub-pocket is only used by the two prostanoids.
The SP3 sub-pocket refers to a large pocket lined by residues Y24, E192, S217, S221, Q222, Y305, and F306 in AKR1C3. These residues surround the indole ring and the carboxylate group of the indomethacin molecule[56]. SP3 is not occupied by other ligands in AKR1C3.
The variety of AKR1C3 ligands in structure and size demonstrate the flexibility of the enzyme’s ligand binding site. The key residues to AKR1C3’s ability to accommodate different ligands are W227, F306, and F311. These residues can assume different conformations and result in “induced-fit” to various ligands. W227 controls the sizes of SP2 and the steroid channel. F306 lines the SP1 site for the AKR1C3•NADP+•flufenamic acid complex, but assumes a different rotamer conformation in the crystal of the AKR1C3•NADP+•indomethacin complex, thereby exposing the SP3 pocket [56]. F311 forms part of SP1 for the binding of flufenamic acid, but lines SP2 for the binding of bimatoprost and PGD2 [55, 56, 58].
3.3 Comparison of Sub-Pockets among AKR1C Isoforms
Comparison of the ligand binding sites of AKR1C1-4 reveals that there are considerable structural differences in subpockets between AKR1C3 and the other isoforms [48, 56, 58, 61, 62]. SP1 is significantly larger for AKR1C3. The difference in the size of SP1 is largely due to the shift in main chain positions of residues 305-311 between AKR1C3 and the other enzymes (Figure 4 left panel). As a result, residue 308 collapses inward and significantly reduces the size of SP1 for AKR1C1, AKR1C2 and AKR1C4. In AKR1C3 the side chain of S308 is not involved inligandbinding (>4 Å), but the corresponding residues of L308 in AKR1C2 and M308 in AKR1C4 would extend into SP1. In addition, different residues at positions 118 and 319 also reduce the size of SP1 in AKR1C1/2 and AKR1C4. In AKR1C3 the side-chains of S118 and Y319 are> 4 Å away from the ligand, whereas the corresponding residues of F118 (1.7 Å)and F319 (~1Å) in AKR1C2 would clash with a long ligand such as PGD2 or bimatoprost[55, 58]. Importantly, the serine residue at position 118and the tyrosine at 319 in AKR1C3 are capable of forming hydrogen-bonding interactions with a ligand, i.e., with the amide group of the 8α-chain of bimatoprost[58]. In contrast, the corresponding residues for AKR1C1 and AKR1C2 are phenylalanines, which are incapable of hydrogen bonding with a ligand.
Figure 4.
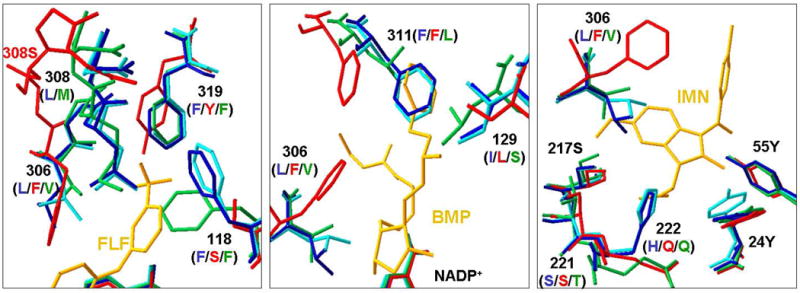
Comparison of SP1, SP2, and SP3 sub-pockets of four human AKR1C enzymes. The crystal structures used for comparisons are AKR1C1•NADP+•20α-hydroxyprogesterone (PDB ID:1MRQ)[62], AKR1C2•NADP+•ursodeoxycholate (PDB ID:1IHI)[61], and AKR1C4•NADP+ (PDB ID:2FVL). The respective AKR1C3 structures in the left, middle and right panels are AKR1C3•NADP+•flufenamic acid (PDB ID:1S2C, for comparison of SP1), AKR1C3•NADP+•bimatoprost (PDB ID:2F38, for comparison of SP2) and AKR1C3•NADP+•indomethacin (PDB ID:1S2A, for comparison of SP3). The residues are shown in stick presentation and in cyan for AKR1C1, blue for AKR1C2, red for AKR1C3, and green for AKR1C4. For clarity, only key residues for binding are shown and are labeled by the number and their respective identities in AKR1C2 (blue), AKR1C3 (red), and AKR1C4 (green). AKR1C1 has the same residues as AKR1C2 at all positions shown. Ligands are shown in gold. FLF = flufenamic acid, BMP = bimatoprost, and IMN = indomethacin.Left, AKR1C3 has a larger SP1 site than the other isoforms due to different mainchain position at 308 and different residues at 118. In addition, S118 of AKR1C3 can form sidechain hydrogen bonding interaction with a ligand, whereas the corresponding residue F118 in other isoforms cannot. Middle, structural differences at positions 129 and 311 make the SP2 pocket shorter for AKR1C1, AKR1C2 and AKR1C4 than for AKR1C3. S129 of AKR1C3 can form sidechain hydrogen bonding interactions with a ligand, whereas the corresponding I129 of AKR1C1/2 and L129 of AKR1C4 cannot. Right, the sidechain of F306 in AKR1C3 assumes a conformation that exposes the SP3 site for indomethacin binding. However, the corresponding L306 of AKR1C1/2 and V306 for AKR1C4 with more rigid sidechains would extend into the indole ring of indomethacin.
No ligands have been observed to occupy the SP2 site in crystals of AKR1C1, AKR1C2, or AKR1C4. Comparison of the SP2 sites of these enzymes shows structural differences at position 129 and 311 that make the SP2 pocket shorter for AKR1C1, AKR1C2, and AKR1C4 than that for AKR1C3 (Figure 4 middle panel). In addition, S129 of AKR1C3 can form hydrogen-bonding interactions with a ligand, i.e., with the carboxylate group of the 8α-chain of PGD2 or with the hydroxyl group on 12β-chain of bimatoprost[55, 58]. In contrast, the corresponding residues in other isoforms are I129 for AKR1C1 and AKR1C2 and L129 for AKR1C4, which are incapable of hydrogen bonding with a ligand.
Structural differences at position 306 also cause the SP3 site of AKR1C3 to differ from those for the other enzymes (Figure 4 right panel). In the AKR1C3•NADP+•indomethacin complex, the sidechain of F306 assumes a conformation that points away from the ligand [56]. However, the corresponding residues, L306 of AKR1C1 and AKR1C2, and V306 of AKR1C4, have more rigid side chains that would clash with the indole ring of indomethacin.
3.4 Use of Crystal Structures in the Rational Design of Isoform-Specific Inhibitors of AKR1C3
To date, the available crystal structures and molecular docking studies of inhibitors using these structures have been mostly used to account for the observed activity of existing inhibitors [48, 53, 56, 58]. These structural studies have revealed features of AKR1C3 that can be exploited in the rational design of new selective inhibitors for this enzyme. However, the complexity of the ligand binding site of AKR1C3 deems the rational design of a new selective inhibitor of AKR1C3 a challenging task. Because of the existence of multiple sites and “induced-fit”, it is not guaranteed that any given inhibitor would bind in the predicted mode. Nonetheless, some progress has been made in using flufenamic acid and indomethacin as lead compounds for rational AKR1C3 inhibitor design [32, 42].
We proposed to use the N-phenylanthranilic acids as templates based on the binding mode of flufenamic acid in AKR1C3. The binding properties of flufenamic acid in AKR1C3 suggest that A-ring substitution can be utilized to eliminate PGHS-inhibition and B-ring substitution may confer selectivity among AKR1C isoforms. Screening of N-phenylanthranilic acid derivatives showed that elimination of the PGHS-inhibition can be readily achieved. However, selectivity among AKR1C isoforms is difficult to obtain [32, 42]. Based on the structural characteristics of the SP1 site of AKR1C3, it is proposed that B-ring substituents larger than the -CF3 group of flufenamic acid and polar groups that would participate in hydrogen-bonding interaction with S118 and/or Y319 of AKR1C3 would provide selectivity against other AKR1C isoforms. Currently N-phenylanthranilic acid derivatives with selectivity for AKR1C3 are being developed by our group.
The lactone moiety was thought to play an important role in the potent inhibition of AKR1C3 by estrogen lactones, including EM1404[48]. In AKR1C3•NADP+•EM1404 complex, the lactone moiety is located at the same site, SP1, as the trifluoromethyl-benzene ring on flufenamic acid. Based on this binding similarity between EM1404 and flufenamic acid, a new non-steroid inhibitor was proposed by replacing the trifluoromethyl-benzene ring of flufenamic acid with a lactone ring. Molecular docking of this new inhibitor into AKR1C3 resulted in the predicted binding conformation [48]. However, experimental data with this N-phenylanthranilic acid-lactone derivative are yet to be obtained and inhibition data for all the lactone containing inhibitors on other AKR1C enzymes are lacking.
Indomethacin is also a promising lead compound as it provided the best selectivity of any NSAID examined for AKR1C3 inhibition against the other AKR1C isoforms. The crystal structure of indomethacin reveals that its binding in AKR1C3 is unique from the other inhibitors in that it is bound mainly in SP3 and only partially uses SP1[48, 56, 58]. It is also interesting that the bridge carbonyl, rather than the terminal carboxylic acid, is anchored near the oxyanion site. Based on the binding mode of indomethacin in AKR1C3, CBM was designed to retain selective inhibition for AKR1C3, but eliminate or reduce inhibition of PGHS enzymes [32]. As predicted, CBM was shown to be a selective AKR1C3 inhibitor, which does not significantly inhibit the other AKR1C enzymes or the PGHS enzymes. However, CBM displayed poor solubility and hence bioavailability. The location of the p-chlorobenzoyl group of indomethacin in the SP1 site also raises the possibility of improving potency and selectivity by fully taking advantage of this pocket.
The binding modes of bimatoprost and PGD2 in AKR1C3 suggest selectivity among AKR1C enzymes may be achieved with cyclopentane derivatives. Both ligands have their long side chains fully extended into SP1 and SP2 sub-pockets of AKR1C3 and form hydrogen bonding interactions with sub-pocket residues. SP1 and SP2 pockets are significantly shorter for AKR1C1, AKR1C2, and AKR1C4 and lack the important residues for hydrogen-bonding interactions. This provides a structural explanation as to why only AKR1C3 displays a sub-micromolar KM for PGD2, and predicts that bimatoprost would not display significant inhibition against AKR1C1, AKR1C2, and AKR1C4. It also explains why no selectivity for AKR1C3 was observed for jasmonic acids [52], since the shorter side chains of these cyclopentane derivativeswould be well accommodated by all AKR1C enzymes. Interestingly, PGD2 and bimatoprost assume reversed cyclopentane and side chain positions, i.e. PGD2 has its 12β-chain in SP1 and 8α-chain in SP2 whereas bimatoprost has its 8α-chain in SP1 and 12β -chain in SP2. Taken together, it would suggest that cyclopentane derivatives with long side chains in trans-configuration containing polar groups may confer selective inhibition for AKR1C3.
4. Conclusion
Accumulating evidence suggests that AKR1C3 plays an important role in the hormone-dependent and hormone-independent cancers. This has led to the increasing interest in the development of AKR1C3 inhibitors. However, selective inhibition is critical, since the other closely related AKR1C enzymes are also ubiquitously expressed and involved in important steroid hormone biotransformation reactions. Among the compounds that have been examined for AKR1C3 inhibition, some inhibitors (e.g. CBM) displayed selectivity against AKR1C but poor bioavailability relative to their potency. Other inhibitors (e.g. EM1404) displayed the desired potent inhibition but their selectivity profile has not been explored. To date, the available crystal structures have been mostly used to account for the observed activity of existing inhibitors, however limited progress has been made in the rational design of new inhibitors using these structures. A potent yet selective AKR1C3 inhibitor remains to be developed.
• AKR1C3 produces steroids and prostaglandins for receptor mediated growth
• AKR1C3 inhibition is desirable for hormone dependent and independent cancers.
• NSAIDs, steroids, flavonoids, cyclopentanes, and benzodiazepines inhibit AKR1C3.
• Crystal structures identify subpockets for rational design of AKR1C3 inhibitors.
• Differences in AKR1C subpockets can be exploited for selective inhibition
Acknowledgments
Funding : Supported by R01-CA90744 and P30-ES013508awarded to T.M.P. M.C.B. was funded by NIH training grants T32-DK007314-25 and T32-HD007305-22. Y.J. was also funded by a FOCUS-Junior Investigator Award from the Kynett foundation.
Abbreviations
- AKR
aldo-keto reductase
- CBM
N-(4-chlorobenzoyl)-melatonin
- 15dPGJ2
15-deoxy- Δ12,14-prostaglandin J2
- 5α-DHT
5α-dihydrotestosterone
- MPA
medroxyprogesterone acetate
- NSAID
non-steroidal anti-inflammatory drug
- PG
prostaglandin
- PGHS
prostaglandin H synthase
Footnotes
The Aldo-Keto Reductase (AKR) protein superfamily nomenclature was developed to annotate family members based on sequence homology rather than enzymatic activity. This is because the same enzyme was given different trivial names based on the enzymatic reaction measured and led to considerable confusion. For example, aldo-keto reductase 1C3 (AKR1C3) has been referred to as: type 5 17β-hydroxysteroid dehydrogenase, prostaglandin F synthase, type 2 3α-hydroxysteroid dehydrogenase; and dihydrodiol dehydrogenase X. The nomenclature was originally adopted by the 6th International Workshop on the Enzymology and Molecular Biology of Carbonyl Metabolism held in Deadwood, South Dakota in 1996 (see, Jez et al., Biochem. Pharmacol. 54 (1997) 639). It has since been adopted by the Human Genome Project (please visit: www.med.upenn.edu/akr).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Penning TM. Human hydroxysteroid dehydrogenases and pre-receptor regulation: Insights into inhibitor design and evaluation. J Steroid Biochem Mol Biol. 2010 doi: 10.1016/j.jsbmb.2011.01.009. current issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marchais-Oberwinkler S, Hennl C, Möller G, Klein T, Negri M, Oster A, Spadaro A, Werth R, Wetzel M, Xu K, Frotscher M, Hartmann RW, Adamski J. 17β-Hydroxysteroid dehydrogenase (17β-HSD): gene, protein structures, novel therapeutic targets and recent progress in inhibitor development. J Steroid Biochem Mol Biol. 2010 doi: 10.1016/j.jsbmb.2010.12.013. current issue. [DOI] [PubMed] [Google Scholar]
- 3.Poirier D. Contribution to the development of inhibitors of 17β-hydroxysteroid dehydrogenase types 1 and 7: key tools for studying and treating estrogen-dependent diseases. J Steroid Biochem Mol Biol. 2010 doi: 10.1016/j.jsbmb.2010.12.007. current issue. [DOI] [PubMed] [Google Scholar]
- 4.Byrns MC, Duan L, Lee SH, Blair IA, Penning TM. Aldo-keto reductase 1C3 expression in MCF-7 cells reveals roles in steroid hormone and prostaglandin metabolism that may explain its over-expression in breast cancer. J Steroid Biochem Mol Biol. 2010;118:177–187. doi: 10.1016/j.jsbmb.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dufort I, Rheault P, Huang X-F, Soucy P, Luu-The V. Characteristics of a highly labile human type 5 17β-hydroxysteroid dehydrogenase. Endocrinology. 1999;140:568–574. doi: 10.1210/endo.140.2.6531. [DOI] [PubMed] [Google Scholar]
- 6.Sharma KK, Lindqvist A, Zhou XJ, Auchus RJ, Penning TM, Andersson S. Deoxycorticosterone inactivation by AKR1C3 in human mineralocorticoid tissues. Mol Cell Endocrinol. 2006;248:79–86. doi: 10.1016/j.mce.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki-Yamamoto T, Nishizawa M, Fukui M, Okuda-Ashitaka E, Nakajima T, Ito S, Watanabe K. cDNA cloning, expression and characterization of human prostaglandin F synthase. FEBS Lett. 1999;462:335–340. doi: 10.1016/s0014-5793(99)01551-3. [DOI] [PubMed] [Google Scholar]
- 8.Koda N, Tsutsui Y, Niwa H, Ito S, Woodward DF, Watanabe K. Synthesis of prostaglandin F ethanolamide by prostaglandin F synthase and identification of bimatoprost as a potent inhibitor of the enzyme: new enzyme assay method using LC/ESI/MS. Arch Biochem & Biophys. 2004;424:128–136. doi: 10.1016/j.abb.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 9.Matsuura K, Shiraishi H, Hara A, Sato K, Deyashiki Y, Nimomiya M, Sakai S. Identification of a principal mRNA species for human 3α-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J Biochem. 1998;124:940–946. doi: 10.1093/oxfordjournals.jbchem.a022211. [DOI] [PubMed] [Google Scholar]
- 10.Jabbour HN, Sales KJ, Boddy SC, Anderson RA, Williams AR. A positive feedback loop that regulates cyclooxygenase-2 expression and prostaglandin F2α synthesis via the F-series-prostanoid receptor and extracellular signal-regulated kinase 1/2 signaling pathway. Endocrinology. 2005;146:4657–4664. doi: 10.1210/en.2005-0804. [DOI] [PubMed] [Google Scholar]
- 11.Ray DM, Akbiyik F, Phipps RP. The peroxisome proliferator-activated receptor γ (PPARγ) ligands 15-deoxy-Δ12,14-prostaglandin J2 and ciglitazone induce human B lymphocyte and B cell lymphoma apoptosis by PPARγ-independent mechanisms. J Immunol. 2006;177:5068–5076. doi: 10.4049/jimmunol.177.8.5068. [DOI] [PubMed] [Google Scholar]
- 12.Diers AR, Dranka BP, Ricart KC, Oh JY, Johnson MS, Zhou F, Pallero MA, Bodenstine TM, Murphy-Ullrich JE, Welch DR, Landar A. Modulation of mammary cell migration by 15-deoxy-Δ12,14-prostaglandin J2: implications for anti-metastatic therapy. Biochem J. 2010;430:69–78. doi: 10.1042/BJ20091193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakata S, Yoshida T, Shiraiahi T, Horinaka M, Kouhara J, Wakada M, Sakai T. 15-Deoxy-Δ12,14-prostaglandin J2 induces death receptor 5 expression through mRNA stabilization independently of PPARγ and potentiates TRAIL-induced apoptosis. Mol Cancer Therapeutics. 2006;5:1827–1835. doi: 10.1158/1535-7163.MCT-06-0023. [DOI] [PubMed] [Google Scholar]
- 14.Butler R, Mitchell SH, Tindall DJ, Young CY. Nonapoptotic cell death associated with S-phase arrest of prostate cancer cells via the peroxisome proliferator-activated receptor γ ligand, 15-deoxy-Δ12,14-prostaglandin J2. Cell Growth Differ. 2000;11:49–61. [PubMed] [Google Scholar]
- 15.Scher JU, Pillinger MH. The anti-inflammatory effects of prostaglandins. J Investig Med. 2009;57:703–708. doi: 10.2310/JIM.0b013e31819aaa76. [DOI] [PubMed] [Google Scholar]
- 16.Penning TM, Jin Y, Rizner TL, Bauman DR. Pre-receptor regulation of the androgen receptor. Mol Cell Endocrinol. 2008;281:1–8. doi: 10.1016/j.mce.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD, Knudsen B, Hess DL, Nelson CC, Matsumoto AM, Bremner WJ, Gleave ME, Nelson PS. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007;67:5033–5041. doi: 10.1158/0008-5472.CAN-06-3332. [DOI] [PubMed] [Google Scholar]
- 19.Smuc T, Rizner TL. Aberrant pre-receptor regulation of estrogen and progesterone action in endometrial cancer. Mol Cell Endocrinol. 2009;301:74–82. doi: 10.1016/j.mce.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 20.Khanim FL, Hayden RE, Birtwistle J, Lodi A, Tiziani S, Davies NJ, Ride JP, Viant MR, Gunther UL, Mountford JC, Schrewe H, Green RM, Murray JA, Drayson MT, Bunce CM. Combined bezafibrate and medroxyprogesterone acetate: potential novel therapy for acute myeloid leukaemia. PLoS One. 2009;4:e8147. doi: 10.1371/journal.pone.0008147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guise CP, Abbattista MR, Singleton RS, Holford SD, Connolly J, Dachs GU, Fox SB, Pollock R, Harvey J, Guilford P, Donate F, Wilson WR, Patterson AV. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res. 2010;70:1573–1584. doi: 10.1158/0008-5472.CAN-09-3237. [DOI] [PubMed] [Google Scholar]
- 22.Lin H-K, Steckelbroeck S, Fung K-M, Jones AN, Penning TM. Characterization of a monoclonal antibody for human aldo-keto reductase AKR1C3 (type 2 3α-hydroxysteroid dehydrogenase/type 5 17β-hydroxysteroid dehydrogenase); immunohistochemical detection in breast and prostate. Steroids. 2004;69:795–801. doi: 10.1016/j.steroids.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 23.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 24.Hofland J, van Weerden WM, Dits NF, Steenbergen J, van Leenders GJ, Jenster G, Schroder FH, de Jong FH. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res. 2010;70:1256–1264. doi: 10.1158/0008-5472.CAN-09-2092. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura Y, Suzuki T, Nakabayashi M, Endoh M, Sakamoto K, Mikami Y, Moriya T, Ito A, Takahashi T, Yamada S, Arai Y, Sasano H. In situ androgen producing enzymes in human prostate. Endocrine-Related Cancer. 2005;12:101–107. doi: 10.1677/erc.1.00914. [DOI] [PubMed] [Google Scholar]
- 26.Jansson AK, Gunnarsson C, Cohen M, Sivik T, Stal O. 17β-hydroxysteroid dehydrogenase 14 affects estradiol levels in breast cancer cells and is a prognostic marker in estrogen receptor-positive breast cancer. Cancer Res. 2006;66:11471–11477. doi: 10.1158/0008-5472.CAN-06-1448. [DOI] [PubMed] [Google Scholar]
- 27.Bauman DR, Steckelbroeck S, Williams MV, Peehl DM, Penning TM. Identification of the major oxidative 3α-hydroxysteroid dehydrogenase in human prostate that converts 5α-androstane-3α,17β-diol to 5α-dihydrotestosterone: A potential therapeutic target for androgen dependent disease. Mol Endocrinol. 2006;20:444–458. doi: 10.1210/me.2005-0287. [DOI] [PubMed] [Google Scholar]
- 28.Smuc T, Hevir N, Ribic-Pucelj M, Husen B, Thole H, Rizner TL. Disturbed estrogen and progesterone action in ovarian endometriosis. Mol Cell Endocrinol. 2009;301:59–64. doi: 10.1016/j.mce.2008.07.020. [DOI] [PubMed] [Google Scholar]
- 29.Breuiller-Fouche M, Leroy MJ, Dubois O, Reinaud P, Chissey A, Qi H, Germain G, Fortier MA, Charpigny G. Differential expression of the enzymatic system controlling synthesis, metabolism, and transport of PGF2α in human fetal membranes. Biol Reprod. 2010;83:155–162. doi: 10.1095/biolreprod.109.080390. [DOI] [PubMed] [Google Scholar]
- 30.Andersson S, Minjarez D, Yost NP, Word RA. Estrogen and progesterone metabolism in the cervix during pregnancy and parturition. J Clin Endocrinol Metab. 2008;93:2366–2374. doi: 10.1210/jc.2007-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loudon JA, Groom KM, Bennett PR. Prostaglandin inhibitors in preterm labour. Best Pract Res Clin Obstet Gynaecol. 2003;17:731–744. doi: 10.1016/s1521-6934(03)00047-6. [DOI] [PubMed] [Google Scholar]
- 32.Byrns MC, Steckelbroeck S, Penning TM. An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3α-HSD, type 5 17β-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem Pharmacol. 2008;75:484–493. doi: 10.1016/j.bcp.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reginato MJ, Krakow SL, Bailey ST, Lazar MA. Prostaglandins promote and block adipogenesis through opposing effects on peroxisome proliferator-activated receptor γ. J Biol Chem. 1998;273:1855–1858. doi: 10.1074/jbc.273.4.1855. [DOI] [PubMed] [Google Scholar]
- 34.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 35.Penning TM, Burczynski ME, Jez JM, Hung C-F, Lin H-K, Ma H, Moore M, Palackal N, Ratnam K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM. Human cytosolic 3α-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3β-hydroxysteroid dehydrogenase activity: Implications for steroid hormone metabolism and action. J Biol Chem. 2003;279:10784–10795. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- 37.DuBois RN, Smalley WE. Cyclooxygenase, NSAIDs, and colorectal cancer. J Gastroenterol. 1996;31:898–906. doi: 10.1007/BF02358623. [DOI] [PubMed] [Google Scholar]
- 38.Janssen A, Maier TJ, Schiffmann S, Coste O, Seegel M, Geisslinger G, Grosch S. Evidence of COX-2 independent induction of apoptosis and cell cycle block in human colon carcinoma cells after S- or R-ibuprofen treatment. Eur J Pharmacol. 2006;540:24–33. doi: 10.1016/j.ejphar.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 39.Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD. Postmenopausal hormone replacement therapy: scientific review. JAMA. 2002;288:872–881. doi: 10.1001/jama.288.7.872. [DOI] [PubMed] [Google Scholar]
- 40.Neuhouser ML. Dietary flavonoids and cancer risk: evidence from human population studies. Nutr Cancer. 2004;50:1–7. doi: 10.1207/s15327914nc5001_1. [DOI] [PubMed] [Google Scholar]
- 41.Penning TM, Talalay P. Inhibition of a major NAD(P)+-linked oxidoreductase from rat liver cytosol by steroidal and nonsteroidal anti-inflammatory agents and by prostaglandins. Proc Natl Acad Sci USA. 1983;80:4504–4508. doi: 10.1073/pnas.80.14.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bauman DR, Rudnick S, Szewczuk L, Sridhar G, Penning TM. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol Pharmacol. 2005;67:60–68. doi: 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]
- 43.Steckelbroeck S, Oyesanmi B, Jin Y, Lee SH, Kloosterboer HJ, Penning TM. Tibolone metabolism in human liver is catalyzed by 3α/3β-hydroxysteroid dehydrogenase activities of the four isoforms of the aldo-keto reductase (AKR)1C subfamily. J Pharmacol Exp Ther. 2006;316:1300–1309. doi: 10.1124/jpet.105.091587. [DOI] [PubMed] [Google Scholar]
- 44.Byrns MC, Penning TM. Type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase (AKR1C3): role in breast cancer and inhibition by non-steroidal anti-inflammatory drug analogs. Chem Biol Interact. 2009;178:221–227. doi: 10.1016/j.cbi.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Higaki Y, Usami N, Shintani S, Ishikura S, El-Kabbani O, Hara A. Selective and potent inhibitors of human 20α-hydroxysteroid dehydrogenase (AKR1C1) that metabolizes neurosteroids derived from progesterone. Chem Biol Interact. 2003;143-144:503–513. doi: 10.1016/s0009-2797(02)00206-5. [DOI] [PubMed] [Google Scholar]
- 46.Poirier D. Inhibitors of 17β-hydroxysteroid dehydrogenases. Curr Med Chem. 2003;10:453–477. doi: 10.2174/0929867033368222. [DOI] [PubMed] [Google Scholar]
- 47.Bydal P, Luu-The V, Labrie F, Poirier D. Steroidal lactones as inhibitors of 17β-hydroxysteroid dehydrogenase type 5: chemical synthesis, enzyme inhibitory activity, and assessment of estrogenic and androgenic activities. Eur J Med Chem. 2009;44:632–644. doi: 10.1016/j.ejmech.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 48.Qiu W, Zhou M, Mazumdar M, Azzi A, Ghanmi D, Luu-The V, Labrie F, Lin SX. Structure-based inhibitor design for an enzyme that binds different steroids: a potent inhibitor for human type 5 17β-hydroxysteroid dehydrogenase. J Biol Chem. 2007;282:8368–8379. doi: 10.1074/jbc.M606784200. [DOI] [PubMed] [Google Scholar]
- 49.Krazeisen A, Breitling R, Moller G, Adamski J. Human 17β-hydroxysteroid dehydrogenase type 5 is inhibited by dietary flavonoids. Adv Exp Med Biol. 2002;505:151–161. doi: 10.1007/978-1-4757-5235-9_14. [DOI] [PubMed] [Google Scholar]
- 50.Skarydova L, Zivna L, Xiong G, Maser E, Wsol V. AKR1C3 as a potential target for the inhibitory effect of dietary flavonoids. Chem Biol Interact. 2009;178:138–144. doi: 10.1016/j.cbi.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 51.Brozic P, Golob B, Gomboc N, Rizner TL, Gobec S. Cinnamic acids as new inhibitors of 17β-hydroxysteroid dehydrogenase type 5 (AKR1C3) Mol Cell Endocrinol. 2006;248:233–235. doi: 10.1016/j.mce.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 52.Davies NJ, Hayden RE, Simpson PJ, Birtwistle J, Mayer K, Ride JP, Bunce CM. AKR1C isoforms represent a novel cellular target for jasmonates alongside their mitochondrial-mediated effects. Cancer Res. 2009;69:4769–4775. doi: 10.1158/0008-5472.CAN-08-4533. [DOI] [PubMed] [Google Scholar]
- 53.Stefane B, Brozic P, Vehovc M, Rizner TL, Gobec S. New cyclopentane derivatives as inhibitors of steroid metabolizing enzymes AKR1C1 and AKR1C3. Eur J Med Chem. 2009;44:2563–2571. doi: 10.1016/j.ejmech.2009.01.028. [DOI] [PubMed] [Google Scholar]
- 54.Usami N, Yamamoto T, Shintani S, Ishikura S, Higaki Y, Katagiri Y, Hara A. Substrate specificity of human 3(20)α-hydroxysteroid dehydrogenase for neurosteroids and its inhibition by benzodiazepines. Biol Pharm Bull. 2002;25:441–445. doi: 10.1248/bpb.25.441. [DOI] [PubMed] [Google Scholar]
- 55.Komoto J, Yamada T, Watanabe K, Takusagawa F. Crystal structure of human prostaglandin F synthase (AKR1C3) Biochemistry. 2004;43:2188–2198. doi: 10.1021/bi036046x. [DOI] [PubMed] [Google Scholar]
- 56.Lovering AL, Ride JP, Bunce CM, Desmond JC, Cummings SM, White SA. Crystal structures of prostaglandin D2 11-ketoreductase (AKR1C3) in complex with the nonsteroidal anti-inflammatory drugs flufenamic acid and indomethacin. Cancer Res. 2004;64:1802–1810. doi: 10.1158/0008-5472.can-03-2847. [DOI] [PubMed] [Google Scholar]
- 57.Qiu W, Zhou M, Labrie F, Lin SX. Crystal structures of the multispecific 17β-hydroxysteroid dehydrogenase type 5: critical androgen regulation in human peripheral tissues. Mol Endocrinol. 2004;18:1798–1807. doi: 10.1210/me.2004-0032. [DOI] [PubMed] [Google Scholar]
- 58.Komoto J, Yamada T, Watanabe K, Woodward DF, Takusagawa F. Prostaglandin F2α formation from prostaglandin H2 by prostaglandin F synthase (PGFS): crystal structure of PGFS containing bimatoprost. Biochemistry. 2006;45:1987–1996. doi: 10.1021/bi051861t. [DOI] [PubMed] [Google Scholar]
- 59.Bennett MJ, Albert RH, Jez JM, Ma H, Penning TM, Lewis M. Steroid recognition and regulation of hormone action: crystal structure of testosterone and NADP+ bound to 3α-hydroxysteroid/dihydrodiol dehydrogenase. Structure. 1997;5:799–812. doi: 10.1016/s0969-2126(97)00234-7. [DOI] [PubMed] [Google Scholar]
- 60.Jin Y, Penning TM. Molecular docking simulations of steroid substrates into human cytosolic hydroxysteroid dehydrogenases (AKR1C1 and AKR1C2): insights into positional and stereochemical preferences. Steroids. 2006;71:380–391. doi: 10.1016/j.steroids.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 61.Jin Y, Stayrook SE, Albert RH, Palackal NT, Penning TM, Lewis M. Crystal structure of human type III 3α-hydroxysteroid dehydrogenase/bile acid binding protein complexed with NADP+ and ursodeoxycholate. Biochemistry. 2001;40:10161–10168. doi: 10.1021/bi010919a. [DOI] [PubMed] [Google Scholar]
- 62.Couture JF, Legrand P, Cantin L, Luu-The V, Labrie F, Breton R. Human 20α-hydroxysteroid dehydrogenase: crystallographic and site-directed mutagenesis studies lead to the identification of an alternative binding site for C21-steroids. J Mol Biol. 2003;331:593–604. doi: 10.1016/s0022-2836(03)00762-9. [DOI] [PubMed] [Google Scholar]