

Abstract
The Ataxia-Telangiectasia mutated (ATM) kinase is regarded as the major regulator of the cellular response to DNA double strand breaks (DSBs). In response to DSBs, ATM dimers dissociate into active monomers in a process promoted by the Mre11-Rad50-Nbs1 (MRN) complex. ATM can also be activated by oxidative stress directly in the form of exposure to H2O2. The active ATM in this case is a disulfide-crosslinked dimer containing two or more disulfide bonds. Mutation of a critical cysteine residue in the FATC domain involved in disulfide bond formation specifically blocks ATM activation by oxidative stress. Here we show that ATM activation by DSB s is inhibited in the presence of H2O2 because oxidation blocks the ability of MRN to bind to DNA . However, ATM activation via direct oxidation by H2O2 complements the loss of MRN/DSB-dependent activation and contributes significantly to the overall level of ATM activity in the presence of both DSB s and oxidative stress.
Key words: ATM, DNA repair, double-strand break, oxidative stress, ROS
Ataxia-Telangiectasia
Ataxia-Telangiectasia (A-T) is a rare autosomal recessive disorder (1 in 40,000 to 100,000 live births worldwide).1 The most striking clinical manifestation of A-T is neurodegeneration, which is characterized by progressive difficulty with motor coordination beginning in early childhood (progressive cerebellar ataxia). A-T patients also have small clusters of enlarged ocular blood vessels (oculocutaneous telangiectasia), a weakened immune system and increased risk of developing cancer, particularly lymphomas and leukemias.2,3
The ATM kinase was identified as the product of the gene that is mutated in A-T.4 Over 400 ATM mutations have been identified in A-T patients, most of which are truncating mutations which generate shorter and unstable forms of the ATM protein. Cells from A-T patients are hypersensitive to ionizing radiation (IR) and deficient in DNA damage-induced checkpoint activation at the G1/S, intra-S and G2/M phases of the cell cycle.3
ATM is a member of the phosphoinositide 3-kinase-related protein kinase (PIKK) family, which also includes ATM and Rad3-related-protein kinase (ATR), the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs), and mammalian target of rapamycin (mTOR).5 ATM regulates the DNA damage response by phosphorylating its downstream targets at specific S/T(Q) sites in response to DSBs. ATM also responds to physiological DNA breaks during the development and differentiation of lymphocytes. Chromosomal abnormalities and accumulation of unrepaired ends during V(D)J recombination and class switch recombination were observed in B cells from A-T patients and ATM-deficient mice, indicating that ATM deficiency impairs site-specific recombination in cells of the immune system.6,7 The increased frequency of defective repair during these processes may explain the high incidence of lymphoid cancer among A-T patients.8
ATM safeguards genome integrity in mammalian cells by regulating the activation of cell cycle checkpoints. p53 was the first substrate of ATM to be identified,9,10 and ATM-mediated phosphorylation of p53 on Ser15 is important for the G1/S cell cycle checkpoint.11 ATM also phosphorylates and activates checkpoint kinase 2 (Chk2),12 which in turn phosphorylates several substrates including p53, BRCA1, CDC25A and CDC25C.13,14 ATM can also regulate the intra-S checkpoint by phosphorylating the SMC1 component of cohesin, and inhibition of this phosphorylation abrogates the S-phase checkpoint.15 A recent combined proteomic and functional checkpoint screen showed that nine proteins of the ubiquitin-proteasome system (UPS) were also phosphorylated by ATM and that they are also required for mammalian DNA damage checkpoint control, particularly the G1/S cell cycle checkpoint.16 A global analysis of protein phosphorylation induced by DSBs showed that over 700 proteins are targeted by ATM following DNA damage, including many proteins involved in DNA repair and cell cycle regulation, but also many with no obvious link to the DNA damage response.17
The primary role of ATM in the DNA damage response is checkpoint signaling, and for many years ATM was thought to have no role in repair because A-T cells show nearly wild-type levels of DNA repair under normal conditions.18 But ATM has been shown to contribute to the repair of DSBs after IR. In the absence of ATM, approximately 10% of DSBs are left unrepaired in A-T cells after IR exposure.19 One explanation for this observation is that a subset of DSBs localized to heterochromatin require ATM to facilitate entry of the DNA-repair machinery by phosphorylating the transcriptional corepressor Kruppel-associated box (KRAB)-associated protein-1 (KAP1). In addition, the high level of unresolved DNA breaks created during V(D)J and class switch recombination in A-T cells suggest that there is also an important role for ATM in non-homologous end joining (NHEJ), but the underlying mechanisms are not understood. DNA breaks that form in A-T cells by any pathway are much more likely to persist through multiple cell cycles compared to wild-type cells since ATM is responsible for directing cells into senescence or apoptosis in response to DSBs.20,21
The neurodegenerative phenotype in A-T patients has been attributed to the genomic instability caused by ATM deficiency. However, in post-mitotic neurons, ATM is not required to stop cell cycle progression in response to DNA damage. In the absence of cell cycle progression, ATM deficiency should have a limited impact on genomic stability because A-T cells show nearly normal levels of DNA repair. Thus, the loss of differentiated non-dividing neuronal cells in A-T patients cannot be explained by genomic instability alone.
ATM Deficiency and Oxidative Stress
The primary site of cellular degeneration in A-T patients is the cerebellum.1 Considering the high level of metabolism in neuronal cells and high oxygen concentration in brain tissue, it is possible that oxidative stress could significantly contribute to the degenerative process. Abnormal control of reactive oxygen species (ROS) has been reported in A-T based on the observations showing that the levels and function of antioxidant systems are lower in A-T cells compared to normal cells.22 Clinical research has shown that A-T patients have significantly reduced levels of total plasma antioxidants to about 77% of normal capacity.23 A-T cells have reduced levels of the important antioxidants vitamin A and E, impaired GSH biosynthesis and reduced levels of NADH and NADPH.22,24,25 A-T fibroblasts are also more sensitive to oxidative stress than healthy fibroblasts,22 and increased levels of ROS generated in ATM-deficient brain tissue under physiological conditions are correlated with increased levels of protein and lipid damage.26 Progressive deterioration of redox balance was also observed in stem cell populations from ATM-deficient mice.25 These mice showed progressive bone marrow failure resulting from a defect in the function of haematopoietic stem cells (HSC) that was associated with elevated reactive oxygen species. Treatment with anti-oxidative agents restored the reconstitutive capacity of HSC and prevented bone marrow failure.27 Observations from several laboratories have shown that oxidative stress in ATM-deficient cells can be alleviated by the antioxidants catalase and N-acetyl cysteine (NAC) which neutralize ROS production.27,28
Why does the loss of ATM, a regulator of the cellular DSBs response, lead to oxidative stress? Our recent work shows that ATM acts as a redox sensor in addition to its role as a sensor of DSBs.29 We hypothesize that the changes in redox balance observed in cells from A-T patients are a result of an inability to sense ROS and may be responsible for the neurodegeneration and other unexplained phenotypes observed in A-T patients.
Results
Direct activation of ATM by oxidation.
There are many observations in the literature that link ATM with oxidative stress but it has not been clear if the association is indirect through the formation of DNA damage.22,30 To test our hypothesis that ATM can be activated by ROS directly, we treated primary human fibroblasts with the stable oxidant H2O2 in comparison to treatment with bleomycin, a genotoxic agent that induces DSBs. The hallmark of ATM activation, ATM autophosphorylation,31 occurred in response to H2O2 as well as to bleomycin treatment.29 p53 and Chk2 phosphorylation by ATM were also observed, but histone H2AX phosphorylation, a marker for DNA DSBs, occurred only with bleomycin treatment. Thus, ATM activation induced by H2O2 can occur in the apparent absence of DSBs. Similar to histone H2AX, the heterochromatin protein Kap1 was phosphorylated after bleomycin exposure but not after H2O2 treatment. ATM-dependent p53 phosphorylation was also observed in ATLD cells which are deficient in DSBinduced ATM signaling.32 These results suggested that ATM can be activated directly by oxidative stress independent of DSBs and the activation mechanism is distinct from MRN/DSB-dependent activation.
In mammalian cells, ATM is initially an inactive, noncovalently-associated dimer31 but converts to an active monomer upon DNA damage in the presence of the Mre11/Rad50/Nbs1 (MRN) complex. Our laboratory has previously reconstituted DNA-dependent ATM activation in vitro with purified recombinant proteins, demonstrating that inactive ATM can be strongly activated by the addition of both MRN complex and linear DNA ends.33,34 In our experiments investigating the effects of oxidation on ATM, we found that the addition of H2O2 to purified dimeric ATM in vitro stimulates its activity on a p53 substrate even in the absence of MRN and DNA.29 The magnitude of this stimulation was similar to that observed with MRN and DNA; however, ATM activation by H2O2 could be completely inhibited by the reducing reagent NAC, while the stimulation by MRN and DNA was unaffected. Unlike the MRN/DNA stimulation that induces ATM monomerization, ATM treated with H2O2 formed covalent dimers that were sensitive to reducing reagents, showing that intermolecular disulfide bonds form between ATM monomers and suggesting that these are important for the activation of ATM by oxidative stress.29
Since ATM responds to DSBs and oxidative stress with distinct activation mechanisms, we reasoned that we might be able to identify a mutation that specifically blocks the oxidation pathway. Mutation of cysteine residue 2991, the only cysteine in the FRAP/ATM/TRRAP C-terminal (FATC) domain (Fig. 1A), resulted in an ATM mutant that could be fully activated by MRN and DNA, but could not be activated by H2O2 in vitro.29 Expression of this allele (C2991L) in human lymphocytes lacking wild-type ATM completely blocked the apoptosis response to ROS while remaining competent for apoptosis in response to DNA damage, confirming that this allele specifically blocks oxidation-induced ATM activation in human cells as well.
Figure 1.

The FATC domain of ATM is critical for its activation. (A) A diagram of ATM domains including the FAT, PI3K kinase and FATC domains, C2991 and deletion point in the R3047X mutant allele. (B) Kinase assays were performed as described in reference 29, except with ATM wild-type or N2963X mutant proteins. Phosphorylation of a GST-p53 substrate was assessed by western blotting with an antibody specific for phospho-p53 serine 15 (Calbiochem, PC461). (C) An alignment of the ATM FATC domain from various species. Cysteine 2991 is marked with an asterisk. (D) The phylogram of ATM sequence alignment generated by ClustalW2. The PHYLIP method was used by ClustalW2 to generate the branching diagram. ClustalW2 phylogenetic calculations are based on the neighbor-joining method.60 Branch lengths are proportional to the amount of inferred evolutionary change.
An oxidation-specific mutation causes A-T.
Similar to the C2991L ATM protein, a mutant lacking the last ten amino acids of the ATM C-terminus results in a mutant protein (R3047X, Fig. 1A) that can be fully activated by MRN and DNA but cannot be activated by oxidation in vitro.29 Interestingly, R3047X has been identified as a causative mutation in several A-T patients.35–37 These patients exhibit ataxia, but in some cases were classified as A-T “variants” because they did not exhibit immunodeficiency and cells from these patients only showed moderate radiosensitivity, perhaps indicating a normal response to DSBs. Consistent with this hypothesis, we found that cells from an A-T patient expressing the R3047X allele were also specifically deficient in oxidative activation of ATM but showed normal ATM activation in response to DNA damage.29 These results suggest that the clinical manifestations of A-T, including neurodegeneration and ataxia, may be primarily due to the loss of oxidationdependent ATM signaling.
The FATC domain of ATM.
The FATC domains comprise the C-terminal regions of PIKKs. In some studies, FATC domains referred to the C-terminal, highly conserved domains of approximately 20–30 amino acids, and the regions between these FATC domains and the kinase domains were called the regulatory domains. Here we consider the entire C terminus adjacent to the kinase domains as FATC domains because they were proposed to be critical for kinase activity of PIKKs.38,39 A recent crystal structure of DNA-PKcs showed that the α-helical FATC domain protrudes from the kinase domain and interacts with the FAT domain, consistent with previous studies with low resolution electron microscopy.40–42 Acetylation of lysine 3016 in the FATC domain of ATM by histone acetyltransferase Tip60 also has been shown to activate the kinase activity of ATM in response to DNA damage.43 To test the role of the FATC domain of ATM, we expressed and purified an ATM mutant that lacks the entire FATC domain (N2963X). In vitro kinase assays comparing the N2963X mutant with wild-type ATM showed that neither DSBs nor H2O2 can activate ATM lacking the entire FATC domain (Fig. 1B).
Our recent work identified cysteine 2991 in the FATC domain as a crucial residue for the activation of ATM via oxidation.29 This residue is conserved in terrestrial vertebrates but not in marine animals, and is not present in lower eukaryotes (Fig. 2C and D). The land invasion by vertebrates during late Paleozoic time period coincides with the spike of atmospheric oxygen to nearly 35 percent,44 perhaps suggesting that gaining the function of oxidative stress sensing by ATM might contribute to the survival of terrestrial vertebrates in a high oxygen environment.
Figure 2.
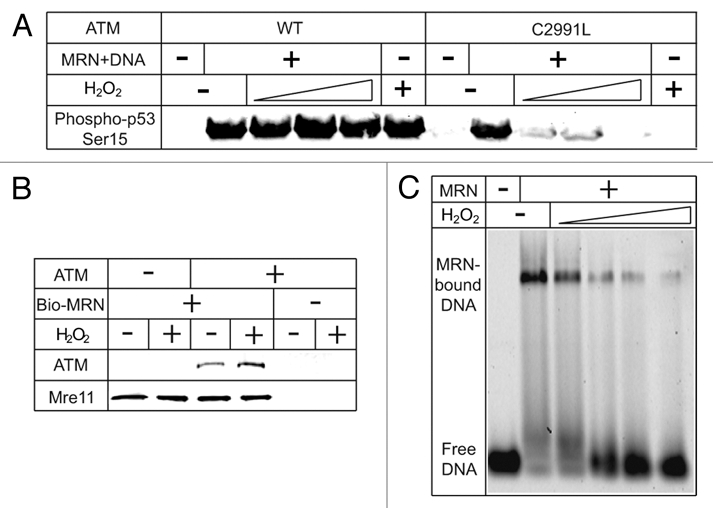
ATM-dependent DNA damage responses in the presence of oxidative stress. (A) Oxidative activation of ATM complements the loss of MRN/DSBs-dependent activation. Kinase assays with ATM wild-type or C2991L were performed as described in reference 29, in the presence of various amounts of H2O2 (0.27, 0.81 and 2.4 mM). (B) H2O2 does not affect MRN binding to ATM. Biotinylated MRN was incubated with ATM in the presence or absence of H2O2 (0.5 mM). MRN was isolated with streptavidin beads and analyzed by western blotting for Mre11 and ATM as indicated. (C) H2O2 inhibits MRN binding to linear DNA in a gel shift assay. Purified MRN was incubated with various amounts of H2O2 (34.4, 68.8, 137.5, 275 µM) before binding to a Cy5-labeled 41 bp dsDNA substrate (37°C, 10 minutes). The binding reaction was resolved on an agarose gel (0.7%, 0.5x TBE) and visualized for Cy5 fluorescence.
The protein kinase mTOR, a member of the PIKK family that regulates cell growth and proliferation in response to cellular nutrition and energy level, was also shown to be oxidized at two conserved cysteines within the C-terminal FATC domain.45 An intramolecular disulfide bond between these cysteines was demonstrated in vitro, and mutation of these cysteines altered the stability of mTOR in budding yeast. These results suggest that cellular redox potential could affect the amount of the mTOR protein by influencing its degradation, and is consistent with the FATC domain playing an important role in regulation of the PIKK family.
Interestingly, the ATM FATC domain was proposed to contain a peroxisome localization signal, and ATM has been observed in the cytoplasm co-localizing with peroxisomes.26 Significantly decreased catalase activity and increased lipid peroxidation in several A-T cell lines suggests that ATM might affect peroxisome functions. Previous reports have also suggested the existence of extranuclear ATM46 but this has been a controversial topic and is not fully resolved. More recent evidence suggests that ATM is predominantly cytoplasmic in neuronal cells, for example, slices of cerebella from mice confirmed the existence of cytoplasmic ATM,47,48 and ATM was shown to translocate from the nucleus to the cytoplasm in neuronal cells upon differentiation.49 Additionally, after the induction of DSBs, a subset of ATM is exported to the cytoplasm in a manner depending on the NF?B essential modulator (NEMO).50,51
ATM oxidative activation after ionizing radiation.
Ionizing radiation (IR) induces DSBs by direct deposition of energy to the sugar-phosphate backbone. IR can also ionize H2O and generate ROS, which accumulate to induce oxidative stress when rapidly produced.52 Many groups use IR to induce DSBs, but the effects of the radiation on oxidation of cellular compounds are rarely investigated. Oxidative stress and DNA damage are generally experienced simultaneously in cell populations, considering that high levels of ROS also cause DSBs as well as a variety of base and backbone modifications.30 DNA damage responses that occur under oxidative stress conditions may be very different from responses under normal conditions, however, because oxidative stress has been shown to reduce the activity of many proteins. The Ku heterodimer, for instance, was shown to be functionally inactivated after oxidation.53
To determine the effect of oxidative stress on MRN/DNA-dependent ATM activation, we performed ATM kinase assays with MRN and DNA in the presence of varying amounts of H2O2. In the reactions with wild-type ATM, H2O2 had no obvious effect on overall ATM activity (Fig. 2A). However, in the reactions with the C2991L ATM mutant, ATM activation was completely inhibited by H2O2. Because C2991L ATM is deficient in oxidative activation and can only be activated by MRN and DNA, the loss of ATM activation is due to the loss of MRN/DSB-dependent ATM activation under oxidative conditions. In the case of wild-type ATM, although H2O2 can inhibit MRN/DNA-dependent ATM activation, it also is capable of being activated through oxidation, which complements the loss of MRN/DNA-dependent activation.
The MRN complex acts as a sensor for DSBs, and is essential for the activation of ATM by DSBs. Shortly after DSBs occur, ATM is recruited to DSB sites.31,54 The MRN complex binds to ATM, recruits ATM to broken DNA ends, and is essential for ATM activation in vivo and in vitro.34,55 A direct pull-down assay was used to determine if the interaction between ATM and MRN is affected by oxidation. This experiment showed that immobilized MRN complex can bind ATM efficiently even in the presence of H2O2 (Fig. 2B), indicating that oxidative stress does not affect the interaction between MRN and ATM.
To determine if the ability of MRN to recognize DSBs is altered by oxidation, we performed a gel mobility shift assay with purified MRN complex in the presence of H2O2 (Fig. 2C). This result shows that, under oxidative conditions, MRN does not bind efficiently to DNA.
Based on these results, we propose that oxidative activation of ATM contributes significantly to overall ATM activity in the presence of both DNA damage and oxidative stress, and its role increases with ROS concentration. During ionizing radiation, the oxidative activation may dominate at very early time points because direct oxidation is instantaneous and does not require other factors to facilitate activation. After a brief exposure to radiation, the reducing environment inside cells could reverse the oxidative activation of ATM after the elimination of the short-lived ROS by cellular antioxidants. Subsequently, the MRN/DSB-dependent activation pathway is likely to generate longer-lasting ATM activation by continual recruitment of DNA damage response proteins to persistent DSB foci. Evidence of such phenomenon may have been shown in previous research; for instance, pan-nuclear ATM autophosphorylation was observed immediately after IR.31 This might be the result of very early ATM activation by oxidative stress induced by IR. At later time points, activated ATM was only observed in DSB foci, indicating primarily MRN/DNA-dependent ATM activation.
Discussion
ATM is thought to safeguard genomic integrity by sensing the generation of DSBs. Our studies with ATM oxidation showed that this enzyme is also an important sensor of ROS.29 In the absence of ATM, mammalian cells exhibit significantly higher levels of ROS,26 suggesting that there may be ATM targets that regulate antioxidant systems. The highly toxic ROS generated in the absence of ATM can damage proteins, lipids and DNA, ultimately leading to cell death in some tissues (Fig. 3).
Figure 3.

Dual activation of ATM by DNA damage or oxidative stress. (A) ATM can be activated by DSBs with the help of MRN complex or by direct oxidation. Oxidation of ATM is proposed to increase the activity of cellular antioxidant systems. (B) Inhibition of ATM oxidative activation results in ROS accumulation, which can damage many cellular components including DNA.
The discovery of the ATM oxidative activation pathway has implications beyond the A-T disorder. Redox balance plays an important role in regulating cell cycle progression, proliferation and survival. ATM functions as a redox sensor to regulate cell cycle checkpoints, apoptosis and very likely the cellular antioxidant systems. Indeed, ATM was recently shown to regulate mTORC1 signaling through LKB1/TSC256 and HIF-1α57 in response to changes in cellular redox state. Oxidative activation of ATM is also known to be critical for the survival of neuronal cells and hematopoietic stem cell populations that are hypersensitive to ROS.27 Tumorigenesis also involves the generation of ROS, and the ability of antioxidants to block formation of T cell lymphomas in ATM-deficient mice suggests that excess ROS contributes to tumorigenesis in the absence of ATM.58 Aneuploidy, a hallmark of cancer cells, was recently shown to induce ROS and the loss of ATM was shown to drastically accelerate aneuploidy-induced tumorigenesis.59 This makes the ATM oxidative activation pathway a potential therapeutic target for cancer as well as oxidative stress-related neurodegeneration diseases.
Materials and Methods
Protein purification and ATM kinase assays were performed as described in reference 29. Biotinylated MRN was purified as described previously in reference 60 and incubated with ATM (100 nM MRN, 7.1 nM ATM) in the presence or absence of H2O2 (0.5 mM) in a buffer containing 25 mM Tris-HCl pH 8.0, 100 mM NaCl, 10% glycerol and 1 mM DTT for 10 min at 30°C before isolating MRN with streptavidin-coated magnetic beads (Invitrogen). Proteins bound to the beads were separated by SDS-PAGE and analyzed by western blotting for Mre11 (Genetex, GTX70212) and ATM (Genetex, GTX70103). Gel shift assays were performed as previously described in reference 61, with purified human MRN and a dsDNA (41 bp, 3′ labeled with Cy5). MRN preincubated with H2O2 was bound to DNA (5 nM) in a buffer containing 25 mM MOPS, pH 7.0, 5 mM MgCl2 and 0.5 mM AMP-PNP for 10 minutes at 37°C, and protein-DNA complexes were separated by native agarose gel electrophoresis. The labeled DNA was analyzed in the gel using a Typhoon imager (GE).
Acknowledgements
We are grateful to the members of the Paull lab for useful suggestions. Funding was provided by the National Institutes of Health, NIH CA132813 and CA92584.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/14323
References
- 1.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 2.Khanna KK. Cancer risk and the ATM gene: a continuing debate. J Natl Cancer Inst. 2000;92:795–802. doi: 10.1093/jnci/92.10.795. [DOI] [PubMed] [Google Scholar]
- 3.Shiloh Y. ATM and related protein kinases: Safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 4.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 5.Abraham RT. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst) 2004;3:883–887. doi: 10.1016/j.dnarep.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006;442:466–470. doi: 10.1038/nature04866. [DOI] [PubMed] [Google Scholar]
- 7.Lumsden JM, McCarty T, Petiniot LK, Shen R, Barlow C, Wynn TA, et al. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. J Exp Med. 2004;200:1111–1121. doi: 10.1084/jem.20041074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gumy-Pause F, Wacker P, Sappino AP. ATM gene and lymphoid malignancies. Leukemia. 2004;18:238–242. doi: 10.1038/sj.leu.2403221. [DOI] [PubMed] [Google Scholar]
- 9.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 10.Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 11.Barlow C, Brown KD, Deng CX, Tagle DA, Wynshaw-Boris A. Atm selectively regulates distinct p53-dependent cell cycle checkpoint and apoptotic pathways. Nat Genet. 1997;17:453–456. doi: 10.1038/ng1297-453. [DOI] [PubMed] [Google Scholar]
- 12.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 13.Buscemi G, Savio C, Zannini L, Micciche F, Masnada D, Nakanishi M, et al. Chk2 activation dependence on Nbs1 after DNA damage. Mol Cell Biol. 2001;21:5214–5222. doi: 10.1128/MCB.21.15.5214-5222.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGowan CH. Checking in on Cds1 (Chk2): A checkpoint kinase and tumor suppressor. Bioessays. 2002;24:502–511. doi: 10.1002/bies.10101. [DOI] [PubMed] [Google Scholar]
- 15.Kim ST, Xu B, Kastan MB. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002;16:560–570. doi: 10.1101/gad.970602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mu JJ, Wang Y, Luo H, Leng M, Zhang J, Yang T, et al. A proteomic analysis of ataxia telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J Biol Chem. 2007;282:17330–17334. doi: 10.1074/jbc.C700079200. [DOI] [PubMed] [Google Scholar]
- 17.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 18.Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 19.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 20.Morgan SE, Kastan MB. p53 and ATM: Cell cycle, cell death and cancer. Adv Cancer Res. 1997;71:1–25. doi: 10.1016/s0065-230x(08)60095-0. [DOI] [PubMed] [Google Scholar]
- 21.Lin WC, Lin FT, Nevins JR. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001;15:1833–1844. [PMC free article] [PubMed] [Google Scholar]
- 22.Barzilai A, Rotman G, Shiloh Y. ATM deficiency and oxidative stress: A new dimension of defective response to DNA damage. DNA Repair (Amst) 2002;1:3–25. doi: 10.1016/s1568-7864(01)00007-6. [DOI] [PubMed] [Google Scholar]
- 23.Reichenbach J, Schubert R, Schwan C, Muller K, Bohles HJ, Zielen S. Anti-oxidative capacity in patients with ataxia telangiectasia. Clin Exp Immunol. 1999;117:535–539. doi: 10.1046/j.1365-2249.1999.01000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dean SW, Sykes HR, Cole J, Jaspers NG, Linssen P, Verkerk A. Re: Impaired glutathione biosynthesis in cultured ataxia-telangiectasia cells. Cancer Res. 1988;48:5374–5376. [PubMed] [Google Scholar]
- 25.Stern N, Hochman A, Zemach N, Weizman N, Hammel I, Shiloh Y, et al. Accumulation of DNA damage and reduced levels of nicotine adenine dinucleotide in the brains of Atm-deficient mice. J Biol Chem. 2002;277:602–608. doi: 10.1074/jbc.M106798200. [DOI] [PubMed] [Google Scholar]
- 26.Watters D, Kedar P, Spring K, Bjorkman J, Chen P, Gatei M, et al. Localization of a portion of extranuclear ATM to peroxisomes. J Biol Chem. 1999;274:34277–4282. doi: 10.1074/jbc.274.48.34277. [DOI] [PubMed] [Google Scholar]
- 27.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 28.Kim J, Wong PK. Loss of ATM impairs proliferation of neural stem cells through oxidative stress-mediated p38 MAPK signaling. Stem Cells. 2009 doi: 10.1002/stem.125. [DOI] [PubMed] [Google Scholar]
- 29.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 30.Barzilai A, Yamamoto K. DNA damage responses to oxidative stress. DNA Repair (Amst) 2004;3:1109–1115. doi: 10.1016/j.dnarep.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 31.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 32.Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell. 1998;93:467–476. doi: 10.1016/s0092-8674(00)81174-5. [DOI] [PubMed] [Google Scholar]
- 33.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 34.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 35.Chessa L, Petrinelli P, Antonelli A, Fiorilli M, Elli R, Marcucci L, et al. Heterogeneity in ataxia-telangiectasia: classical phenotype associated with intermediate cellular radiosensitivity. Am J Med Genet. 1992;42:741–746. doi: 10.1002/ajmg.1320420524. [DOI] [PubMed] [Google Scholar]
- 36.Toyoshima M, Hara T, Zhang H, Yamamoto T, Akaboshi S, Nanba E, et al. Ataxia-telangiectasia without immunodeficiency: Novel point mutations within and adjacent to the phosphatidylinositol 3-kinase-like domain. Am J Med Genet. 1998;75:141–144. [PubMed] [Google Scholar]
- 37.Gilad S, Chessa L, Khosravi R, Russell P, Galanty Y, Piane M, et al. Genotype-phenotype relationships in ataxia-telangiectasia and variants. Am J Hum Genet. 1998;62:551–561. doi: 10.1086/301755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peterson RT, Beal PA, Comb MJ, Schreiber SL. FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J Biol Chem. 2000;275:7416–7423. doi: 10.1074/jbc.275.10.7416. [DOI] [PubMed] [Google Scholar]
- 39.Morita T, Yamashita A, Kashima I, Ogata K, Ishiura S, Ohno S. Distant N- and C-terminal domains are required for intrinsic kinase activity of SMG-1, a critical component of nonsense-mediated mRNA decay. J Biol Chem. 2007;282:7799–7808. doi: 10.1074/jbc.M610159200. [DOI] [PubMed] [Google Scholar]
- 40.Sibanda BL, Chirgadze DY, Blundell TL. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature. 2010;463:118–121. doi: 10.1038/nature08648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spagnolo L, Rivera-Calzada A, Pearl LH, Llorca O. Three-dimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Mol Cell. 2006;22:511–519. doi: 10.1016/j.molcel.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 42.Rivera-Calzada A, Maman JD, Spagnolo L, Pearl LH, Llorca O. Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) Structure. 2005;13:243–255. doi: 10.1016/j.str.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 43.Sun Y, Xu Y, Roy K, Price BD. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol. 2007;27:8502–8509. doi: 10.1128/MCB.01382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berner RA. Atmospheric oxygen over Phanerozoic time. Proc Natl Acad Sci USA. 1999;96:10955–10957. doi: 10.1073/pnas.96.20.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dames SA, Mulet JM, Rathgeb-Szabo K, Hall MN, Grzesiek S. The solution structure of the FATC domain of the protein kinase target of rapamycin suggests a role for redox-dependent structural and cellular stability. J Biol Chem. 2005;280:20558–20564. doi: 10.1074/jbc.M501116200. [DOI] [PubMed] [Google Scholar]
- 46.Lim DS, Kirsch DG, Canman CE, Ahn JH, Ziv Y, Newman LS, et al. ATM binds to beta-adaptin in cytoplasmic vesicles. Proc Natl Acad Sci USA. 1998;95:10146–10151. doi: 10.1073/pnas.95.17.10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J, Han YR, Plummer MR, Herrup K. Cytoplasmic ATM in neurons modulates synaptic function. Curr Biol. 2009;19:2091–2096. doi: 10.1016/j.cub.2009.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barlow C, Ribaut-Barassin C, Zwingman TA, Pope AJ, Brown KD, Owens JW, et al. ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proc Natl Acad Sci USA. 2000;97:871–876. doi: 10.1073/pnas.97.2.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boehrs JK, He J, Halaby MJ, Yang DQ. Constitutive expression and cytoplasmic compartmentalization of ATM protein in differentiated human neuron-like SH-SY5Y cells. J Neurochem. 2007;100:337–345. doi: 10.1111/j.1471-4159.2006.04254.x. [DOI] [PubMed] [Google Scholar]
- 50.Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NFkappaB signaling in response to genotoxic stimuli. Science. 2006;311:1141–1146. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- 51.Wu ZH, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, et al. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell. 40:75–86. doi: 10.1016/j.molcel.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mena S, Ortega A, Estrela JM. Oxidative stress in environmental-induced carcinogenesis. Mutat Res. 2009;674:36–44. doi: 10.1016/j.mrgentox.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 53.Andrews BJ, Lehman JA, Turchi JJ. Kinetic analysis of the Ku-DNA binding activity reveals a redox-dependent alteration in protein structure that stimulates dissociation of the Ku-DNA complex. J Biol Chem. 2006;281:13596–13603. doi: 10.1074/jbc.M512787200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 55.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci USA. 2010;107:4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cam H, Easton J, High A, Houghton P. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. Mol Cell. 2010;40:509–520. doi: 10.1016/j.molcel.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yan M, Shen J, Person MD, Kuang X, Lynn WS, Atlas D, et al. Endoplasmic reticulum stress and unfolded protein response in Atm-deficient thymocytes and thymic lymphoma cells are attributable to oxidative stress. Neoplasia. 2008;10:160–167. doi: 10.1593/neo.07935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, et al. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci USA. 107:14188–14193. doi: 10.1073/pnas.1005960107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee JH, Goodarzi AA, Jeggo PA, Paull TT. 53BP1 promotes ATM activity through direct interactions with the MRN complex. EMBO. 29:574–585. doi: 10.1038/emboj.2009.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee JH, Ghirlando R, Bhaskara V, Hoffmeyer MR, Gu J, Paull TT. Regulation of Mre11/Rad50 by Nbs1: Effects on nucleotide-dependent DNA binding and association with ATLD mutant complexes. J Biol Chem. 2003;278:45171–45181. doi: 10.1074/jbc.M308705200. [DOI] [PubMed] [Google Scholar]
- 62.Saitou N, Nei M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]