

Abstract
CC3/TIP30 is a metastasis and tumor suppressor, with reduced or absent expression in a variety of aggressive tumors. Overexpression of CC3 in tumor cells predisposes them to apoptosis in response to different death signals. We found that silencing of CC3 expression does not increase apoptotic resistance of cells. However, it strongly improves survival of tumor cells in response to glucose limitation. HeLa cells with silenced CC3 survive long-term in low glucose, and in comparison to control HeLa cells, show superior metabolic adaptation to glucose limitation. First, unlike the parental HeLa cells, HeLa with silenced CC3 activate and maintain high levels of mitochondrial respiration that is critical for their ability to thrive in low glucose. Second, silencing of CC3 leads to higher expression levels of mitochondrial proteins in respiration complexes when cells are continuously cultured in limiting glucose. Third, HeLa cells with silenced CC3 maintain higher levels of c-MYC and the M2 isoform of pyruvate kinase in low glucose, contributing to more efficient glycolysis. Fourth, HeLa cells with silenced CC3 fail to fully activate AMPK in response to glucose limitation. Inhibition of AMPK, either pharmacologic or via siRNA, protects control HeLa cells from death in low glucose. The metabolic flexibility acquired by cells after silencing of CC3 could be directly relevant to the development of metastatic and aggressive human tumors that frequently have low or absent expression of CC3.
Key words: CC3/TIP30, glycolysis, OXPHOS, MYC, AMPK
Introduction
The human gene CC3/TIP30 was originally identified as a metastasis-suppressor of variant small cell lung carcinoma (vSCLC).1 CC3 is a phylogenetically conserved protein whose expression is absent or much reduced in a variety of aggressive or metastatic tumors such as vSCLC,1 neuroblastoma and glioblastoma,2,3 metastatic cancers of breast,4 and many others.5–7 Forced expression of CC3 in vSCLC,1 mouse melanoma, breast carcinoma,8 hepatocellular carcinoma,6 and gastric carcinoma cell lines5 inhibits metastatic behaviour in vitro and/or metastasis in vivo. Published results show that deletion of CC3 in germline results in spontaneous tumorigenesis in mice,9 and CC3-null mammary epithelial cells undergo immortalization in vitro,10 indicating that CC3 could be a tumor suppressor.11 High levels of acutely expressed exogenous CC3 induce apoptosis,1,3 while stable expression of exogenous CC3 results in sensitization of cells to apoptosis after variety of treatments, such as serum withdrawal, cytotoxic drugs, γ-irradiation and oxidative agents,3,12 Expression of CC3 in CC3-negative tumor cells has an inhibitory effect on the ability of these cells to produce angiogenic factors in vitro,2 consistent with the conclusion that downregulation of CC3 contributes to the development of aggressive metastatic phenotypes.
CC3/TIP30 has a significant sequence homology with short-chain dehydrogenases-reductases or SDRs,13,14 although an enzymatic activity was not demonstrated for CC3. The structural analysis of CC3 protein confirmed the prediction that CC3 contains a NADP(H) binding site.15 The cellular function of CC3 remained unknown, but it was found that CC3 plays an inhibitory role in the regulation of nuclear transport.16 CC3 binds directly to the karyopherins of the importin β family in a RanGTP-insensitive manner, and associates with nucleoporins in vivo. CC3 inhibits nuclear import of proteins with either the classic nuclear localization signal (NLS) recognized by importin α:β1, or the M9 signal recognized by transportin (importin β2). Cells forced to express high levels of CC3 have a slower rate of nuclear import.16 CC3 protein with mutated NADP(H) binding site lacks pro-apoptotic activity, is displaced from transportin by RanGTP, and fails to inhibit nuclear import in vitro and in vivo. Our results suggest that ability of CC3 to form a RanGTP resistant complex with importins and the NPC is central to its ability to inhibit nuclear import and induce apoptosis.16 The function of CC3 in nuclear transport is likely to be evolutionarily conserved because the S. cerevisiae homolog of CC3, YER004w, interacts with exportin CRM1 and with NTF2, the import factor for RanGDP.17
The role of the inhibition of nuclear transport by CC3 in apoptosis was highlighted in a study demonstrating that exogenously expressed CC3/TIP30 blocks nuclear import of mRNA-binding protein HuR, which leads to the stabilization of tp53 mRNA under conditions of oxidative stress and contributes to apoptosis.12 An independent confirmation for the function of CC3 as a negative regulator of nuclear transport came from studies showing aberrant expression of CC3 in oligodendrocyte precursors in multiple sclerosis lesions. This causes arrest of the nuclear import of the intracellular domain of Notch (NICD) and abnormal accumulation of a complexes of importin, its cargo NICD and CC3/TIP30 in the cytoplasm.18 The observed lack of the nuclear translocation of NICD leads to the failure of remyelinaiton that plays a causative role in pathogenesis of multiple sclerosis.18 Finally, overexpression of CC3 also has a negative effect on DNA damage repair by affecting nuclear transport of proteins relevant to this process.19
We have derived human cell lines where expression of CC3 was permanently silenced. Two of these lines were analyzed in a study that showed a mild impairment in DNA damage repair without affecting cell survival.19 Because multiple publications show that overexpression of exogenous CC3 has a strong proapoptotic effect in response to DNA damage, oxidative insults and more, we expected that silencing of CC3 might increase resistance to cell death. However, we observed that silencing of CC3 in three cell lines had no significant effect on their apoptotic resistance in response to treatments with a variety of apoptosis-inducing agents. In this study, we have examined the effect of CC3 silencing on cellular responses to glucose deprivation, and found that silencing of CC3 expression dramatically increases the ability of cells to survive under limiting glucose availability. Our results show that the absence of CC3/TIP30 expression promotes mitochondrial oxidative phosphorylation (OXPHOS) in tumor cells maintained in low glucose without diminishing activity of the glycolytic pathway. These findings could be directly relevant to the documented role of CC3 as a tumor suppressor, because absence of CC3 might confer to tumor cells the metabolic adaptability necessary to survive in adverse environment.
Results
Silencing of CC3 improves cell survival in response to glucose deprivation.
Multiple reports, including ours,1,3,12,18 made a casual connection between forced overexpression of CC3 and cellular susceptibility to apoptosis. However, the expression of exogenous CC3 from a strong promoter frequently far exceeds levels of endogenous CC3. High levels of CC3 might inhibit nuclear transport even under normal conditions by sequestering nuclear transport receptors and inducing apoptosis.16 We were interested in possible anti-apoptotic effects of CC3 silencing. Expression of endogenous CC3 was silenced in two cancer cell lines that contain relatively high levels of CC3 protein, HeLa and MCF7 (the levels of CC3 in control and silenced cells are shown in Figs. 1A and 2A). Treatment of these lines with a variety of death inducing agents had not produced convincing results to support the hypothesis that elimination of CC3 could significantly improve resistance of cells to apoptotic signals. Cells were treated with UV,19 chemotherapeutic drugs etoposide, cisplatinum and taxol, serum withdrawal and kinase inhibitors, and we have not observed any significant effect of CC3 silencing on the extent of cell death in response to these treatments (Sup. Fig. 1). We have then examined the potential effect of metabolic stress on proliferation and survival of cells lacking endogenous CC3 protein.
Figure 1.
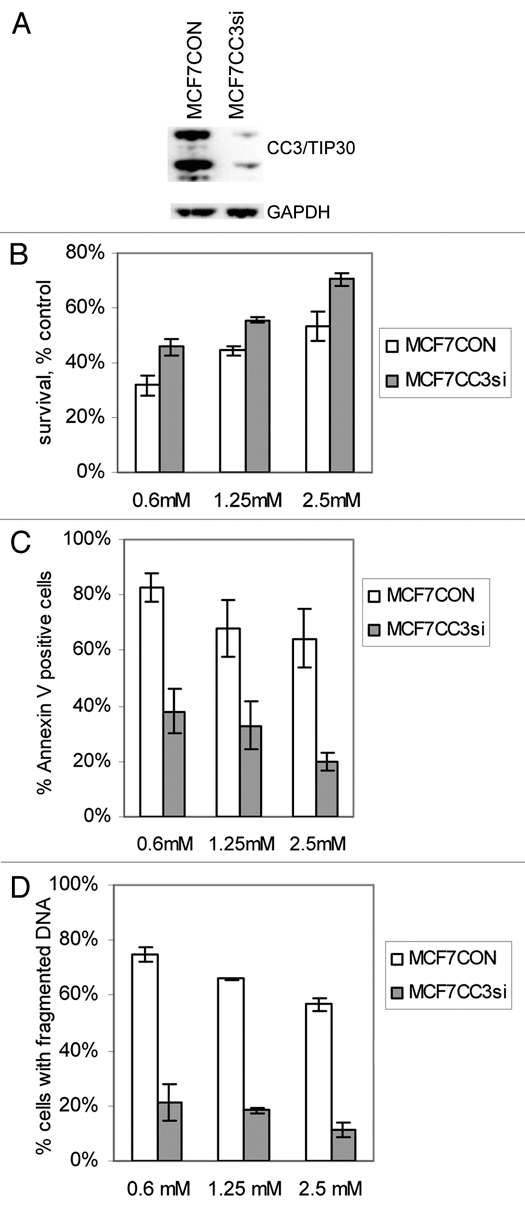
Silencing of CC3 promotes survival of MCF7 cells under limiting glucose conditions. (A) Western blot analysis of CC3 expression in MCF7 cells transduced with a control lentivirus and lentivirus coding for siRNA to CC3. (B) MCF7 cells with or without endogenous CC3 protein were grown in media with the indicated concentrations of glucose for three days. The chart shows quantification of viable cells in low glucose as percentages of control cultures grown in full medium with 25 mM glucose. Cell viability was quantified using CCK-8 kit from Dojindo. Data are expressed as mean ± SE (n = 4, for all three groups p < 0. 001; paired t-test). (C) Apoptosis in MCF7 cells subjected to reduced glucose conditions for three days. Cells were stained with Annexin V and propidium iodide (PI) followed by FACS analysis. The chart shows percentages of all apoptotic cells (i.e., all cells positive for ANNV binding) as average ± SE of three experiments (p < 0.002 for all three conditions). (D) Apoptotic DNA fragmentation in MCF7 cultures maintained in low glucose for three days. The p values are 0.0006, 0.0007 and 0.0026 (n = 3) for the three groups.
Figure 2.
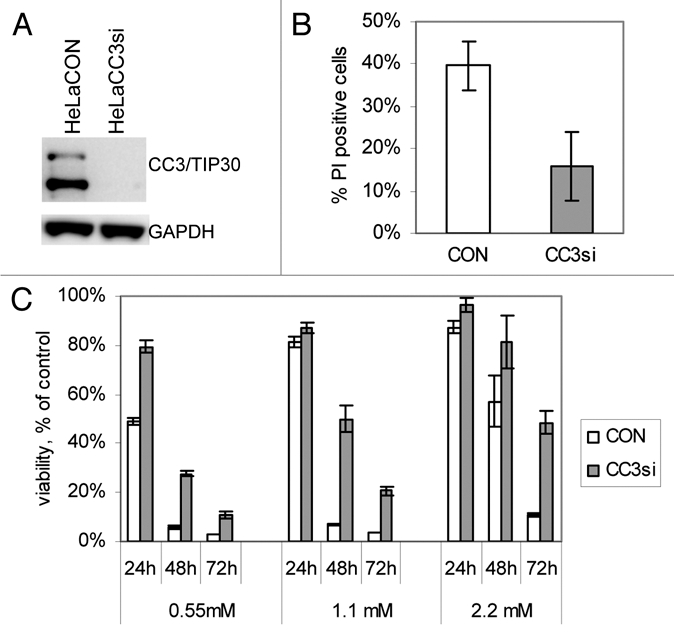
Silencing of CC3 promotes survival of HeLa cells under limiting glucose conditions. (A) Western blot analysis of CC3 expression in HeLa cells transduced with a control lentivirus and a lentivirus coding for siRNA to CC3. (B) Cell death in HeLa cell lines cultured in media with 0.55 mM glucose for 24 hours. Cultures were stained with Annexin V and propidium iodide (PI) followed by FACS analysis. Chart shows percentage of PI-positive (dead) cells; average and s. e. of three experiments, p = 0.0097. (C) Control and CC3-silenced HeLa cells were grown in media with the indicated concentrations of glucose for 3 days. Cell viability was quantified on each day of incubation. Data are expressed as in Figure 1B (n = 3, p values are < 0.005 for all except 24 hour incubations in 1.1 mM and 2.2 mM glucose (p < 0.1).
MCF7 cells stably transduced with control lentivirus (MCF7CON) and with CC3 silencing virus (MCF7CC3si) were subjected to growth in media with normal glucose concentration (25 mM) or reduced glucose concentrations (0.6, 1.25 and 2. 5 mM glucose). Quantification of cell proliferation under reduced glucose conditions showed a significant, concentration-dependent decrease in cell numbers within the first three days of glucose limitation (Fig. 1B). The inhibitory effect on cell growth was significantly more pronounced in MCF7CON cells than in MCF7CC3si cells. We examined if the inhibition of cell growth is due to arrested proliferation or cell death. Analysis of cultures kept in limiting glucose concentrations showed that the reduction in cell numbers was mostly due to apoptosis, in particular in MCF7CON cultures grown in low glucose concentrations, which contained very high numbers of cells positive for Annexin V binding and for propidium iodide uptake (Fig. 1C). Most of these cells also had fragmented DNA, further confirming the apoptotic nature of cell death in MCF7CON cells growing in low glucose (Fig. 1D). We conclude that silencing of CC3 reduces apoptosis of MCF7 cells subjected to glucose limitation.
If the cultures of MCF7 lines were kept continually in 0.6 mM glucose, both lines could adapt to growth under these conditions within a week. The glucose-deprived cultures grew slower then cells in full medium, and contained somewhat higher numbers of dead cells (not shown), but they retained the proliferative potential. Analysis of BrdU incorporation in both cell lines kept in low glucose for five days to eight weeks showed a higher percentage of cells in the G1 phase of the cell cycle, and a decrease in cells in the S phase compared to cells cultured in full medium (not shown). We have maintained MCF7 lines in culture for at least three months (not shown). This process of “adaptation” to growth in low glucose took a somewhat longer time in MCF7CON cells compared to CC3si cells, apparently because initially there was more cell loss due to increased death (Fig. 1C). However, after the first week of transfer to 0.6 mM glucose, growth rates of both lines were practically same (not shown).
Metabolic flux analyses were conducted with HeLa cell lines, control and with silenced CC3 expression. Figure 2B shows that in general HeLa cells were much more sensitive to glucose deprivation than MCF7 cells, with HeLaCON cells undergoing massive loss of cell viability in low glucose already after 24 hour incubation. Nevertheless, similar to MCF7 cells, silencing of CC3 in HeLa also greatly improved short-term survival of cells in low glucose conditions. Analysis of cultures kept in low glucose for one to three days showed that HeLaCC3si cells survived glucose limitation much better than HeLaCC3CON cells (Fig. 2C). Only a small fraction of dead cells in glucose-starved cultures contained fragmented DNA (not shown) indicating that apoptosis might be not the predominant mode of death in HeLa cells deprived of glucose. Several independent attempts for long-term culture of HeLaCON cells in media with 2.2 mM glucose (1/5 of glucose content in full medium) failed because all HeLaCON died within first three to five days. However, HeLaCC3si cells were maintained for over four months in medium with 2.2 or even 1.1 mM glucose after surviving the first acute death response (not shown).
Cells with silenced CC3 show a superior metabolic adaptability.
We have considered the possibility that the increased survival of cells with silenced endogenous CC3 in glucose limiting conditions could be due to the ability of CC3 deficient cells to modify their metabolism in order to survive in low glucose. We have therefore examined the metabolic characteristics of cells with silenced CC3 versus control cells under conditions of limited glucose availability. Cell metabolism was analyzed on the Seahorse XF96 extracellular flux analyzer for extracellular acidification rate (ECAR, a measure of lactate output and thus a measure of the glycolytic activity of the cells) and oxygen consumption rate (OCR). Cells were cultured either in normal full media or in media completely lacking glucose for four hours prior to analysis.
Figure 3A shows that in MCF7CON cells glycolysis (ECAR) is inhibited by about 20% within the first four hours of glucose withdrawal, but the change in ECAR was not significant in MCF7CC3si cells within this time (ECAR is much reduced in both cell lines after longer incubation times without glucose; not shown). We have also observed a reduction in the oxygen consumption rate in MCF7CON cells upon removal of glucose, probably as a consequence of the inhibition of glycolysis that normally provides pyruvate for the TCA cycle to feed into mitochondrial oxidative phosphorylation (OXPHOS). However, no significant reduction in OCR was observed in MCF7CC3si cells indicating that these cells might still rely on intracellular pyruvate and/or glutamine in the growth media as substrate for OXPHOS.
Figure 3.

Cells with silenced CC3 expression show increased metabolic flexibility and higher production of ATP. (A) Analysis of metabolic fluxes in MCF7 cells cultured without glucose for four hours. The chart shows quantification of ECAR and OCR in CON and CC3si cells kept in glucose-free or complete media for four hours. Normalized ECAR and OCR values of cells in glucose deprived media cells are shown as percentage of values measured in cells kept in full media in other wells of the same experimental plates. In each experiment, each treatment group was composed of 6 to 10 replica wells. Data are average and SE of four experiments. For MCF7CON versus CC3si groups, p = 0.046 (ECAR) and 0.008 (OCR). (B) Same as (3A), with HeLa cell lines. P value = 0.009 (ECAR) and 0.011 (OCR). (C) ATP levels were measured in cells cultured in medium lacking glucose for 4 and 8 hours. Chart shows ATP levels as percent of ATP in parallel control cultures kept in full medium. Results are representative of one of two experiments performed. (D) ATP levels in MCF7 lines kept in the indicated concentrations of glucose for three days. Results are average of three experiments ± SE (p = 0.0099, 0.0025 and 0.035 for 0.6 mM, 1.25 mM and 2.5 mM glucose respectively). (E) Same as in (D), with HeLa cell lines cultured in media with indicated concentrations of glucose for 1–3 days. Results are representative of one of two experiments performed.
HeLa cells with and without CC3 behaved similarly to MCF7 lines, but the decrease in ECAR and OCR induced by low glucose supply was significantly larger in HeLaCON cells than in MCF7CON (Fig. 3B). HeLaCC3si cells maintained a higher rate of oxygen consumption in media without glucose compared to HeLaCON cells, similarly to MCF7 cell lines. Apparently, elimination of CC3 allows cells to maintain normal glycolytic rates at least during the first hours of glucose deprivation, perhaps by more efficient utilization of intracellular glucose. These cells also maintain higher respiration levels (Fig. 3A and B). That could obviously contribute to the superior survival of cells with silenced CC3 in low glucose. We conclude that silencing of CC3 allows cells to at least temporarily maintain higher rates of both glycolysis and mitochondrial respiration when glucose levels are critically lowered, and thus contributes to cell survival in limiting glucose.
To verify this conclusion, we have examined the consequences of the shift to culture without glucose on the energy (ATP) production in the cell lines with and without CC3 expression. Figure 3C shows that cells with silenced expression of CC3 show an improved ability to maintain higher ATP levels in media lacking glucose, suggesting that their improved metabolic flexibility helps to maintain higher energy under metabolic stress and promotes cell survival. Analysis of ATP levels in cell lines maintained under conditions of limiting glucose concentration for three days clearly shows that cells lacking CC3 produce more ATP, and that ATP levels parallel the observed decreases in cell viability (Fig. 3D and E; compare to Figs. 1B and 2C).
Silencing of CC3 increases the mitochondrial respiration in HeLa cells under conditions of limiting glucose.
We have described above that only HeLa cells deficient in CC3 could continuously grow in medium containing low glucose amounts. We hypothesized that HeLaCC3si cells, unlike HeLaCON, could have successfully switched to oxidative phosphorylation (OXPHOS) for energy production when subjected to limiting glucose availability, utilizing glutamine as a substrate. To address this possibility, we have performed metabolic flux analysis in HeLaCC3si cells continuously cultured in limiting glucose conditions.
Both HeLa cell lines were cultured in 2.2 mM glucose for 24 hours, a time point when there is very little cell death even in HeLaCON cells (Fig. 2C). Figure 4A shows that oxygen consumption was not significantly altered in HeLaCON cells within first 24 hours after transfer to 2.2 mM glucose. However, OCR (and ECAR) were increased in CC3si cells. HeLaCON cells could not be analyzed at later times because half of the cells died after the first two days (Fig. 2C). At days 5, 10 and 20 after glucose limitation, HeLaCC3si cells showed a very significant (two- to three-fold) increase in their mitochondrial respiration compared to cells cultured in full medium. The change in ECAR was insignificant through this time (Fig. 4A). These observations strongly suggest that the ability of HeLa cells with silenced CC3 to survive in limiting glucose conditions is due to their ability to strongly increase mitochondrial respiration while maintaining normal levels of glycolysis. To confirm the critical role of mitochondria in survival in limiting glucose, we have selected HeLaCC3si variant line lacking functional mitochondria (rho-) by culturing them in medium with 50 ng/ml ethidium bromide for 30 days. In full medium, these cells were viable even though their mitochondrial oxygen consumption was dramatically reduced (not shown). However, upon transfer to media with 2.2 mM glucose, cells did not survive beyond 48 hours (data not shown), unlike original HeLACC3si cells (80% of which were viable after 48 hours in 2.2 mM glucose, Fig. 2C). This observation confirmed that silencing of CC3 allows survival in limiting glucose by stimulating mitochondrial respiration.
Figure 4.
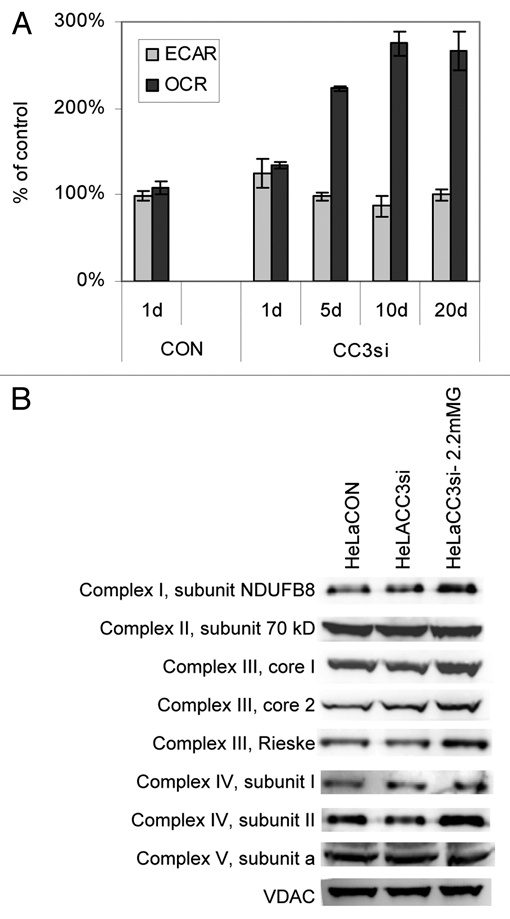
Silencing of CC3 leads to higher consumption of oxygen in HeLa cells in low glucose and higher levels of mitochondrial respiration proteins. (A) Metabolic fluxes in HeLaCC3si cells cultured in medium with 2.2 mM glucose for the indicated number of days. Data are normalized to cell numbers and presented as percentage of control cultures grown in full medium with 11 mM glucose. Data are average ± SE of three experiments. The p values for OCR in HeLaCC3si cells in low glucose versus normal glucose are <0.01 for all four times measurements were performed. (D) Cells were harvested from cultures kept in full media or in 2.2 mM glucose for 30 days (HeLaCC3si-2.2 mMG). HeLaCON cells did not survive beyond 72 hours in 2.2 mM glucose and therefore could not be analyzed. Extracts were analyzed by western blotting with the indicated antibodies to mitochondrial proteins.
We have asked what substrate is used as a fuel for mitochondrial respiration in HeLaCC3si cells in low glucose. We observed that presence or lack of pyruvate in low glucose medium does not affect survival of HeLa cell lines, suggesting that extracellular pyruvate is not efficiently utilized by cells deprived of glucose (not shown). We have then examined effect of glutamine on metabolic fluxes of HeLa cells. Glutamine (4 mM) was normally present in all media used in our experiments, and could be used as a substrate for OXPHOS. We have cultured cells without glutamine overnight, and examined how addition of glutamine would affect the metabolic fluxes. Supplemental Figure 2 shows that addition of glutamine greatly increased OCR in HeLaCC3si cells, and to a lesser degree in HeLaCON cells. This strongly indicates that glutamine is indeed a substrate of mitochondrial respiration utilized more efficiently in CC3-deficient cells under low glucose.
We have examined one possible reason for the increased OCR in glucose-starved HeLaCC3si cells, specifically, the expression levels of some of the protein subunits that compose the mitochondrial electron transport complexes. Levels of several mitochondrial proteins, components of respiration complexes, were examined by western blot analysis. We have observed an increase in levels of proteins in complex I, III and IV, but not in II or V (Fig. 4B). The upregulated subunits were encoded either in nuclear DNA (complex I NDUFB8, and complex III core I, II and Rieske) or in mitochondrial genome (complex IV subunits I and II).
Cells with silenced CC3 maintain higher levels of MYC protein and its targets in the glucose utilization pathway when glucose is limited.
We have explored the possible effects of CC3 on levels of MYC protein during glucose limitation, guided by two published findings: that CC3/TIP30 has a negative effect on MYC expression,10,20 and that MYC plays a prominent role in regulation of expression of genes involved in both glycolysis and oxidative phosphorylation.21–23 While levels of c-MYC were not different in HeLa cells with or without CC3 under normal conditions, a much stronger downregulation of MYC protein was observed in HeLaCON cells cultured in low glucose (Fig. 5A). A recent publication24 implicated c-MYC in control of aerobic glycolysis through its positive effect on expression of pyruvate kinase isoform M2 (PKM2), a central regulator of aerobic glycolysis in tumor cells.25 The transcriptional targets of MYC, hnRNP proteins A2/B1, A1 and I were shown to be instrumental in enforcing alternative splicing event specifically producing the PKM2 RNA.25,26 In correlation with the higher levels of MYC protein in CC3si cells, we have observed higher levels of hnRNP A2/B1 and I, as well as PKM2 proteins in HeLaCC3si than in HeLaCON cells under low glucose (Fig. 5A).
Figure 5.
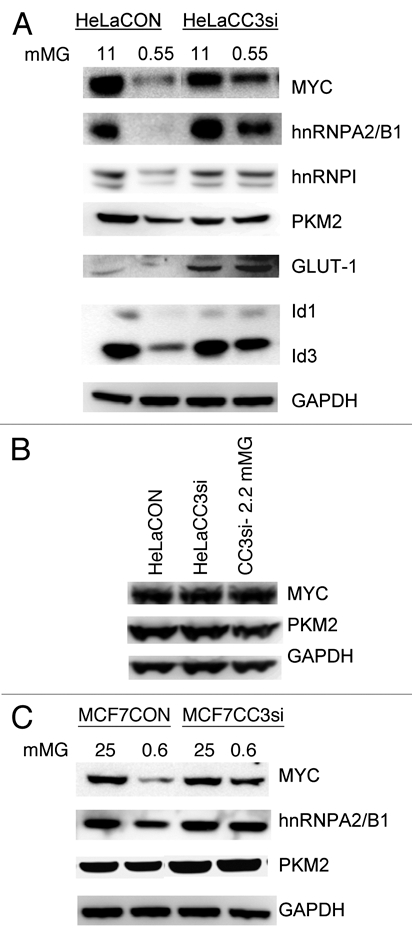
Analysis of expression of proteins related to glucose metabolism in cells with silenced CC3. (A) HeLa cells were cultured in full medium (11 mM glucose) or in 0.55 mM glucose for 18 hours prior to cell harvest and protein analysis by western blotting. (B) HeLa cells were harvested from cultures kept in full media or in 2.2 mM glucose for 30 days (CC3si-2.2 mM). Extracts were analyzed by western blotting with the indicated antibodies. (C) MCF7 cells were cultured in normal media (25 mM glucose) or low glucose (0.6 mM) for 72 hours prior to protein extraction and analysis.
To confirm the increased c-MYC activity in HeLACC3si cells in low glucose, we have examined expression of other genes subject to transcriptional regulation by c-MYC. The glucose transporter GLUT-1 that is regulated in part by MYC27 is expressed at higher levels in HeLaCC3si cells, and its expression levels are maintained under low glucose unlike in HeLaCON cells (Fig. 5A). The maintenance of GLUT-1 levels could also be relevant to the higher glycolytic activity of HeLaCC3si cells in low glucose (Fig. 4A). Two small HLH proteins Id1 and Id3, known targets of c-MYC, were strongly downregulated in HeLaCON cells cultures in low glucose, but less so in HeLaCC3si cells, in correlation with c-MYC expression (Fig. 5A). These data support the conclusion that the stronger downregulation of c-MYC protein in HeLaCON cells has functional consequences for expression of its target genes, some of which are relevant to glucose metabolism.
HeLaCC3si cells cultured for one month in medium with low glucose expressed MYC and PKM2 proteins at levels similar to cells cultured in full medium (Fig. 5B). Obviously, the initial reduction in expression of both proteins (Fig. 5A) was “corrected” in cells that survived and proliferated in low glucose. This indicates that part of the process of adjustment of these cells to growth in reduced glucose might rely on their ability to restore normal levels of at least some proteins involved in glycolysis and its regulation.
Only some of the changes in MYC targets observed in HeLa lines were also observed in MCF7 cells in limiting glucose (Fig. 5C). Decreases in c-MYC and hnRNPA2/B1 proteins were seen in MCF7CON cells in low glucose, but levels of PKM2 were only slightly affected (Fig. 5C). This was not entirely unexpected considering that the both metabolic and survival responses to glucose starvation in MCF7 cell lines were less acute than in HeLa lines (Figs. 1–3), and both control and CC3si MCF7 cells eventually adapt to growth in very low glucose concentration. It appears that silencing of CC3 expression has a strong but only transient pro-survival effect in MCF7 cells, while in HeLa cells it dramatically improves both the short- and long-term survival.
HeLa cells lacking CC3 fail to fully activate AMPK in response to glucose limitation which contributes to their increased survival.
Protein kinase AMPK is central in the control of cellular metabolic and energy producing pathways.28 We have therefore examined activation of AMPK in response to nutrient deprivation in cell lines with normal and reduced CC3 expression. As shown in Figure 6A, AMPK is heavily phosphorylated (and presumably activated) in control HeLa cells but phosphorylated only weakly in HeLaCC3si cells in response to lower glucose levels. Withdrawal of glutamine, most consumed amino acid in cultured cells (reviewed in ref. 29), also failed to induce phosphorylation of AMPK in HeLaCC3si cells, while a robust phosphorylation of AMPK was observed in HeLaCON cells (Fig. 6B). In contrast to these differences observed between HeLa cell lines, MCF7 cells, both with and without CC3 expression, had elevated AMPK phosphorylation in response to low levels of glucose (Fig. 6C) and to glutamine withdrawal (not shown).
Figure 6.
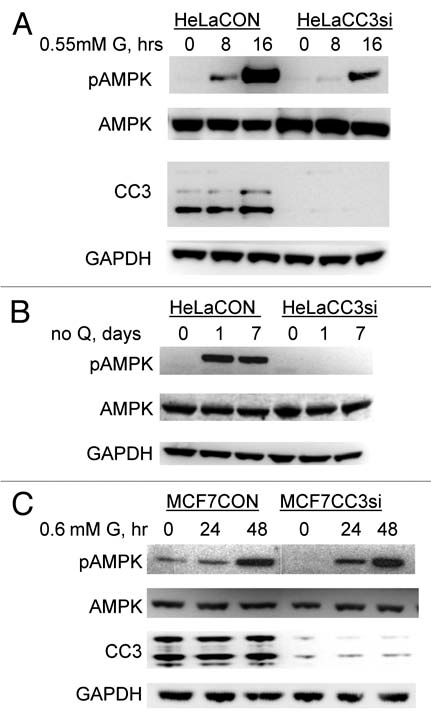
Analysis of AMPK activation in MCF7 and HeLa cell lines cultured in media with limited glucose (G) or without glutamine (Q). (A) HeLa cells were treated as described previously and analyzed for presence of activated AMPK with an antibody to phospho Thr172 (pAMPK) or total AMPK (AMPK), as well as for expression of CC3. (B) HeLa cells were cultured in media lacking glutamine (Q) for indicated times and analyzed as in (A). (C) Similar analysis conducted with MCF7 cells treated with low glucose for indicated times (hrs).
A major difference in regulation of AMPK in MCF7 versus HeLa cells could be due to the fact that HeLa cells lack kinase LKB1,30 which is the central activator of AMPK in response to increasing AMP levels, such as induced in cells by glucose withdrawal. Indeed, we have observed that, as already reported,31 treatment of HeLa cells with AICAR (AMP mimetic and AMPK activator via LKB1) failed to induce AMPK phosphorylation (not shown). However, LKB1 is not the only upstream kinase for AMPK. Multiple mechanisms of AMPK regulation have been recognized (reviewed in ref. 32). Among them are the elevation of cellular calcium that activates AMPK via CaMKKβ,31,33,34 and inhibition of AMPK by PKA (cAMP activated kinase) in certain cellular contexts.35 Inhibition of AMPK activity by high levels of cAMP was also documented to proceed via CaMKKβ.36
We have examined possible effects of pharmacological AMPK inhibition as well as of the cAMP levels, calcium levels and a selective CaMKKβ inhibitor on survival of HeLa cells with and without endogenous CC3 in low glucose. Figure 7A shows that specific inhibition of AMPK by Compound C increased survival of HeLaCON cells almost to the levels seen in HeLaCC3si cells, in spite of the fact that Compound C had significant toxicity under normal culture conditions. This clearly indicates that observed strong activation of AMPK in HeLa cells under low glucose is detrimental for their survival. Treatment of cells with forskolin, an inducer of adenylate cyclase activity and cAMP levels also improved survival of HeLaCON cells. STO-609, a direct inhibitor of the AMPK activating kinase CaMKKβ, had a strong cytotoxic effect on HeLa cells in complete media, but still rescued HeLaCON cells from glucose limitation induced death. Similarly, 1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (BAPTA), a calcium chelating agent, and an inhibitor of, among other kinases, CaMKKβ, also had a positive effect on survival of HeLaCON cells. Supplemental Figure 3 shows that increased survival of cells in presence of FSK and BAPTA is indeed due to prevention of cell death and not increased proliferation. These results suggest that inhibition of AMPK by different pharmacological treatments contributes to cell survival in low glucose. Therefore, failure of HeLaCC3si cells to activate AMPK could be indeed relevant to their improved survival.
Figure 7.
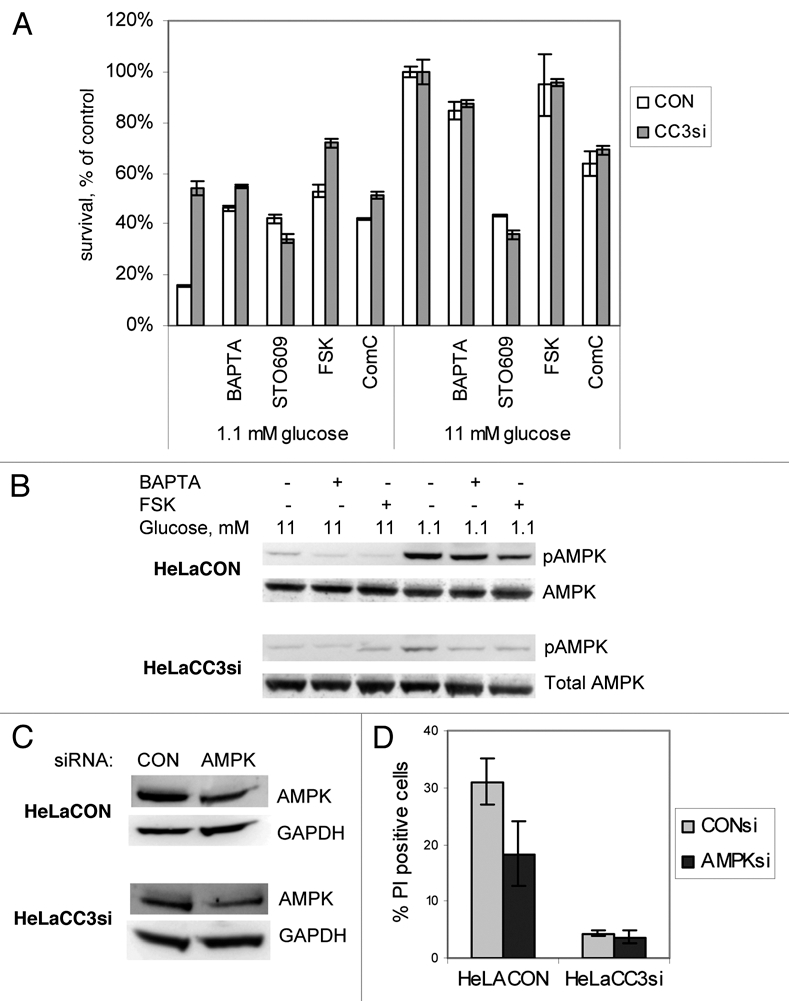
Inhibition of AMPK by different means increases survival of HeLaCON cells in low glucose. (A) HeLa cell lines were cultured in either low glucose (1.1 mM) or full medium (11 mM) for 48 hours in presence of 4 mM BAPTA, 5 mM FSK (forskolin), 10 mM STO-609 or 5 mM compound C. Cell viability was quantified with CCK-8 assay. Results are representative of one of two experiments conducted in quadruplicates. (B) Cells were cultured for 36 hours as in (A) and harvested for western blot analysis with antibodies to phospho Thr172 AMPK or total AMPK. (C) Cells were transfected with control non-targeted siRNA or siRNAs to AMPK and analyzed for levels of AMPK 72 hours later. (D) Transfected cultures were transferred to media with 1.1 mM glucose 48 hours after transfection and incubated for 30 hours prior to analysis. Dead cells were identified by flow cytometry of Annexin V/PI stained cells; percentages show PI positive (dead) cells only. Results are average and SE of three experiments (p = 0.0065).
To verify that the pro-survival effects of treatments described above are mediated through AMPK, we first examined activating phosphorylation on Thr 172 of AMPK in HeLa cell lines treated with BAPTA and FSK (both had a strong positive effect on survival of HeLaCON cells in 1.1 mM glucose without inducing cell death in full media (Fig. 7A). Figure 7B shows that BAPTA and FSK indeed inhibit AMPK activation in cells cultured in 1.1 mM glucose. These data strongly indicate that activation of AMPK could contribute to the increased rate of HeLaCON cells death in response to glucose limitation. To support this hypothesis, we have partially silenced expression of AMPK using specific siRNAs (Fig. 7C). While silencing of AMPK predictably had no effect on survival of HeLaCC3si cells, HeLaCON cells with silenced AMPK showed improved viability under conditions of limiting glucose (Fig. 7D). We conclude that activation of AMPK contributes to the loss of viability in HeLa cells in low glucose.
Discussion
Our analysis of CC3/TIP30 in responses to metabolic stress was prompted by the first set of observations that silencing of CC3 strongly protects tumor cells from apoptosis induced by glucose deprivation. However, cells with silenced CC3 showed no survival advantages in response to treatments with a wide variety of death inducing agents. This indicated that a function of CC3 distinct from its pro-apoptotic quality when overexpressed, could be involved in the improved survival of CC3-silenced cells under conditions of limited glucose concentration. Our results show that the increased survival of cells with silenced CC3 is largely due to their increased metabolic flexibility, i.e., their capacity to switch energy production pathways to accommodate the diminished supply of glucose.
Withdrawal of glucose allows MCF7 and HeLa cells without CC3 to sustain higher levels of both glycolysis and mitochondrial respiration in short term experiments (Fig. 3A and B). A simple explanation for the observed inhibition of respiration in glucose deprived cells is the shortage of pyruvate, the intracellular end product of glycolysis that, after decarboxylation by pyruvate dehydrogenase, enters TCA cycle for oxidation. Because glycolysis is inhibited much less in cells with silenced CC3, their OCR is also affected much less. An alternative explanation would be that cells with silenced CC3 might be able to utilize substrates other than pyruvate to fuel OXPHOS such as amino acids or fatty acids.
Indeed, the mitochondrial activity of HeLaCC3si cells cultured long-term in limiting glucose proved to be highly increased (Fig. 4A). There is little doubt that the increased OXPHOS allows HeLaCC3si cells to survive and proliferate in low glucose, a conclusion supported by higher levels of ATP in glucose-starved CC3-deficient cells (Fig. 3C–E). These results actually suggest that endogenous CC3 limits the ability of glycolysis-addicted cells like HeLa to switch to mitochondrial energy production when sugar is limited.
The mechanism through which lack of CC3 releases the apparent block of OXPHOS in HeLa cells remains to be explored. However, the observed increase in protein subunits of respiration complexes I, III and IV in HeLaCC3si cells growing in limiting glucose (Fig. 4B) is most likely relevant to their increased respiration rate. It is of interest that the three complexes affected by CC3 silencing contain proteins encoded in the mitochondrial genome, as well as nuclear encoded proteins. In this regard, the well known cross talk between changes in levels of the mitochondrially encoded proteins and nuclear transcriptional changes (retrograde response) could be relevant. If CC3 expression leads to a decrease in the levels of mitochondrially encoded proteins, a retrograde response could lead to a decrease in expression of nuclear encoded proteins of the relevant complexes. We have reported previously that CC3 interacts with LRPPRC,16 a RNA binding protein mutated in Leigh syndrome French Canadian variant (LSFC), a cytochrome oxidase deficiency.37 LRPPRC is a major positive regulator of expression of mitochondrially encoded mRNAs.37–39 The consequences of the interaction between CC3 and LRPPRC are certainly worth exploring. A distinct possibility is that binding of CC3 to LRPPRC inhibits its function in RNA transport, similar to the described inhibitory role of CC3 in nuclear transport through binding to importins. LRPPRC and importins are characterized by the presence of multiple HEAT repeats, which seems to be a unifying structural feature of CC3-binding proteins.16
We have examined levels of MYC in cells with silenced CC3 in view of reports linking CC3 expression to that of MYC,10 and also in view of the accumulating evidence that MYC acts as a master regulator of activity of both glycolysis and mitochondrial respiration.21,22,24,40,41 We have found that silencing of CC3 allows for higher MYC expression when glucose is limited, as well as higher expression of MYC target genes PKM2, a key enzyme promoting aerobic glycolysis in tumor cells,25 and Glut-1,27 a major glucose transporter with elevated expression in many cancers. This could be directly relevant to the ability of CC3 silenced cells to sustain glucose uptake and glycolytic activity when glucose is limited. It is likely that MYC could contribute to the observed high mitochondrial respiration in HeLaCC3si cells, considering its reported role in OXPHOS.22,42
We have observed major metabolic differences between MCF7 and HeLa cells. Silencing of CC3 increases OCR in both lines when they are subjected to glucose withdrawal. However, both MCF7 lines survive long-term in glucose-low media, while only CC3-deficient HeLa cells do. How could one explain that silencing of CC3 in two, admittedly very different genetically cell lines has different effects on survival under metabolic stress? We found that MCF7 cells rely less on glycolysis than HeLa, and actively engage in oxidative phosphorylation. Supplemental Figure 4 shows that the ratio of OCR to ECAR is significantly higher in MCF7 cells than in HeLa cells under both normal and glucose-deprived conditions, and silencing of CC3 further increases OCR/ECAR ratio in MCF7 cells. The higher basal OCR/ECAR ratio, i.e., higher OXPHOS contribution to energy production, is most likely involved in better survival of MCF7 in low glucose in short term, as well as their ability to adapt to chronic shortage of glucose. The lower OCR/ECAR ratio in HeLa cells is typical for highly glycolytic cells, in line with their enhanced sensitivity to glucose withdrawal. Silencing of CC3 allows for a strong increase in OCR during long term incubation in limiting glucose (Fig. 4A). Therefore, the differences in the metabolic responses of HeLa versus MCF7 cells to silencing of CC3 could depend on the inherent differences in their reliance on glycolysis versus OXPHOS for energy production.
We have considered other factors to explain the pro-survival effects of CC3 silencing on the short term survival of MCF7 and HeLa cells in absence of glucose. In particular, oxidative stress was reported to play a role in the glucose deprivation induced cell death. Induction of ROS in glucose deprived cells used in this study was examined at different times in low glucose. Flow cytometry with ROS indicator CM-H2DCFDA showed that ROS accumulation was not significant for the first six hours after glucose deprivation, and increased up to four-fold after 16 hours, without significant differences between control cells and cells with silenced CC3 (not shown). Therefore, at present we have no indications that a better control of oxidative stress contributes to survival of cells lacking CC3 expression.
We have found that central regulator of cellular metabolism, AMPK, is involved in survival of HeLaCC3si cells cultured in low glucose media. Unlike control cells, they fail to fully activate AMPK in limiting glucose and when glutamine is withdrawn (Fig. 6). However, silencing of CC3 does not affect AMPK activation in MCF7 cells (Fig. 6C). We suggest that this is because MCF7 cells contain functional LKB1, while HeLa cells do not express LKB1.30 Therefore, activation of AMPK in HeLaCON cells must involve regulators other than LKB1, which fail to be in engaged in absence of CC3. Our results indicate that activation of AMPK in HeLa cells involves calcium responsive kinase, most likely CaMKKβ, and is inhibited by increase in cAMP. It follows then that CC3 might play a role in cellular metabolic pathways governed by calcium and/or cAMP, which would be of interest to analyze. A different possibility exists for the observed lack of activation of AMPK by glucose deprivation in HeLa cells lacking CC3: these cells are able to maintain mitochondrial respiration in low glucose, and inhibition of mitochondrial metabolism was shown to be a very common mechanism for activation of AMPK by a variety of conditions and drugs.32
The role of AMPK activity in cell survival has been reported to be either protective or death-promoting, depending on cellular contexts and treatments.43–49 However, AMPK is currently considered to act as a tumor suppressor,50–53 and activation of AMPK emerged as a potential therapeutic goal in cancer treatment.53 Our data expand the understanding of CC3 function as tumor/metastasis suppressor through its contribution to AMPK activation.
Many types of advanced cancers have a shift in energy production from OXPHOS to aerobic glycolysis, termed the Warburg effect, which describes the increased uptake and conversion of glucose to lactate by cancer cells under adequate oxygen tension. In the last several years the importance of aerobic glycolysis in tumor progression has become a focus of intense research, with a general understanding that it universally promotes tumor growth by increasing energy production, while OXPHOS, even though it is by far more efficient pathway, is generally dispensable in tumors. However, a number of publications have recently challenged the generality of this conclusion. A more evolved understanding underscores the fact that tumor cells have increased demands not only for energy but other metabolic needs to sustain both growth and increased proliferation.21,42,54 In addition, oncogenes such as c-MYC55 and mutant H-Ras56 were shown to promote OXPHOS. Mitochondrial metabolism and generation of ROS were shown to be essential for Kras induced tumorigenicity.57 Importantly, active OXPHOS could be indispensable for the in vivo growth of highly glycolytic tumors, which could re-use secreted lactate to fuel mitochondrial activity.58 Very recently, a transcriptional profiling approach revealed that epithelial cancer cells upregulate expression of genes involved in oxidative metabolism.59 A recent study60 has demonstrated that a protein frequently overexpressed in cancer cells actually promotes OXPHOS and tumor growth. This protein, p32(C1QBP), also negatively affects the glycolytic activity of tumor cells while promoting tumor growth.
We show here that CC3/TIP30, protein frequently absent or diminished in aggressive cancers might actually, by its absence, promote OXPHOS without diminishing glycolytic activity. Silencing of CC3 in tumor cells highly dependent on glycolysis gives them the metabolic flexibility to utilize both OXPHOS and glycolysis for their metabolic needs, which confers growth advantage to these aggressive tumors. Our data support the notion that low or absent expression of CC3, found in a variety of metastatic and aggressive tumors, could be directly relevant to cancer progression through modifications in cellular bioenergetics.
Materials and Methods
Reagents and antibodies.
BAPTA, FSK, compound C, AICAR and STO-609 were purchased from Sigma. FITC-conjugated Annexin V was from BioVision. Antibodies to c-MYC, Glut-1, phospho Thr172-AMPK, AMPK and GAPDH were from Santa Cruz Biotechnology; to subunits of mitochondrial electron transport complexes from Mitosciences, to PKM2, hnRNPI and hnRNPA2/B1 from Cell Signaling, to Id1 and Id3 from Biocheck. Polyclonal antibodies to CC3/TIP30 were described previously in reference 16.
Cell culture and treatments.
MCF7 and HeLa cell lines were obtained from the ATCC, and propagated in either DMEM with 10% FBS (MCF7) or RPMI with 10% FBS (HeLa), supplemented with 1 mM pyruvate and 4 mM glutamine. Cells were transferred to corresponding media with low glucose content for the experiments. Glucose free DMEM and RPMI (Mediatech) were supplemented with full media to achieve a desired concentration of glucose (full DMEM contains 25 mM glucose and was diluted in glucose-free DME 40, 20 and 10 fold to achieve concentrations of 0.625, 1.25 ad 2.5 mM respectively. Similarly, RPMI (11 mM glucose) was diluted in glucose free RPMI to achieve glucose concentrations of 0.55, 1.1 and 2.2 mM).
Lentivirus-mediated siRNA expression and transient siRNA transfection.
For CC3 silencing, shRNA constructs in the LKO plasmid vector were purchased from Open Biosystems/Thermo. Viruses were produced in HEK293 cells according to manufacturer's instructions and used to transduce cells, followed by selection in pre-determined concentration of puromycin. For transient silencing of AMPK, a pool of five siRNAs targeting AMPK was purchased from Open Biosystems/Thermo (catalog number D-005027-02-0005) along with a non-targeting siRNA control (D-001810-05). siRNAs were transfected into HeLaCON and HeLaCC3si cells using Lipofectamine 2000 (Invitrogene). Cells were treated with low glucose 48 hours after transfection, or analyzed by western blotting for AMPK levels 72 hours after transfection.
Measurements of cell viability and apoptosis.
Cell viability was determined using CCK-8 kit (Dojindo). Apoptosis/cell death analysis was done using the Annexin V/propidium iodide staining followed by flow cytometry. Cells were collected by trypsinization into their culture media, washed with PBS and stained with Annexin V-FITC (BioVision) and propidium iodide (PI), and analyzed immediately after 5 minute incubation with the use of the CellQuest software on the FACScan (Becton Dickinson). ATP levels were quantified using ATP Bioluminescence assay kit HS II (Roche Applied Science).
Metabolic analyses with seahorse XF96 extracellular flux analyzer.
Cells were plated overnight on XF96 PET 96 well plates at experimentally predetermined numbers (12 × 103 cells per well for HeLa lines and 14 × 103 cells per well for MCF7 lines). Metabolic fluxes were analyzed on Seahorse XF96 analyzer per manufacturer instructions as previously described in reference 61. The basal ECAR values (in mpH/min) and OCR values (in pMoles O2/min) were measured for 4 cycles. Each cycle consisted of a 3 minutes media mixing time and a 5 minutes measurements time. After measuring the basal levels of ECAR and OCR for 4 cycles, the inhibitor of glycolysis/ECAR (2-deoxy-glucose at 100 mM) or inhibitors of mitochondrial respiration (rotenone at 1 µM or Antimycin A at 10 µM) were automatically injected into the experimental wells, and another two measurement cycles were performed. The injections were used in order to confirm that the observed values for the media acidification rate (ECAR) and depletion of oxygen (OCR) from the media were truly reflecting the glycolytic activity (lactate production) and mitochondrial respiration (oxygen depletion from the media), respectively. Each experimental plate contained cells seeded into wells in full medium overnight, and half of the wells were changed into media lacking glucose for the last 4 hours of culture. Some experiments used HeLaCC3si cells that were cultured in medium with 2.2 mM glucose for the times indicated (HeLaCON cells did not survive in 2.2 mM glucose for longer that 1–2 days). The experimental media for the metabolic flux assays contained 1 mM glucose. Each experimental point is an average of 6 to 10 replica wells, and experiments were repeated at least 4 times. Normalization of ECAR and OCR values obtained in XF96 assays was performed using quantification of cellular DNA with the CyQuant assay (Promega) on experimental plates. All the XF96 data are expressed as ECAR or OCR values normalized to DNA content, or as percent of activities relative to these measured in the same experiment in cells kept in full media (after normalization of both data sets).
Western blot analysis.
Whole cell lysates were electrophoresed on SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blotted with antibodies at recommended concentrations overnight at 4°C and the bound primary antibodies were detected using peroxidase-conjugated secondary antibodies. Blots were developed using SuperSignal enhanced chemiluminescence kit (Pierce) and imaged on Kodak Imager ISR2000.
Acknowledgements
We are grateful to Dr. Sylvia Fong for the critical reading of the manuscript. This work was supported by the grant to E.S. (R01CA114430) from the National Cancer Institute.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/14230
Authors' Contributions
V.C. performed experiments and analyzed data. E.S. designed and performed experiments, analyzed data and wrote the manuscript. V.C. read and approved the manuscript.
Supplementary Material
References
- 1.Shtivelman E. A link between metastasis and resistance to apoptosis of variant small cell lung carcinoma. Oncogene. 1997;14:2167–2173. doi: 10.1038/sj.onc.1201059. [DOI] [PubMed] [Google Scholar]
- 2.NicAmhlaoibh R, Shtivelman E. Metastasis suppressor CC3 inhibits angiogenic properties of tumor cells in vitro. Oncogene. 2001;20:270–275. doi: 10.1038/sj.onc.1204075. [DOI] [PubMed] [Google Scholar]
- 3.Whitman S, Wang X, Shalaby R, Shtivelman E. Alternatively spliced products CC3 and TC3 have opposing effects on apoptosis. Mol Cell Biol. 2000;20:583–593. doi: 10.1128/mcb.20.2.583-593.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao J, Ni H, Ma Y, Dong L, Dai J, Zhao F, et al. TIP30/CC3 expression in breast carcinoma: relation to metastasis, clinicopathologic parameters and p53 expression. Hum Pathol. 2007;38:293–298. doi: 10.1016/j.humpath.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Li X, Zhang Y, Cao S, Chen X, Lu Y, Jin H, et al. Reduction of TIP30 correlates with poor prognosis of gastric cancer patients and its restoration drastically inhibits tumor growth and metastasis. Int J Cancer. 2009;124:713–721. doi: 10.1002/ijc.23967. [DOI] [PubMed] [Google Scholar]
- 6.Lu B, Ma Y, Wu G, Tong X, Guo H, Liang A, et al. Methylation of Tip30 promoter is associated with poor prognosis in human hepatocellular carcinoma. Clin Cancer Res. 2008;14:7405–7412. doi: 10.1158/1078-0432.CCR-08-0409. [DOI] [PubMed] [Google Scholar]
- 7.Tong X, Li K, Luo Z, Lu B, Liu X, Wang T, et al. Decreased TIP30 expression promotes tumor metastasis in lung cancer. Am J Pathol. 2009;174:1931–1939. doi: 10.2353/ajpath.2009.080846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Thor A, Shtivelman E, Cao Y, Tu G, Heath TD, et al. Systemic gene delivery expands the repertoire of effective antiangiogenic agents. J Biol Chem. 1999;274:13338–13344. doi: 10.1074/jbc.274.19.13338. [DOI] [PubMed] [Google Scholar]
- 9.Xiao H, Palhan V, Yang Y, Roeder RG. TIP30 has an intrinsic kinase activity required for upregulation of a subset of apoptotic genes. EMBO J. 2000;19:956–963. doi: 10.1093/emboj/19.5.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pecha J, Ankrapp D, Jiang C, Tang W, Hoshino I, Bruck K, et al. Deletion of Tip30 leads to rapid immortalization of murine mammary epithelial cells and ductal hyperplasia in the mammary gland. Oncogene. 2007;26:7423–7431. doi: 10.1038/sj.onc.1210548. [DOI] [PubMed] [Google Scholar]
- 11.Ito M, Jiang C, Krumm K, Zhang X, Pecha J, Zhao J, et al. TIP30 deficiency increases susceptibility to tumorigenesis. Cancer Res. 2003;63:8763–8767. [PubMed] [Google Scholar]
- 12.Zhao J, Chen J, Lu B, Dong L, Wang H, Bi C, et al. TIP30 induces apoptosis under oxidative stress through stabilization of p53 messenger RNA in human hepatocellular carcinoma. Cancer Res. 2008;68:4133–4141. doi: 10.1158/0008-5472.CAN-08-0432. [DOI] [PubMed] [Google Scholar]
- 13.Baker ME. TIP30, a cofactor for HIV-1 Tat-activated transcription, is homologous to short-chain dehydrogenases/reductases. Curr Biol. 1999;9:471. doi: 10.1016/s0960-9822(99)80297-8. [DOI] [PubMed] [Google Scholar]
- 14.Baker ME, Yan L, Pear MR. Three-dimensional model of human TIP30, a coactivator for HIV-1 Tat-activated transcription and CC3, a protein associated with metastasis suppression. Cell Mol Life Sci. 2000;57:851–858. doi: 10.1007/s000180050047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El Omari K, Bird LE, Nichols CE, Ren J, Stammers DK. Crystal structure of CC3 (TIP30): implications for its role as a tumor suppressor. J Biol Chem. 2005;280:18229–18236. doi: 10.1074/jbc.M501113200. [DOI] [PubMed] [Google Scholar]
- 16.King FW, Shtivelman E. Inhibition of nuclear import by the proapoptotic protein CC3. Mol Cell Biol. 2004;24:7091–7101. doi: 10.1128/MCB.24.16.7091-7101.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakahara J, Kanekura K, Nawa M, Aiso S, Suzuki N. Abnormal expression of TIP30 and arrested nucleocytoplasmic transport within oligodendrocyte precursor cells in multiple sclerosis. J Clin Invest. 2009;119:169–181. doi: 10.1172/JCI35440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fong S, King F, Shtivelman E. CC3/TIP30 affects DNA damage repair. BMC Cell Biol. 11:23. doi: 10.1186/1471-2121-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang C, Ito M, Piening V, Bruck K, Roeder RG, Xiao H. TIP30 interacts with an estrogen receptor alpha-interacting coactivator CIA and regulates c-myc transcription. J Biol Chem. 2004;279:27781–27789. doi: 10.1074/jbc.M401809200. [DOI] [PubMed] [Google Scholar]
- 21.Dang CV. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res. 70:859–862. doi: 10.1158/0008-5472.CAN-09-3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15:6479–6483. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108–113. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 26.Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci USA. 2010;107:1894–1899. doi: 10.1073/pnas.0914845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797–1800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- 28.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 29.DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tiainen M, Ylikorkala A, Makela TP. Growth suppression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci USA. 1999;96:9248–9251. doi: 10.1073/pnas.96.16.9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 32.Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 11:554–565. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 34.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell metabolism. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 35.Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, et al. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J. 2010;29:469–481. doi: 10.1038/emboj.2009.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hurley RL, Barre LK, Wood SD, Anderson KA, Kemp BE, Means AR, et al. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J Biol Chem. 2006;281:36662–36672. doi: 10.1074/jbc.M606676200. [DOI] [PubMed] [Google Scholar]
- 37.Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci USA. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gohil VM, Nilsson R, Belcher-Timme CA, Luo B, Root DE, Mootha VK. Mitochondrial and nuclear genomic responses to loss of LRPPRC expression. J Biol Chem. 285:13742–13747. doi: 10.1074/jbc.M109.098400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sasarman F, Brunel-Guitton C, Antonicka H, Wai T, Shoubridge EA. LRPPRC and SLIRP interact in a ribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria. Mol Biol Cell. 2010;21:1315–1323. doi: 10.1091/mbc.E10-01-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morrish F, Neretti N, Sedivy JM, Hockenbery DM. The oncogene c-Myc coordinates regulation of metabolic networks to enable rapid cell cycle entry. Cell Cycle. 2008;7:1054–1066. doi: 10.4161/cc.7.8.5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dang CV. p32 (C1QBP) and cancer cell metabolism: is the Warburg effect a lot of hot air? Mol Cell Biol. 2010;30:1300–1302. doi: 10.1128/MCB.01661-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 44.Chhipa RR, Wu Y, Mohler JL, Ip C. Survival advantage of AMPK activation to androgen-independent prostate cancer cells during energy stress. Cell Signal. 2010;22:1554–1561. doi: 10.1016/j.cellsig.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Concannon CG, Tuffy LP, Weisova P, Bonner HP, Davila D, Bonner C, et al. AMP kinase-mediated activation of the BH3-only protein Bim couples energy depletion to stress-induced apoptosis. J Cell Biol. 2010;189:83–94. doi: 10.1083/jcb.200909166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 47.Kim MJ, Park IJ, Yun H, Kang I, Choe W, Kim SS, et al. AMP-activated protein kinase antagonizes pro-apoptotic extracellular signal-regulated kinase activation by inducing dual-specificity protein phosphatases in response to glucose deprivation in HCT116 carcinoma. J Biol Chem. 2010;285:14617–14627. doi: 10.1074/jbc.M109.085456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 49.Zang Y, Yu LF, Nan FJ, Feng LY, Li J. AMP-activated protein kinase is involved in neural stem cell growth suppression and cell cycle arrest by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside and glucose deprivation by downregulating phospho-retinoblastoma protein and cyclin D. J Biol Chem. 2009;284:6175–6184. doi: 10.1074/jbc.M806887200. [DOI] [PubMed] [Google Scholar]
- 50.Jansen M, Ten Klooster JP, Offerhaus GJ, Clevers H. LKB1 and AMPK family signaling: the intimate link between cell polarity and energy metabolism. Physiol Rev. 2009;89:777–798. doi: 10.1152/physrev.00026.2008. [DOI] [PubMed] [Google Scholar]
- 51.Luo Z, Zang M, Guo W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol. 2010;6:457–470. doi: 10.2217/fon.09.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang W, Guan KL. AMP-activated protein kinase and cancer. Acta Physiol (Oxf) 2009;196:55–63. doi: 10.1111/j.1748-1716.2009.01980.x. [DOI] [PubMed] [Google Scholar]
- 53.Fogarty S, Hardie DG. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim Biophys Acta. 2010;1804:581–591. doi: 10.1016/j.bbapap.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 54.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O'Donnell KA, et al. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol. 2005;25:6225–6234. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Telang S, Lane AN, Nelson KK, Arumugam S, Chesney J. The oncoprotein H-RasV12 increases mitochondrial metabolism. Mol Cancer. 2007;6:77. doi: 10.1186/1476-4598-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010;9:3506–3514. doi: 10.4161/cc.9.17.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fogal V, Richardson AD, Karmali PP, Scheffler IE, Smith JW, Ruoslahti E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol Cell Biol. 2010;30:1303–1318. doi: 10.1128/MCB.01101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:125–136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.