

Summary
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal human malignancies. To investigate the cellular origin(s) of this cancer, we determined the effect of PDAC-relevant gene mutations in distinct cell types of the adult pancreas. We show that a subpopulation of Pdx1-expressing cells is susceptible to oncogenic K-Ras induced transformation without tissue injury, whereas insulin-expressing endocrine cells are completely refractory to transformation under these conditions. However, chronic pancreatic injury can alter their endocrine fate and allow them to serve as the cell of origin for exocrine neoplasia. These results suggest that one mechanism by which inflammation and/or tissue damage can promote neoplasia is by altering the fate of differentiated cells that are normally refractory to oncogenic stimulation.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer death in the United States (Li et al., 2004). Identification of the cell(s)-of-origin of PDAC and defining the biochemical and biological changes that accompany their transformation are essential for the development of early detection and treatment tools.
The pancreas is composed of four parenchymal cell types: acinar, ductal, centroacinar (exocrine) and islet (endocrine) cells. The acinar cells synthesize and secrete zymogens into the ductal lumen. The ducts carry the enzymes to the duodenum and generate bicarbonate. The centroacinar cells (CACs) have been suggested to be progenitor cells in the adult pancreas. The islets of Langerhans are composed of endocrine cell types, which secrete insulin and other proteins that regulate glucose homeostasis (Bardeesy and DePinho, 2002). Rare cells expressing endocrine markers such as insulin can also be found associated with the acini and the ductal epithelium (Mills, 2007).
PDAC arises from in situ precursor lesions termed pancreatic intraepithelial neoplasia (PanIN) (Hruban et al., 2001). PanIN lesions include a spectrum of abnormal proliferative ductal structures (termed PanIN 1-3) that are recognized by transformation from a cuboidal to columnar epithelium, mucin production, and nuclear atypia.
Mutations that result in a constitutively active K-RAS are found in over 95% of PDACs and are believed to be an initiating event for this type of cancer (Jones et al., 2008; Rozenblum et al., 1997). KRAS encodes a small GTPase that in its active GTP-bound form promotes a wide range of cellular responses including proliferation, survival, migration, and metabolism (Shields et al., 2000). The CDKN2A locus encodes the Ink4A and ARF tumor suppressor genes and is also frequently mutated in PDAC. INK4A inactivation mutations are present in virtually all sporadic PDAC, suggesting that INK4A/ARF normally constrains the malignant potential of mutant K-RAS. Other frequent alterations include loss of function mutations of the SMAD4 and p53 tumor suppressor genes (Jaffee et al., 2002). Moreover, developmental regulatory pathways, in particular the Notch, Sonic Hedgehog and Wnt pathways, are often activated in PDACs (Berman et al., 2003; Miyamoto et al., 2003; Thayer et al., 2003; Wang et al., 2009).
PDAC is commonly believed to arise from transformation of ductal cells, although experimental studies have suggested other differentiated cells or progenitor/stem cells as potential origin(s) for this type of cancer. For instance, mouse models expressing various oncogenes in acinar cells during development, such as the Elastase-Tgfα (Wagner et al., 1998) and Elastase-KrasG12D (Grippo et al., 2003) transgenic strains and the knock-in Mist1-Kras4BG12D animals (Tuveson et al., 2006), have implicated acinar cells as being susceptible to transformation and giving rise to tumors containing ductal elements. Transplantation of mouse islet cell cultures expressing the polyoma virus middle T (PyMT) oncogene into histocompatible mice resulted in the induction of pancreatic cancer with ductal histology (Yoshida and Hanahan, 1994), suggesting a potential endocrine origin for PDAC. A role of CACs has been proposed based on studies of mice that harbor a pancreas-specific deletion of the PTEN gene; these animals exhibit a proliferative expansion of CACs that can progress to carcinoma (Stanger et al., 2005). Nestin+ exocrine progenitor cells have been shown to be highly responsive to K-Ras oncogenic activation and were suggested to represent a progenitor origin for PDAC (Carriere et al., 2007).
The close developmental relationships of the different pancreatic cell types and the capacity of endodermal lineages to transdifferentiate (Slack, 1995) add complexity to the PDAC cell-of-origin question. PanIN lesions can express markers of gastric and foregut differentiation (Prasad et al., 2005), suggesting that some degree of transdifferentiation occurs routinely during PDAC progression. This developmental plasticity is evident in some human PDACs that show focal expression of non-ductal lineage markers, including endocrine and exocrine factors. More recently, Habbe et al. (2008) and De La O et al. (2008) have shown that activation of oncogenic K-Ras in acinar cells of the adult mouse leads to efficient mPanIN formation. These groups have also demonstrated in vivo acinar-to-ductal transdifferentiation by lineage tracing studies in these models (De La O et al., 2008; Habbe et al., 2008). Given this plasticity, it is possible that there is no exclusive cell-of-origin for PDAC and that specific genetic alteration(s) define the resulting malignant phenotype regardless of the cell in which they occur. Moreover, the physiological context, including tissue damage and inflammation, may alter a given cell's susceptibility to transformation. For example, Guerra et al. (2007) have reported that K-Ras activation in acinar cells (using the K-Ras+/LSLG12Vgeo; Elas-tTA/tetO-Cre system) resulted in adult onset malignancy only following treatment with caerulein, which causes tissue injury and inflammation (Guerra et al., 2007).
In this study, we investigate whether PDAC arises from an exclusive cell-of-origin and whether a combination of genetic and non-genetic events can collaborate to induce neoplasia in a range of cell types. We describe the use of genetic manipulation of the mouse to systematically determine the effects of oncogenic K-Ras in distinct subsets of pancreatic cells of the adult mouse in a context dependent manner.
Results
Temporally-restricted activation of oncogenic K-Ras in distinct cell populations of the adult pancreas
Activation of oncogenic K-Ras in the common pancreatic embryonic progenitor in the Pdx1Cre;LSL-KrasG12D model results in mPanIN formation and spontaneous progression to PDAC (Aguirre et al., 2003; Hingorani et al., 2003). Because human PDAC is an adult-onset malignancy, we sought to engineer PDAC mouse models in which oncogenic K-Ras (KrasG12D) is activated in the adult animal. We crossed the LSL-KrasG12D strain to different cell-specific CreER lines and compared the phenotypic effect of KrasG12D activation on these distinct cell populations in the adult pancreas (Fig. 1A). The CreER lines used were: (i) the Pdx1CreERTM transgenic strain (Gu et al., 2002), which marks the earliest pancreatic progenitor cell during development as well as adult endocrine β cells, some ductal, acinar cells and possibly adult progenitor/stem cells (Koizumi et al., 2003; Stoffers et al., 1999; Swift et al., 1998; Wu et al., 1997); (ii) the RipCreERTM transgenic strain (Dor et al., 2004), which is selective for insulin+ cells; and (iii) the acinar procarboxypeptidase A1 CreERT2 (proCPA1CreERT2) knock-in strain (Zhou et al., 2007). The recombination specificity and efficiency of the different CreER strains was determined by crossing them to the LSL-LacZ reporter mouse and analyzing the X-gal stained pancreata of their Tamoxifen (TM)-treated compound progeny (Fig. 1B-E and Table S1). As shown in Fig. 1B, in the Pdx1CreERTM transgenic strain recombination was achieved in a mosaic fashion in the islets, acinar and ductal cells. The pro-CPA1CreERT2 strain exhibited recombination mainly in acinar cells and possibly CACs; however, recombination was evident also in a subset of ductal and islet cells. Recombination in this strain was variable, with half of the treated mice showing no evidence of recombination and the other half ranging from 0-49% in acinar cells (Fig. 1C, Table S1 and data not shown). With TM administration, the RipCreERTM transgenic strain was very specific and efficient in causing recombination in both insulin-producing β cells located in the islets of Langerhans and in single insulin+ cells found scattered throughout the pancreas parenchyma, as shown by X-gal and co-immunofluorescent (CoIF) staining (Fig. 1D, E respectively and Table S1).
Figure 1. KrasG12D activation in distinct cell populations of the adult pancreas.
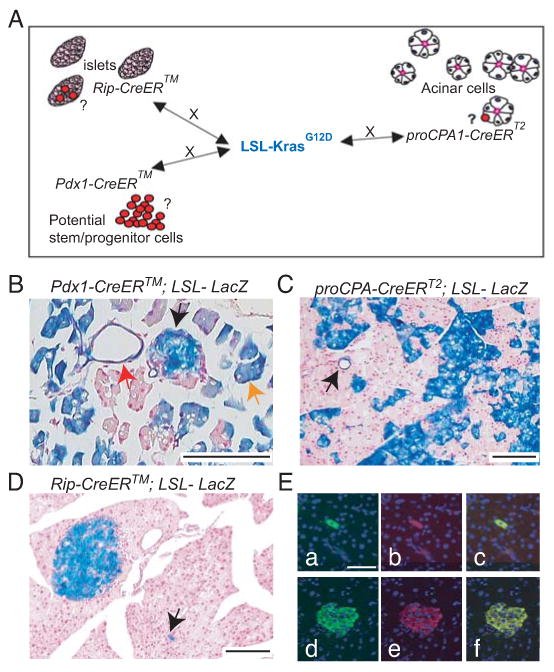
A: Diagram describing the different mouse strains used in the study. The LSL-KrasG12D strain was crossed to the insulin+ cells specific RipCreERTM strain (Dor et al., 2004); to the acinar proCPA1CreERT2 strain (Zhou et al., 2007) and to the putative stem/ progenitor cell specific Pdx1CreERTM strain (Gu et al., 2002).
B-D: X-gal stained pancreata of TM-treated Pdx1CreERTM;LSL-LacZ (B), proCPA1CreERT2;LSL-LacZ (C), RipCreERTM;LSL-LacZ (D). Note specific recombination in islet (black arrow), acinar (orange arrow) and ductal (red arrow) cells (B), acinar cells and a duct (C and arrow in C, respectively) and in the islets of Langerhans and scattered extrainsular endocrine cells (D and arrow in D, respectively).
E: LacZ and insulin CoIF staining of pancreas derived from a TM-treated RipCreERTM;LSL-LacZ. Note double staining of a single cell (top row) and of cells within the islets of Langerhan (lower row) (a)+(d): Insulin, (b)+(e) LacZ, (c)+(f) merge
Bars: 100 μm.
Expression of KrasG12D in different cell types of the adult pancreas was achieved by systemic TM administration in mice at 14-56 days of age. Because newly transformed cells would be expected to resemble their cell-of-origin both genetically and morphologically, we sought to define the earliest time point when transformed cells could be detected after TM administration. Therefore, mice were sacrificed at different times following the last dose, ranging from 4-240 days.
KrasG12D induces transformation in Pdx1+ cells of the adult pancreas
Pdx1CreERTM;LSL-KrasG12D mice were treated with TM at day 14, 21, 24, 27 or 56 after birth and sacrificed after 4-120 days. mPanINs and ductal metaplasia were observed in all age groups (Fig. 2 and Table S2). Low-grade mPanIN 1A lesions were the most prevalent and were observed to a comparable extent across the three main age groups, while mPanIN 1B were less frequent in the 56 day old group. Although mPanIN lesions were predominantly grade 1, grade 2 lesions were also identified, mostly at 120 days after TM administration and only in the 14- and 27-day old treated mice (Table S2, Fig. 2E). These results suggest that the Pdx1+ target cell for transformation is either less susceptible or less abundant in 56 day old mice. mPanIN 3 was found only in one mouse that was analyzed 188 days after TM administration and it was also the only mouse that developed PDAC. This result is consistent with observations in humans that it is unusual to find PanIN 3 in pancreata lacking invasive carcinoma (Mills, 2007).
Figure 2. KrasG12D activation by Pdx1CreERTM causes mPanIN formation and ductal metaplasia.
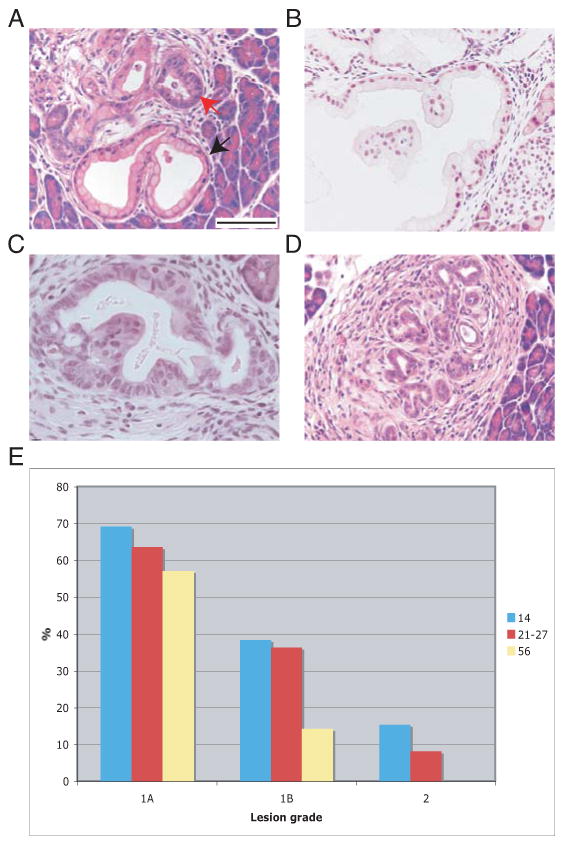
A-D: H&E-stained sections of Pdx1CreERTM;LSL-KrasG12D mice 20 (A) and 120 (B-D) days after TM administration. A. mPanIN1A (black arrow). Note flat columnar epithelium with mucin accumulation and basally oriented nuclei. Red arrow: ductal metaplasia. B. mPanIN1B. Note papillary architecture with small, basally oriented nuclei. C. mPanIN2. Note papillary architecture, moderate nuclear pleomorphism and partial loss of polarity. D. Ductal metaplasia. Bars: 100 μm.
E: Percentage of mice that developed different grades of mPanINs upon KrasG12D activation at the described postnatal dates
TM-treated 14 or 27 day old mice exhibited additional phenotypes (N=23). One of the most notable was the appearance of acinar cells and ductal structures “embedded” in the islets of Langerhans as soon as 10 days following TM treatment (Fig. 3A, B, respectively). The ductal structures grew over time (Fig. 3C), and remained proliferative (data not shown). These lesions became elongated and produced mucin, reminiscent of mPanINs (Fig. 3D). On the other hand, some mPanIN in the exocrine compartment harbored cells that positively stained for endocrine markers such as glucagon, insulin, somatostatin and PYY (Fig. S1A,B and data not shown). Other histologic findings at 120 days after TM treatment included robust atrophy of the pancreas with a hypoplastic exocrine compartment, large cystically dilated areas, and strong stromal reaction (Fig. S1C,D). Importantly, no lesions arose in vehicle-treated mice (N=15) (Table S2). These results exclude the possibility of leaky Cre recombinase activity and TM-independent K-Ras activation during development and/or adulthood. In addition, we observed no evidence of chronic pancreatitis in treated mice. Since TM is dissolved in corn oil, which by itself causes a mild form of peritonitis (Fig. S1E), we treated Pdx1CreERTM;LSL-KrasG12D mice with TM dissolved in carboxymethyl cellulose (CMC) by oral gavage. Dissolving TM in corn oil or CMC did not affect the specificity and efficiency of Cre-induced recombination as confirmed by X-gal staining in mice harboring a LacZ reporter (data not shown). As shown in Fig. S1F, pancreata of mice treated with TM dissolved in CMC were free of peritonitis yet still developed grade 1 mPanINs (N=5). This result precludes the possibility that the corn oil-induced peritonitis was essential for formation of mPanINs and ductal metaplasia in the Pdx1CreERTM;LSL-KrasG12D model.
Figure 3. KrasG12D activation by Pdx1CreERTM causes intra-islet ductal/ acinar lesions.
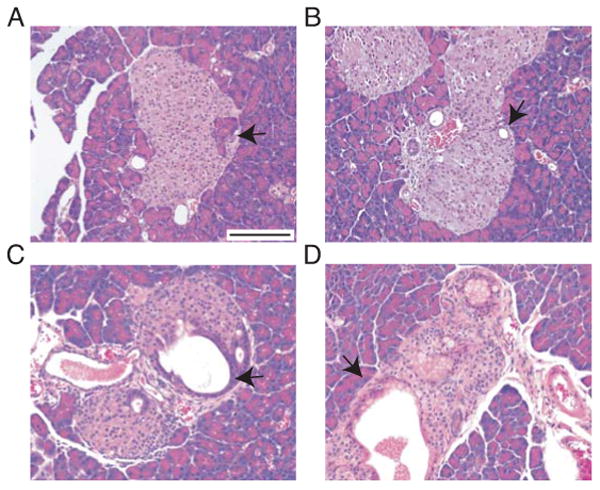
A-D: H&E-stained sections of Pdx1CreERTM;LSL-KrasG12D mice 10 (A, B) 20 (C) and 40 (D) days after TM administration. Acinar cells (arrow in A) and small ducts (arrow in B) embedded within the islets. Enlarged ducts within the islets (arrow in C). Mucin-producing elongated cells in ductal structures in the islets (arrow in D).
Bars: 50 μm.
Activation of KrasG12D in combination with mutations of Ink4A/Arf (Aguirre et al., 2003) or Trp53 (Jonkers et al., 2001) in Pdx1+ cells in the adult mouse resulted in formation of moderately to poorly differentiated PDAC in 10/13 mice tested (Fig. 4 and Table S2). Many of the tumors contained undifferentiated areas with sarcomatoid or anaplastic features, and extension beyond the pancreas was common. We observed direct invasion into the stomach and small intestine as well as metastasis to mediastinal lymph nodes, diaphragm and peritoneal adipose tissue (Fig. 4 and data not shown). We also identified a full range of mPanIN lesions in these mice, including grade 3 (Table S2, Fig. 4A, B). Thus, Pdx1+ cells, or at least a subpopulation of Pdx1+ cells, represent an excellent candidate to be the/a cell-of-origin for PDAC in the mouse and in humans.
Figure 4. KrasG12D activation in combination with p53 or Ink4A/Arf loss in Pdx1+ cells of the adult pancreas results in mPanIN3 and PDAC development.
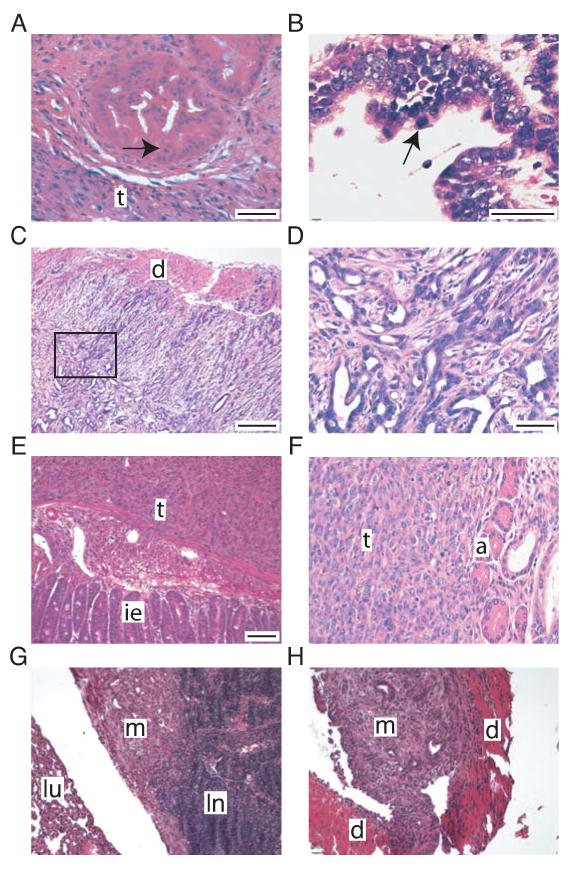
A-E, G-H: H&E-stained sections of mPanIN3 and PDACs derived from Pdx1CreERTM;LSL-KrasG12D; Trp53flox/flox mice.
A-B: mPanIN3 lesions. Note adjacent tumor (t) and high grade features, including cribriform architecture (A) and apical mitosis (B). Arrows point to mitotic figures.
C-D: Invasive, moderately differentiated PDAC (Note invasion through the duodenal wall (d) in C). D: enlarged box in C.
E: Undifferentiated PDAC. Tumor (t) invades submucosa of the small intestine and abuts intestinal epithelium (ie).
F: H&E-stained section of undifferentiated PDAC (t) and residual normal acini (a) derived from a Pdx1CreERTM;LSL-KrasG12D; Ink4A/Arfflox/flox mouse.
G-H: Metastatic PDAC. G: Metastasis (m) to mediastinal lymph node (ln) with adjacent lung (lu). H: Metastasis to diaphragm (d).
Bars: A, B: 100 μm; C 200 μm; D, F 50 μm; E,G,H: 200 μm.
Targeted expression of KrasG12D in proCPA1+ cells
Targeting the expression of KrasG12D to pancreatic acinar cells and other cell types by treating proCPA1CreERT2;LSL-KrasG12D mice with TM resulted in low grade mPanIN-1A formation in only 2/20 mice (Table S3 and Fig. S2A). All mice were tested and confirmed for LSL-KrasG12D recombination by PCR analysis of DNA extracted from tissue sections (Table S3). These results suggest that proCPA1+ cells are not efficiently transformed by KrasG12D into mPanIN lesions in the adult mouse. It has been recently shown that KrasG12D activation in acinar cells of the adult mouse leads to efficient mPanIN formation in the absence of tissue injury in the Ela-CreERT2Tg/+, Mist1CreERT2/+ and ElaCreERT strains (De La O et al., 2008; Habbe et al., 2008). The phenotypic diversity observed between these models is discussed below (see also Table S7).
To determine whether loss of p53 or Ink4A/Arf could affect the neoplastic potential of proCPA1+ cells, we combined the LSL-KrasG12D allele with the Trp53flox or Ink4A/Arfflox alleles on the background of the proCPA1CreERT2 strain. The pancreata derived from TM-treated proCPA1CreERT2;LSL-KrasG12D;Ink4A/Arfflox/flox mice (N=7) showed no overt cancer phenotype when examined at 50, 83, 113, 158, 188 and 309 days after TM administration. However, one proCPA1CreERT2;LSL-KrasG12D;Trp53flox/flox mouse (N=5) developed a moderately to poorly differentiated PDAC 81 days after TM injection at 53 days of age. The tumor contained undifferentiated areas, was locally invasive, and metastasized to the liver and diaphragm (Table S3; Fig. S2B). As described earlier, all mice in this study were analyzed for DNA recombination by PCR (Table S3). We conclude that highly aggressive PDAC can be initiated in proCPA1+ cells in adult mice without chronic pancreatitis, albeit with low penetrance in this system.
In a previous study, Guerra et al. (2007) have reported that selective expression of an endogenous K-Ras(G12V) oncogene in adult cells of the acinar/centroacinar lineage using the K-Ras+/LSLG12Vgeo; Elas-tTA/tetO-Cre mouse model resulted in no phenotype unless the mice were pre-treated with caerulein (Guerra et al., 2007). Caerulein induces chronic pancreatitis that results in regeneration of normal tissue and formation of metaplastic lesions of a ductal phenotype in the exocrine compartment (Strobel et al., 2007). The authors concluded that chronic pancreatitis can be a co-factor in the induction of PDAC by the K-Ras(G12V) oncogene in adult mice (Guerra et al., 2007). Because the proCPA1CreERT2;LSL-KrasG12D mice described above were largely refractory to KrasG12D induced transformation, we set out to test whether chronic pancreatitis could facilitate PDAC formation in these mice.
We treated 34-60-day-old mice (N=9) with caerulein for a total of 44-193 days. One month after caerulein treatment initiation, the mice were treated with TM to activate Cre. In addition to caerulein-induced inflammation and ductal metaplasia, 3/9 proCPA1CreERT2;LSL-KrasG12D and proCPA1CreERT2;LSL-KrasG12D;LSL-LacZ mice developed grade 1 mPanINs (Fig. 5A and Table S4). We confirmed LSL-KrasG12D recombination by PCR analysis of DNA extracted from laser-captured microdissected (LCM) lesions (Fig. 5C and Table S4). Hence, the mPanINs arose from ProCPA+ cells. Importantly, 3 of 6 proCPA1CreERT2;LSL-KrasG12D;p53flox/flox developed PDAC when treated first with caerulein and then with TM (Fig. S3B and Table S4). mPanIN grades 1 and 2 were observed, and the neoplastic lesions and advanced tumors were similar to those arising in Pdx1CreERTM compound mutant mice with respect to histologic appearance, local invasion and distant metastasis. In addition, a single proCPACreERtT2;LSL-KrasG12D;Ink4A/Arff/f; mouse that was treated first with TM and then with caerulein developed PDAC (Fig. S3C and Table S4). To rule out false-positive results due to non-specific Cre recombinase activity upon caerulein-induced injury, proCPACreERT2;LSL-KrasG12D;LSL-LacZ mice (N=3) were treated with caerulein without prior TM administration for 8, 15 and 22 days. LacZ+ cells were not observed in these mice (data not shown). Thus, the proCPACreERT2 allele remains tightly controlled under chronic pancreatitis conditions. Immunofluorescence for CPA revealed that most mPanIN cells in the proCPA1CreERT2;LSL-KrasG12D mouse model failed to express this marker (Fig. 6D). This result supports the hypothesis that proCPA+ cells can undergo injury-induced transdifferentiation and give rise to mPanIN formation in the context of KrasG12D activation. In addition, our results provide evidence that pancreatic injury synergizes with KrasG12D in transforming proCPA+ cells and initiating PDAC.
Figure 5. Chronic pancreatitis promotes mPanIN formation in mouse models largely refractory to KrasG12D activation alone.
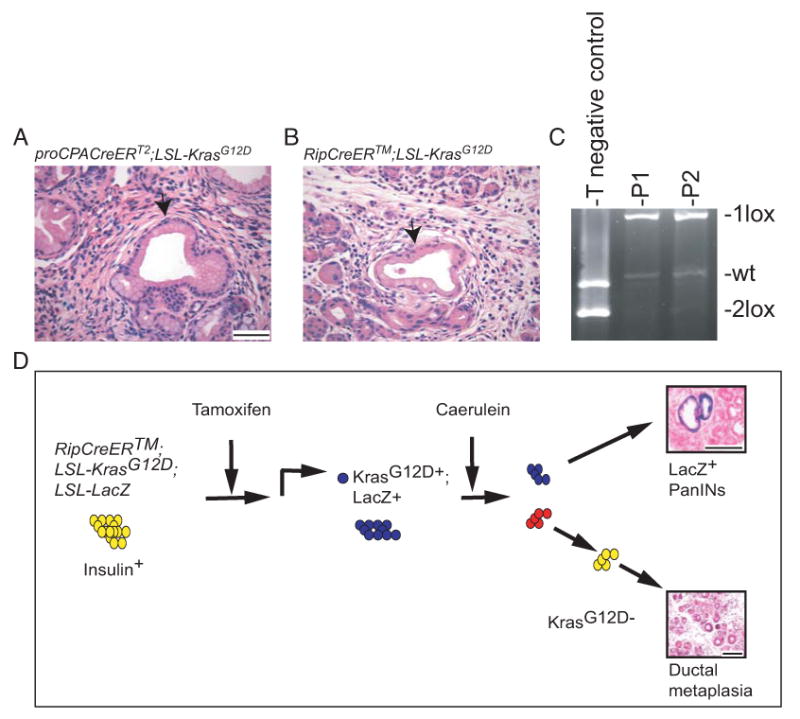
A-B: H&E staining of pancreata derived from Caerulein and TM-treated proCPA1CreERT2;LSL-KrasG12D (A) and RipCreERTM;LSL-KrasG12D mice (B). Note mPanIN formation (arrows). Bars: 50 μm
C: PCR of tail (T) and laser captured microdissected mPanINs DNA from Caerulein+ TM-treated proCPA1CreERT2;LSL-KrasG12D (P1) and RipCreERTM;LSL-KrasG12D (P2) mice. The recombined allele (1lox) is present in the mPanINs DNA but not in the tail DNA of compound mutant mice.
D: Suggested model of Caerulein/ inflammation–induced mPanIN formation in TM-treated RipCreERTM;LSL-KrasG12D;LSL-LacZ mice. TM administration results in recombination-induced activation of KrasG12D and LacZ expression in insulin+ cells of the adult mouse. This recombination is permanent and marks both the cells and their progeny. Upon caerulein treatments, if insulin+ cells are targeted for transformation by KrasG12D, they will give rise to LacZ+ mPanINs. Otherwise, only LacZ- caerulein-induced metaplasia will form.
Bars: A, B 50 μm; D, 100 μm.
Figure 6. Characterization of mPanINs derived from TM and then caerulein treated proCPA1CreERT2;LSL-KrasG12D and RipCreERTM;LSL-KrasG12D mice.
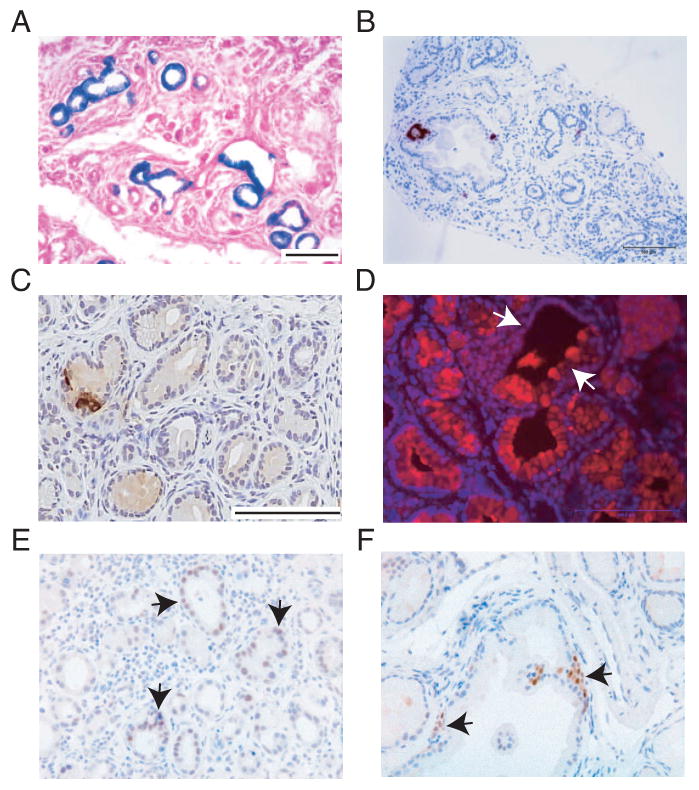
A-C, E: X-gal (A) insulin (B, C) and Pdx1 (E) staining of pancreata derived from RipCreERTM;LSL-LacZ;LSL-KrasG12D mice. Note X-gal and Pdx1 positive staining in mPanINs (A and black arrows in E, repectively) and negative staining for insulin in most mPanINs (B-C)
D, F: IF staining for CPA1 and IHC staining for Pdx1 of pancreata derived from proCPA1CreERT2;LSL-KrasG12D mice. Note positive and negative CPA1 staining in the same mPanIN (white arrows in D) and positive staining for Pdx1 (black arrows in F).
Bars: A, B 100 μm; C-F 100 μm.
Targeted expression of KrasG12D in insulin+ cells
Targeting the expression of KrasG12D to insulin+ cells by treating RipCreERTM;LSL-KrasG12D mice with TM did not result in mPanIN formation in any of the different age groups (N=30), even after 8 months (Table S3). Although these results suggest that insulin+ cells are not targeted for transformation by KrasG12D, we nonetheless combined the KrasG12D allele with the conditional Trp53 or Ink4A/Arf alleles on the background of the RipCreERTM strain. None of the RipCreERTM;LSL-KrasG12D; Trp53flox/flox mice or the RipCreERTM;LSL-KrasG12D; Ink4A/Arfflox/flox mice developed mPanINs or PDAC (N=14). As described above, recombination of LSL-KrasG12D was confirmed by PCR analysis of DNA extracted from tissue sections of all TM-treated mice in this study (Table S3). These results strongly suggest that insulin+ cells are highly refractory to transformation by multiple oncogenic mutations.
We next set out to test the effect of chronic pancreatitis on PDAC formation in the RipCreERTM;LSL-KrasG12D mice. Two RipCreERTM;LSL-KrasG12D;LSL-LacZ mice were treated with caerulein for a total of 84 and 89 days. One month after caerulein treatment initiation, the mice were treated with TM to activate Cre. Interestingly, grade 1 mPanINs were detected in one of these mice (Fig. 5B and Table S4). We confirmed LSL-KrasG12D recombination in the pancreas of the mouse that did not show mPanIN formation as well as in mPanINs derived from the second mouse by PCR analysis of DNA extracted from tissue section and of DNA extracted from LCM lesions, respectively (Fig. 5C and Table S4). Hence, the mPanINs observed arose from cells in which the transgenic Rat insulin promoter was active. Importantly, all three RipCreERTM;LSL-KrasG12D; p53flox/flox;LSL-LacZ mice developed poorly- and undifferentiated PDAC (N=2 and N=1, respectively) when treated first with caerulein and then with TM (Fig. S3D and Table S4). As in the Pdx1CreERTM;LSL-KrasG12D;p53flox/flox mutant mice, these were highly aggressive tumors that exhibited a capacity for local invasion and distant metastasis.
One interpretation of these results (as well as those of Guerra et al. (2007)) is that caerulein treatment or inflammation caused a change in differentiated cells that made them susceptible to K-Ras activation. To investigate this possibility, we crossed the RipCreERTM;LSL-KrasG12D mice to the LacZ reporter strain and pulsed the triple compound progeny of these mice with TM 7-14 days prior to caerulein treatment initiation (N=6) (Table S4). In this way, differentiated insulin+ cells and their progeny would be marked by the LacZ expression and their fate could be identified after caerulein treatment by X-gal staining (Fig. 5D). Strikingly, LacZ+ mPanINs were found in 4/6 pancreata of these mice (Fig. 6A, and Table S4). In the remaining 2 mice mPanINs were not observed, which might be explained by the small pancreatic area analyzed (less than 2 mm2, Table S6). In addition, 2 mice developed LacZ+ poorly to undifferentiated tumors with local invasion to adjacent tissues. These results suggest that KrasG12D expressing insulin+ cells can be transformed and give rise to mPanINs and PDAC specifically in the context of chronic pancreatitis.
To explore the possibility of non-specific Cre recombinase activity upon caerulein-induced injury, RipCreERTM;LSL-KrasG12D;LSL-LacZ mice (N=5) were treated with caerulein without prior TM administration. Surprisingly, despite the absence of TM, LacZ+ cells were observed in the islets of Langerhans and in scattered cells throughout the parenchyma as early as 2 days after caerulein treatment initiation (data not shown). Thus, although the RipCreERTM strain is tightly controlled and does not normally show leaky expression of the Rat insulin promoter in insulin- cells or TM-independent activation of Cre recombinase (Table S1), Cre activity was more promiscuous under chronic pancreatitis conditions. This result raised the possibility that the mPanIN lesions observed in the experiment described above arose in non-insulin expressing cells following inappropriate Cre activation during the caerulein treatment. To address this possibility directly, RipCreERTM;LSL-LacZ mice (N=5) were treated with TM for 5-7 days followed by caerulein treatment for 65 days and then analyzed for coexpression of LacZ and insulin by CoIF (Table S5). In all cases, LacZ+insulin+ cells were observed within the islet of Langerhans. In addition, we scored a total of 83 LacZ+insulin+ and 6 LacZ+insulin- single cells scattered throughout the pancreata. We then calculated the frequency of mPanIN lesions per mm2 in the TM/caerulein treated RipCreERTM;LSL-KrasG12D;LSL-LacZ and RipCreERTM;LSL-KrasG12D mice. Importantly, the frequency of mPanIN lesions in these mice was 6-fold higher than the frequency of LacZ+insulin- cells in TM/caerulein treated RipCreERTM;LSL-LacZ mice (P=0.04 by Wilcoxon Rank Sum Test) (Tables S5 and S6). Thus, a minority of the mPanINs observed in the TM and caerulein treated RipCreERTM;LSL-KrasG12D;LSL-LacZ and RipCreERTM;LSL-KrasG12D mice might have arisen from insulin- cells. However, based on this statistical analysis, it is likely that the vast majority of the mPanINs can be attributed to transformation of insulin+ cells. Thus, our results provide evidence that insulin+ cells can be made susceptible to KrasG12D and give rise to mPanIN and PDAC formation following tissue injury and inflammation. Interestingly, immunohistochemistry (IHC) revealed that most mPanIN cells in this model failed to express insulin (Fig. 6B and C). Although the mechanism through which mature insulin-producing cells undergo this fate change is unclear, it is notable that these mPanINs harbored Pdx1+ cells and cells that positively stain for the Notch intracellular domain (NotchIC) (Fig. 6E and data not shown).
To further study transformation of insulin+ cells, four RipCreERTM;LSL-KrasG12D;p53flox/flox;LSL-LacZ mice were treated with TM and 14-40 days later with caerulein for additional 42-52 days. These mice developed high-grade mPanIN and poorly differentiated to undifferentiated carcinoma capable of local invasion and distant metastasis (Fig. 7A, B and Table S4). The undifferentiated areas were morphologically identical to those observed in Pdx1CreERTM;LSL-KrasG12D; Trp53flox/flox mice and also resembled human undifferentiated pancreatic carcinoma (Hoorens et al., 1998). Tumors in these mice were negative for insulin as well as the neuroendocrine marker synaptophysin by IHC (Fig. 7C, D). Furthermore, no preneoplastic lesions were observed in the islets of these mice (data not shown). In conclusion, our results provide evidence that in combination with pancreatic injury, KrasG12D-expressing insulin+ cells of the endocrine lineage, which are refractory to transformation under normal conditions, can also serve as a cell-of-origin of PDAC, a malignancy with an exocrine phenotype.
Figure 7. KrasG12D activation and p53 loss followed by chronic pancreatitis results in mPanIN2-3 and PDAC development from insulin+ cells.
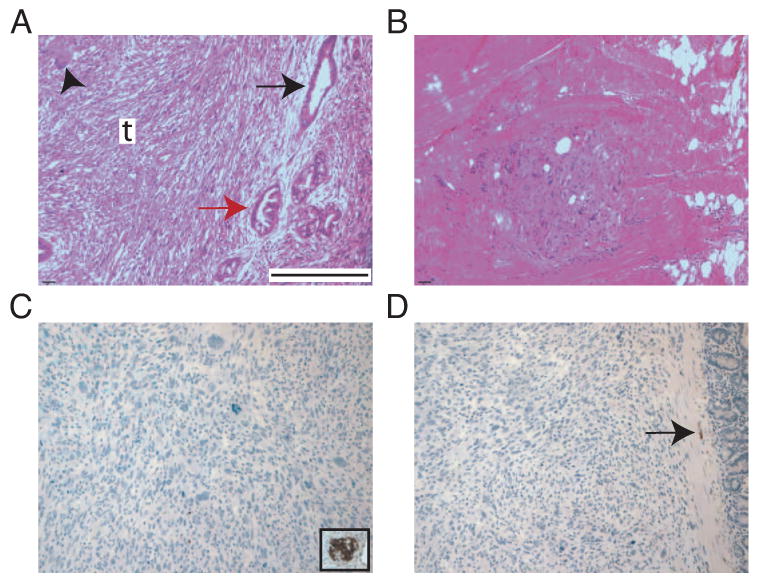
A-D: Pancreata derived from RipCreERTM;LSL-KrasG12D; Trp53flox/flox mice treated with TM followed by caerulein.
A: Multiple mPanINs (grade 2-3, black and red arrows, respecively) and undifferentiated carcinoma (t). Arrowhead: giant cell of invasive carcinoma.
B: Metastatic carcinoma in the diaphragm.
C: Negative immunostain for insulin in carcinoma. Inset: positive insulin immunostain in non-neoplastic islet.
D: Negative immunostain for synaptophysin in carcinoma. Note positive staining in axon located in the smooth muscle of the small intestine (arrow).
Bars: A-D: 400 μm.
Discussion
We have studied the susceptibility of adult pancreatic cells to transformation by KrasG12D. We show that Pdx1+ cells can efficiently give rise to mPanIN and ductal metaplasia and, in combination with p53 or Ink4A/Arf deletion, can develop into PDAC. In contrast, in the absence of tissue injury and inflammation, insulin+ cells (which also express Pdx1) did not show any overt phenotype, even in the context of tumor suppressor gene mutation. Likewise, proCPA1+ cells were inefficiently transformed. Thus, we conclude that under non-inflammatory conditions, a subpopulation of Pdx1+ cells of the adult pancreas have heightened sensitivity to the KrasG12D oncogenic effects and represent a cell-of-origin of PDAC. Of note, Habbe et al. (2008) have reported that Pdx1+ cells are relatively resistant to KrasG12D-induced transformation in 42 day-old mice. Our data suggest that younger mice are more susceptible to transformation of this cell type. It is also possible that the different observations are attributable to background differences between the mice used in both studies.
Four Cre lines have been used to target the expression of oncogenic K-Ras to adult acinar cells: K-Ras+/LSLG12Vgeo(KRasG12V); Elas-tTA/tetO-Cre, (Guerra et al., 2007); Ela-CreERT2Tg/+ and Mist1CreERT2/+ (Habbe et al., 2008); ElaCreERT (De La O et al., 2008) and the proCPA1CreERT2 studied here. Different phenotypes, ranging from no lesion to high-grade mPanIN-3 formation, were observed in these strains (Table S7). The phenotypic diversity might be explained by several factors, including: 1) The K-Ras+/LSLG12Vgeo strain lacks the 3′ UTR element which contains several regulatory sequences, including binding sites for the let-7 miRNA family that regulates both N-Ras and K-Ras (Johnson et al., 2005) and has been suggested to function in tumor suppression (Lee and Dutta, 2007; Mayr et al., 2007; Takamizawa et al., 2004; Yu et al., 2007). Thus, the more abundant lesion formation in the 10 day-old K-Ras+/LSLG12Vgeo; Elas-tTA/tetO-Cre mice compared to the proCPA1CreERT2;LSL-KrasG12D may result from more tightly controlled translational repression of KrasG12D versus K-Ras(G12V). 2) The models utilize distinct K-ras activating mutations (G12V versus G12D) which might have an effect on the oncogenic potency of K-Ras (Bardeesy and Sharpless, 2006; Collado et al., 2005; Seeburg et al., 1984). 3) Recombination efficiency differences between the different strains may exist, either because of differences between Cre-driving promoters per se, mouse background, or different TM and doxycycline administration protocols. 4) The different mouse models may reflect a previously uncharacterized heterogeneity of acinar cells with respect to function and/or susceptibility to transformation. Nevertheless, the collective observations provide evidence that there are acinar cells in the adult pancreas sensitive to oncogenic K-Ras activation that can progress to at least mPanIN in the absence of overt tissue injury and that highly aggressive PDAC can be initiated from proCPA1+ cells, albeit with low penetrance.
When injury was induced before recombination, the proCPA1CreERT2;LSL-KrasG12D and RipCreERTM;LSL-KrasG12D mice became sensitive to KrasG12D activation. Importantly, when we activated KrasG12D first and then induced chronic pancreatitis, RipCreERTM;LSL-KrasG12D mice still developed mPanINs. We found that under caerulein-only treatment conditions Cre recombinase activity was not detectable in the proCPA1CreERT2 strain, although, surprisingly, it could be detected in insulin+ and rare insulin- cells in the RipCreERTM strain. This result might reflect a differential sensitivity to caerulein of CreERT2 (Indra et al., 1999) in the proCPA1CreERT2 versus CreER™ (Danielian et al., 1998) in the RipCreERTM strain. Alternatively, an increased amount of Cre protein might be produced by the RipCreERTM transgene in comparison with the proCPA1CreERT2 knockin strain that might result in its greater sensitivity to caerulein. Regardless of the effects of caerulein, the vast majority of the mPanINs can be attributed to transformation of insulin+ cells, although it is possible that a few mPanINs are derived from insulin- cells as well. Furthermore, 2/6 of the RipCreERTM;LSL-KrasG12D;LSL-LacZ and all RipCreERTM;LSL-KrasG12D;p53 flox/flox;LSL-LacZ mice (N=4) developed high grade mPanIN and tumors that resembled the most aggressive PDAC that arose in Pdx1CreERTM and proCPA1CreERT2 compound mice. These tumors exhibited no morphologic or immunohistochemical evidence of neuroendocrine differentiation. In addition, the pre-invasive changes in these mice were of an exocrine phenotype (i.e. mPanIN) rather than endocrine. Taken together, these results show that insulin+ cells of the endocrine lineage of the adult pancreas can transdifferentiate and give rise to highly aggressive exocrine neoplasia (PDAC). Of note, LacZ+ cells were not detected in ductal metaplasia in TM/caerulein treated RipCreERTM;LSL-LacZ mice (data not shown), suggesting that the transdifferentiation of the insulin+ cells requires both injury and oncogenic K-Ras. Similar results were observed by Strobel et al. in RipCreERTM;Z/AP mice (Strobel et al., 2007). Interestingly, oncogenic K-Ras activation is rarely observed in human endocrine tumors (Jonkers et al., 2007). This may in part be explained by reprogramming of endocrine cells to an exocrine fate upon injury and K-Ras activation.
The transdifferentiated cells in the RipCreERTM;LSL-KrasG12D mice may be insulin+ cells that reside either in and/or outside the islets of Langerhans. The physiological role of the insulin+ cells that reside outside of the islets is largely unknown. However, the mPanINs in the caerulein+TM-treated mice were always observed outside the islets suggesting that these cells may be more susceptible to KrasG12D than those that reside in the islets. Interestingly, neoplastic ducts were found within the islets of TM-treated Pdx1CreERTM;LSL-KrasG12D mice but not when KrasG12D was activated during development using the non-conditional Pdx1Cre strain (Hingorani et al., 2003). This may reflect a developmental compensation mechanism or result from mouse background differences.
Caerulein might promote KrasG12D-induced transformation by several mechanisms. It is possible that a rare population of proCPA1- and/or insulin-expressing progenitor cells that are normally susceptible to KrasG12D oncogenic effect needs to be amplified by tissue injury for the appearance of the phenotype. Another possibility is that injury-induced regeneration or the secretion of inflammatory cytokines leads to the proliferation of stem cell/facultative stem cells that are most probably Pdx1+. Upon the initiation of differentiation of such cells, a subset might transiently express either proCPA1CreERT2 or RipCreERTM and, thus, activate KrasG12D upon TM administration. However, our results provide evidence that proCPA1- and insulin-expressing cells can form mPanINs, favoring the hypothesis that inflammation and tissue injury facilitate PDAC development by promoting reprogramming of differentiated cells rather than by stem cell mobilization. The mechanism through which such a reprogramming event might occur requires further investigation. However, our study and those of others suggest a role for the Notch signaling pathway in this process. Caerulein-derived acinar-ductal metaplasia is associated with Notch pathway up-regulation (Gomez et al., 2004; Jensen et al., 2005; Siveke et al., 2008). We also found Notch to be activated in mPanINs derived from Caerulein/TM –RipCreERTM;LSL-KrasG12D mice. Re-activation of the Notch signaling pathway, which is important for cell fate decisions and maintenance of undifferentiated stem/progenitor cells during embryogenesis (Artavanis-Tsakonas et al., 1999), may be a mechanism by which differentiated cells can revert to a stem/ progenitor fate or to convert to a cell type that is sensitive to KrasG12D activity. It has been recently shown that Notch and K-Ras reprogram acinar cells to mPanINs (De La O et al., 2008). The precise mechanism of, and requirement for, interaction between chronic pancreatitis, Notch and K-Ras activation for mPanIN formation from different pancreatic cells still need to be determined.
These data add to the increasing body of evidence pointing to a remarkable plasticity of pancreatic adult differentiated cells (De La O et al., 2008; Habbe et al., 2008; Shen et al., 2000; Zhou et al., 2008) and questions the existence of a pancreatic “stem cell” as well as its relevance for PDAC. Our findings suggest that KrasG12D regulates the differentiation status of pancreatic epithelial progenitor/differentiated cells to a certain extent and that in combination with non-genetic stress such as inflammation, this is further accelerated. More specifically, a multipotent Pdx1+ Insulin- cell may exist in the adult pancreas. This cell type may be the source of the endocrine cells within mPanINs and ductal metaplasia observed in the TM treated Pdx1CreERTM;LSL-KrasG12D mice and the endocrine cells arising from the ductal lining after pancreatic duct ligation (Xu et al., 2008). It is possible that this cell type is activated only in certain settings such as injury and neoplasia and may be particularly susceptible to KrasG12D-induced proliferation and differentiation. It might also be the source of the ductal structures located in the islets, if it resides both in the ductal lining as well as the islets or if it migrates to the islets under certain conditions. Alternatively, these structures may arise from a differentiated cell, such as the carbonic anhydrase II+ cell that give rise to endocrine and exocrine cells after birth and injury (Inada et al., 2008), upon KrasG12D activation.
Our study reveals the involvement of different cell types in PDAC initiation and provides a series of models to explore the biology and treatment of PDAC. With the advances in the ability to reprogram adult pancreatic cells in a controlled manner (Zhou et al., 2008), we expect that a better understanding of the reprogramming and transforming events attributed to KrasG12D activation in combination with chronic inflammation will lead to the development of new therapeutic modalities to prevent PanIN formation or to repair the diseased/damaged tissue.
Experimental Procedures
Mouse strains
All animal studies and procedures were approved by the MIT Institutional Animal Care and Use Committee. Research was conducted in compliance with the Animal Welfare Act Regulations and other Federal statutes relating to animals and experiments involving animals and adheres to the principles set forth in the Guide for Care and Use of Laboratory Animals, National Research Council, 1996. The LSL-KrasG12D strain (Jackson et al., 2001) was crossed to the following strains: Pdx1CreERTM (Gu et al., 2002), proCPA1CreERT2 (Zhou et al., 2007), RipCreERTM (Dor et al., 2004), Trp53flox (Jonkers et al., 2001) and Ink4A/Arfflox (Aguirre et al., 2003). Primers used for genotyping by PCR are listed in Table S8 and details of reactions are available upon request. Cre strains were crossed to R26-LSL-LacZ mice (Jackson Laboratories) to determine Cre expression patterns.
Histopathology, Immunohistochemistry (IHC) and immunofluorescence (IF)
Histopathologic analysis of pancreata was carried out by two pathologists (G.C.C. and E.L.S.). mPanIN lesions and PDAC were graded according to consensus criteria for mouse models of pancreatic cancer (Hruban et al., 2006). We carried out IHC and IF analyses according to manufacturer's recommendations, typically using a modified citric acid unmasking protocol followed by standard detection with 3,3-diaminobenzidine (DAB) using a kit from Vector Laboratories. In some cases, secondary antibodies were conjugated to AlexaFluor 594 (Invitrogen) and nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI). We used the following primary antibodies: insulin (Zymed), glucagon (Abcam), PYY (RDI), Pdx1 (Gift of Dr. Christopher V Wright), CPA1 (Chemicon) and β-galactosidase (AbD Serotec). IF staining was performed as described (Jackson et al., 2001). Triple-color microscopy and imaging were performed with a Nikon Eclipse E600 and a Spot cooled CCD camera and software. Images were processed with Adobe Photoshop.
β-Galactosidase histochemistry
Pancreata were dissected and fixed in 4% PFA, incubated in 0.5M Sucrose and frozen in OCT. β-galactosidase activity was assayed as describe (Paratore et al., 2002).
Tamoxifen treatment
Mice were treated with TM (Sigma) by intraperitoneal injections (I.P.) of 9mg/40g body weight in corn oil, one every other day for a total of 3 injections. For oral delivery, TM was dissolved in a 0.5% Carboxymethyl cellulose, 0.4% Tween 80, 0.9% NaCl solution.
Caerulein treatment
Mice were I.P. injected with 0.1 ml of a 50μg/ml Caerulein (Sigma) in saline solution 5 times per week.
Pancreata size measurement
Pancreata areas were determined using Bioquant Image Analysis software in manual measurement mode.
Molecular analysis
For verification of Cre-mediated recombination, DNA was prepared from wild-type tails and LCM mPanINs of Caerulein &TM treated proCPA1CreERT2;LSL-KrasG12D and RipCreERTM;LSL-KrasG12D mice. PCR was performed with primers flanking the Lox–Stop–Lox cassette (sequences available in Table S8 and on http://web.mit.edu/jacks-lab/protocols_table.html). Wild-type K-ras, 2Lox and 1Lox -K-rasG12D alleles were detected, yielding 620-bp, 510-bp and ∼1100-bp products, respectively. LCM and DNA isolation were performed using the Veritas Microdissection System and the PicoPure™ DNA Extraction Kit from Molecular Devices, respectively.
Supplementary Material
Acknowledgments
We are grateful to D. Melton for gifts of the Pdx1CreERTM, proCPA1CreERT2 and RipCreERTM strains and stimulating discussions. We thank A. Berns (Netherlands Cancer Institute) for providing the Trp53flox mice; C. Wright for the anti-Pdx1 antibody; Sebastian Hoersch for statistical analysis and N. Bardeesy for critical reading of the manuscript. T.J. is a Howard Hughes Medical Institute Investigator and a Daniel K. Ludwig Scholar. S.Y.G.F. is an Anna Fuller fund of New Haven Fellow. R.A.D is an American Cancer Society Research Professor. This work was supported by Grant Number 1-PO1-CA117969-01 from the National Institutes of Health (NIH) and in part by Cancer Center Support (core) grant P30-CA14051 from the National Cancer Institute (NCI). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NCI or the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- Bardeesy N, Sharpless NE. RAS unplugged: negative feedback and oncogene-induced senescence. Cancer Cell. 2006;10:451–453. doi: 10.1016/j.ccr.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, Beachy PA. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–851. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- Carriere C, Seeley ES, Goetze T, Longnecker DS, Korc M. The Nestin progenitor lineage is the compartment of origin for pancreatic intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2007;104:4437–4442. doi: 10.1073/pnas.0701117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- De La O JP, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, Murtaugh LC. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2008;105:18907–18912. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- Gomez G, Englander EW, Wang G, Greeley GH., Jr Increased expression of hypoxia-inducible factor-1alpha, p48, and the Notch signaling cascade during acute pancreatitis in mice. Pancreas. 2004;28:58–64. doi: 10.1097/00006676-200401000-00009. [DOI] [PubMed] [Google Scholar]
- Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003;63:2016–2019. [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, Feldmann G, Stoffers DA, Konieczny SF, Leach SD, Maitra A. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008;105:18913–18918. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- Hoorens A, Prenzel K, Lemoine NR, Kloppel G. Undifferentiated carcinoma of the pancreas: analysis of intermediate filament profile and Ki-ras mutations provides evidence of a ductal origin. J Pathol. 1998;185:53–60. doi: 10.1002/(SICI)1096-9896(199805)185:1<53::AID-PATH45>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Hruban RH, Adsay NV, Albores-Saavedra J, Anver MR, Biankin AV, Boivin GP, Furth EE, Furukawa T, Klein A, Klimstra DS, et al. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res. 2006;66:95–106. doi: 10.1158/0008-5472.CAN-05-2168. [DOI] [PubMed] [Google Scholar]
- Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, Kern SE, Klimstra DS, Kloppel G, Longnecker DS, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–586. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- Inada A, Nienaber C, Katsuta H, Fujitani Y, Levine J, Morita R, Sharma A, Bonner-Weir S. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci U S A. 2008;105:19915–19919. doi: 10.1073/pnas.0805803105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, Metzger D. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 1999;27:4324–4327. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffee EM, Hruban RH, Canto M, Kern SE. Focus on pancreas cancer. Cancer Cell. 2002;2:25–28. doi: 10.1016/s1535-6108(02)00093-4. [DOI] [PubMed] [Google Scholar]
- Jensen JN, Cameron E, Garay MV, Starkey TW, Gianani R, Jensen J. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology. 2005;128:728–741. doi: 10.1053/j.gastro.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- Jonkers YM, Ramaekers FC, Speel EJ. Molecular alterations during insulinoma tumorigenesis. Biochim Biophys Acta. 2007;1775:313–332. doi: 10.1016/j.bbcan.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Koizumi M, Doi R, Toyoda E, Masui T, Tulachan SS, Kawaguchi Y, Fujimoto K, Gittes GK, Imamura M. Increased PDX-1 expression is associated with outcome in patients with pancreatic cancer. Surgery. 2003;134:260–266. doi: 10.1067/msy.2003.231. [DOI] [PubMed] [Google Scholar]
- Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–1030. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills SE. Histology for Pathologists. third. Philadelphia: Lippincott Williams & Wilkins; 2007. [Google Scholar]
- Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, Sriuranpong V, Iso T, Meszoely IM, Wolfe MS, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3:565–576. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- Paratore C, Eichenberger C, Suter U, Sommer L. Sox10 haploinsufficiency affects maintenance of progenitor cells in a mouse model of Hirschsprung disease. Hum Mol Genet. 2002;11:3075–3085. doi: 10.1093/hmg/11.24.3075. [DOI] [PubMed] [Google Scholar]
- Prasad NB, Biankin AV, Fukushima N, Maitra A, Dhara S, Elkahloun AG, Hruban RH, Goggins M, Leach SD. Gene expression profiles in pancreatic intraepithelial neoplasia reflect the effects of Hedgehog signaling on pancreatic ductal epithelial cells. Cancer Res. 2005;65:1619–1626. doi: 10.1158/0008-5472.CAN-04-1413. [DOI] [PubMed] [Google Scholar]
- Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M, Goodman SN, Sohn TA, Hruban RH, Yeo CJ, Kern SE. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997;57:1731–1734. [PubMed] [Google Scholar]
- Seeburg PH, Colby WW, Capon DJ, Goeddel DV, Levinson AD. Biological properties of human c-Ha-ras1 genes mutated at codon 12. Nature. 1984;312:71–75. doi: 10.1038/312071a0. [DOI] [PubMed] [Google Scholar]
- Shen CN, Slack JM, Tosh D. Molecular basis of transdifferentiation of pancreas to liver. Nat Cell Biol. 2000;2:879–887. doi: 10.1038/35046522. [DOI] [PubMed] [Google Scholar]
- Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: ‘it ain’t over ‘til it's over’. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- Siveke JT, Lubeseder-Martellato C, Lee M, Mazur PK, Nakhai H, Radtke F, Schmid RM. Notch signaling is required for exocrine regeneration after acute pancreatitis. Gastroenterology. 2008;134:544–555. doi: 10.1053/j.gastro.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Slack JM. Developmental biology of the pancreas. Development. 1995;121:1569–1580. doi: 10.1242/dev.121.6.1569. [DOI] [PubMed] [Google Scholar]
- Stanger BZ, Stiles B, Lauwers GY, Bardeesy N, Mendoza M, Wang Y, Greenwood A, Cheng KH, McLaughlin M, Brown D, et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8:185–195. doi: 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Heller RS, Miller CP, Habener JF. Developmental expression of the homeodomain protein IDX-1 in mice transgenic for an IDX-1 promoter/lacZ transcriptional reporter. Endocrinology. 1999;140:5374–5381. doi: 10.1210/endo.140.11.7122. [DOI] [PubMed] [Google Scholar]
- Strobel O, Dor Y, Stirman A, Trainor A, Fernandez-del Castillo C, Warshaw AL, Thayer SP. Beta cell transdifferentiation does not contribute to preneoplastic/metaplastic ductal lesions of the pancreas by genetic lineage tracing in vivo. Proc Natl Acad Sci U S A. 2007;104:4419–4424. doi: 10.1073/pnas.0605248104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift GH, Liu Y, Rose SD, Bischof LJ, Steelman S, Buchberg AM, Wright CV, MacDonald RJ. An endocrine-exocrine switch in the activity of the pancreatic homeodomain protein PDX1 through formation of a trimeric complex with PBX1b and MRG1 (MEIS2) Mol Cell Biol. 1998;18:5109–5120. doi: 10.1128/mcb.18.9.5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernandez-del Castillo C, Yajnik V, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson DA, Zhu L, Gopinathan A, Willis NA, Kachatrian L, Grochow R, Pin CL, Mitin NY, Taparowsky EJ, Gimotty PA, et al. Mist1-KrasG12D knock-in mice develop mixed differentiation metastatic exocrine pancreatic carcinoma and hepatocellular carcinoma. Cancer Res. 2006;66:242–247. doi: 10.1158/0008-5472.CAN-05-2305. [DOI] [PubMed] [Google Scholar]
- Wagner M, Luhrs H, Kloppel G, Adler G, Schmid RM. Malignant transformation of duct-like cells originating from acini in transforming growth factor transgenic mice. Gastroenterology. 1998;115:1254–1262. doi: 10.1016/s0016-5085(98)70098-8. [DOI] [PubMed] [Google Scholar]
- Wang L, Heidt DG, Lee CJ, Yang H, Logsdon CD, Zhang L, Fearon ER, Ljungman M, Simeone DM. Oncogenic function of ATDC in pancreatic cancer through Wnt pathway activation and beta-catenin stabilization. Cancer Cell. 2009;15:207–219. doi: 10.1016/j.ccr.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KL, Gannon M, Peshavaria M, Offield MF, Henderson E, Ray M, Marks A, Gamer LW, Wright CV, Stein R. Hepatocyte nuclear factor 3beta is involved in pancreatic beta-cell-specific transcription of the pdx-1 gene. Mol Cell Biol. 1997;17:6002–6013. doi: 10.1128/mcb.17.10.6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, D'Hoker J, Stange G, Bonne S, De Leu N, Xiao X, Van De Casteele M, Mellitzer G, Ling Z, Pipeleers D, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132:197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Hanahan D. Murine pancreatic ductal adenocarcinoma produced by in vitro transduction of polyoma middle T oncogene into the islets of Langerhans. Am J Pathol. 1994;145:671–684. [PMC free article] [PubMed] [Google Scholar]
- Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, Song E. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Law AC, Rajagopal J, Anderson WJ, Gray PA, Melton DA. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell. 2007;13:103–114. doi: 10.1016/j.devcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.