

Abstract
Oxygenic photosynthesis is the basis for aerobic life on earth. The catalytic Mn4OxCaYZ center of photosystem II (PSII), after fourfold oxidation, extracts four electrons from two water molecules to yield dioxygen. This reaction cascade has appeared as a single four-electron transfer that occurs in typically 1 ms. Inevitable redox intermediates have so far escaped detection, probably because of very short lifetime. Previous attempts to stabilize intermediates by high O2-back pressure have revealed controversial results. Here we monitored by membrane-inlet mass spectrometry (MIMS) the production of  from 18O-labeled water against a high background of
from 18O-labeled water against a high background of  in a suspension of PSII-core complexes. We found neither an inhibition nor an altered pattern of O2 production by up to 50-fold increased concentration of dissolved O2. Lack of inhibition is in line with results from previous X-ray absorption and visible-fluorescence experiments, but contradictory to the interpretation of previous UV-absorption data. Because we used essentially identical experimental conditions in MIMS as had been used in the UV work, the contradiction was serious, and we found it was not to be resolved by assuming a significant slowdown of the O2 release kinetics or a subsequent slow conformational relaxation. This calls for reevaluation of the less direct UV experiments. The direct detection of O2 release by MIMS shows unequivocally that O2 release in PSII is highly exothermic. Under the likely assumption that one H+ is released in the S4 → S0 transition, the driving force at pH 6.5 and atmospheric O2 pressure is at least 220 meV, otherwise 160 meV.
in a suspension of PSII-core complexes. We found neither an inhibition nor an altered pattern of O2 production by up to 50-fold increased concentration of dissolved O2. Lack of inhibition is in line with results from previous X-ray absorption and visible-fluorescence experiments, but contradictory to the interpretation of previous UV-absorption data. Because we used essentially identical experimental conditions in MIMS as had been used in the UV work, the contradiction was serious, and we found it was not to be resolved by assuming a significant slowdown of the O2 release kinetics or a subsequent slow conformational relaxation. This calls for reevaluation of the less direct UV experiments. The direct detection of O2 release by MIMS shows unequivocally that O2 release in PSII is highly exothermic. Under the likely assumption that one H+ is released in the S4 → S0 transition, the driving force at pH 6.5 and atmospheric O2 pressure is at least 220 meV, otherwise 160 meV.
Keywords: water oxidation, oxygen evolution, isotope-ratio mass spectrometry, bioenergetics
Oxygenic photosynthesis is the metabolic basis for the most successful forms of life on earth. In cyanobacteria, algae and plants photosystem II (PSII) uses sunlight to split water into dioxygen and reducing equivalents, and it generates proton motive force. The catalytic centre of O2 production in PSII, coined the OEC (oxygen evolving complex), comprises the manganese–oxygen–calcium (Mn4OxCa) complex and its ligands, which include two substrate “water” molecules of undefined protonation state. It also includes a redox-active tyrosine residue, coined tyrosine Z(YZ), which is the essential electron transfer link to the photoactive reaction center of PSII. Over 40 years ago Kok et al. (1) proposed—on the basis of flash-induced O2-evolution patterns (2)—that PSII, clocked and driven by four quanta of light, cycles through five different redox states, named Si (i = 0,…,4), where i is the number of stored oxidizing equivalents. Once four oxidizing equivalents are accumulated O2 is produced in the spontaneous reaction from S4 to yield S0.
| [1] |
In this Si state transition four electrons are transferred from two bound substrate water molecules that were partially or fully deprotonated (m = 0–2) during the Si state cycle. In addition, one or two new substrate water molecules (n = 1,2) bind to the Mn4OxCa cluster under the release of np protons into the medium (p = 0–2) (3–5). In general the release of two protons is observed for the S3 → S4 → S0 transition (6), of which one is expelled after  formation (7, 8), the highest oxidation state characterized so far by spectroscopy. It appears that reaction 1 occurs significantly faster than the formation of S4 from
formation (7, 8), the highest oxidation state characterized so far by spectroscopy. It appears that reaction 1 occurs significantly faster than the formation of S4 from  (see ref. 3 for a recent review).
(see ref. 3 for a recent review).
For decades it has appeared as if the four electrons were transferred in reaction 1 in one batch, because three different events proceed with the same half-rise time, typically 1–1.5 ms, namely (i) the appearance of O2 in solution, (ii) the reduction of  , and (iii) the reduction of the Mn4OxCa cluster in the S3 state. This coincidence has been well documented by a wealth of independent techniques. (i) The appearance of O2 in solution was time resolved by four different techniques, namely continuous flow (9), EPR (10, 11), polarography with a bare Pt electrode (2, 12–14) and via absorption changes of intracellular cytochrome c oxidase (15). (ii) The reduction of
, and (iii) the reduction of the Mn4OxCa cluster in the S3 state. This coincidence has been well documented by a wealth of independent techniques. (i) The appearance of O2 in solution was time resolved by four different techniques, namely continuous flow (9), EPR (10, 11), polarography with a bare Pt electrode (2, 12–14) and via absorption changes of intracellular cytochrome c oxidase (15). (ii) The reduction of  was detected by time-resolved EPR (10, 16, 17), and (iii) the reduction of the Mn4OxCa cluster by UV absorption (7, 12, 18–20), delayed chlorophyll fluorescence (DF) (21), and time-resolved Mn-K-edge spectroscopy (8, 22). Based on general considerations, however, a direct four-electron transfer from water to the
was detected by time-resolved EPR (10, 16, 17), and (iii) the reduction of the Mn4OxCa cluster by UV absorption (7, 12, 18–20), delayed chlorophyll fluorescence (DF) (21), and time-resolved Mn-K-edge spectroscopy (8, 22). Based on general considerations, however, a direct four-electron transfer from water to the  moiety is improbable, redox intermediates inevitably exist, and it is pivotal for understanding the mechanism of water oxidation to characterize their chemical nature.
moiety is improbable, redox intermediates inevitably exist, and it is pivotal for understanding the mechanism of water oxidation to characterize their chemical nature.
In an attempt to stabilize putative redox intermediates ( states) of the reaction
states) of the reaction  two of us (23) applied elevated O2 back pressure and found that the characteristic millisecond component of UV-absorption transients [that has been attributed to Mn oxidoreduction (18)] was half suppressed if the O2 pressure was raised only 10-fold above the atmospheric level (2.3 bars versus 0.21 bar). DF studies corroborated these effects of moderately elevated O2 pressure on the OEC (21). Experiments by time-resolved K-edge X-ray absorption spectroscopy (TR-XAS), which directly monitored the Mn-oxidoreduction, indicated a changed dark-equilibrium between Si states or changes in the flash-induced turnover efficiencies of PSII, but neither a blockade nor a significant slowing of the S3 → S0 transition in response to elevated O2 levels (22). All the above cited studies were carried out with isolated PSII-core complexes (PSII-cc) of Synechocystis sp. PCC 6803 (Syn. sp.) or spinach PSII membrane fragments (PSII-mf).
two of us (23) applied elevated O2 back pressure and found that the characteristic millisecond component of UV-absorption transients [that has been attributed to Mn oxidoreduction (18)] was half suppressed if the O2 pressure was raised only 10-fold above the atmospheric level (2.3 bars versus 0.21 bar). DF studies corroborated these effects of moderately elevated O2 pressure on the OEC (21). Experiments by time-resolved K-edge X-ray absorption spectroscopy (TR-XAS), which directly monitored the Mn-oxidoreduction, indicated a changed dark-equilibrium between Si states or changes in the flash-induced turnover efficiencies of PSII, but neither a blockade nor a significant slowing of the S3 → S0 transition in response to elevated O2 levels (22). All the above cited studies were carried out with isolated PSII-core complexes (PSII-cc) of Synechocystis sp. PCC 6803 (Syn. sp.) or spinach PSII membrane fragments (PSII-mf).
If the inhibition by elevated O2 pressure holds true for whole cells or intact leafs, photosynthesis could not raise the atmospheric O2 content of the atmosphere much above its present level (23). The prospect of such a rather strict boundary condition for past and future evolution of life on earth has provoked vivid discussion (24–28), and a recent study on the operation of the OEC in various plants, algae, and cyanobacteria has shed doubt on such limitation (29). The matter has remained controversial because in all the above cited studies on the effects of elevated O2 pressure, the release of O2 was not detected directly, and different sample materials (species and type of preparation) were used. This has prompted us to monitor O2 production from H218O by membrane-inlet mass spectrometry (MIMS) as a function of the  pressure (na, natural abundance, hereafter simply denoted as O2). Scrutinizing the specific results and conclusions of previous studies (21–23, 30), the experiments were carried out under basically identical experimental boundary conditions (pH, temperature, and buffer composition), and by using samples from the same stock of frozen Syn. sp. PSII-cc as employed in ref. 23.
pressure (na, natural abundance, hereafter simply denoted as O2). Scrutinizing the specific results and conclusions of previous studies (21–23, 30), the experiments were carried out under basically identical experimental boundary conditions (pH, temperature, and buffer composition), and by using samples from the same stock of frozen Syn. sp. PSII-cc as employed in ref. 23.
Results
Enrichment of PSII Suspension with O2.
The equilibration of PSII suspensions in a buffer containing 100 mM sucrose, 25 mM CaCl2, 10 mM NaCl, 1 M glycine betaine, 50 mM MES (pH 6.7 or 5.5) and 30–50% H218O with O2 or N2 at the applied pressure in the gas phase of a specially developed reaction cell (Fig. 1) was monitored by MIMS. Fig. 2 shows the time course of the enrichment of the PSII suspension with O2 at 20 bars pressure. After about 35 min exposure to 20 bars, the  concentration increased 50–60 times over the ambient initial
concentration increased 50–60 times over the ambient initial 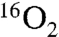 level (∼0.21 bar). Assuming a linear response to the dissolved
level (∼0.21 bar). Assuming a linear response to the dissolved  of the MIMS output signal and the validity of Henry’s law the saturation level corresponds to at least 10 bars, i.e., to about 50% of the expected level. This is a lower estimate because calibration measurements (see Materials and Methods and Fig. S1) show that the sensitivity of our MIMS setup is reduced by 20–40% at 20 bars. Although it is difficult to determine the exact concentration of dissolved O2 at high pressure, it is important to emphasize that even this lowest estimate (equivalent to 10 bars) is high enough to probe the previously reported effects, which suggest that half-inhibition of O2 evolution by PSII occurs at 2.3 bars O2. In the following, we therefore simply report the admitted pressure in the gas phase, and not the achieved partial pressure in solution.
of the MIMS output signal and the validity of Henry’s law the saturation level corresponds to at least 10 bars, i.e., to about 50% of the expected level. This is a lower estimate because calibration measurements (see Materials and Methods and Fig. S1) show that the sensitivity of our MIMS setup is reduced by 20–40% at 20 bars. Although it is difficult to determine the exact concentration of dissolved O2 at high pressure, it is important to emphasize that even this lowest estimate (equivalent to 10 bars) is high enough to probe the previously reported effects, which suggest that half-inhibition of O2 evolution by PSII occurs at 2.3 bars O2. In the following, we therefore simply report the admitted pressure in the gas phase, and not the achieved partial pressure in solution.
Fig. 1.

Pressure cell for the MIMS measurements of flash-induced O2-evolving activity of PSII samples at O2/N2 pressures up to 20 bars. For details, see Materials and Methods.
Fig. 2.

Rise of dissolved  (m/z = 32) in the suspension of PSII-cc from Syn. sp. upon application of 20 bars O2 pressure (closed arrow) monitored by MIMS. (Inset) The zoomed region of
(m/z = 32) in the suspension of PSII-cc from Syn. sp. upon application of 20 bars O2 pressure (closed arrow) monitored by MIMS. (Inset) The zoomed region of  evolution (m/z = 36) induced by 30 Xe flashes (2 Hz; open arrow). Values in brackets indicate MS Faraday cup amplification. Other conditions: ∼50% H218O, [Chl] = 5 μM Chl, 250 μM DCBQ, pH 6.7, 20 °C.
evolution (m/z = 36) induced by 30 Xe flashes (2 Hz; open arrow). Values in brackets indicate MS Faraday cup amplification. Other conditions: ∼50% H218O, [Chl] = 5 μM Chl, 250 μM DCBQ, pH 6.7, 20 °C.
Fig. 2 also shows that although the signal for  slowly increased, the level of the m/z = 36 signal remained almost stable at a very low level. The initial amplitude of the 36 signal is dominated by the 36Ar isotope (0.337% na) in air. The concentration of Ar declines with time by degassation into the mass spectrometer, but this is almost compensated by the increasing level of
slowly increased, the level of the m/z = 36 signal remained almost stable at a very low level. The initial amplitude of the 36 signal is dominated by the 36Ar isotope (0.337% na) in air. The concentration of Ar declines with time by degassation into the mass spectrometer, but this is almost compensated by the increasing level of  , which occurs with low natural abundance (∼0.0004%). The rise of the O2 concentration at m/z = 34 was always simultaneously recorded and gave consistent data (Fig. S2, Inset). To separate pressure-induced effects on the MIMS apparatus or on the OEC from O2-specific effects, control experiments were performed under
, which occurs with low natural abundance (∼0.0004%). The rise of the O2 concentration at m/z = 34 was always simultaneously recorded and gave consistent data (Fig. S2, Inset). To separate pressure-induced effects on the MIMS apparatus or on the OEC from O2-specific effects, control experiments were performed under  pressure (hereafter N2). The enrichment of the PSII suspension with
pressure (hereafter N2). The enrichment of the PSII suspension with  was also monitored by MIMS (Fig. S2).
was also monitored by MIMS (Fig. S2).
Photosynthetic O2 Evolution at Atmospheric and Elevated O2/N2 Pressures.
The experiments reported below were performed after the saturation of the gas solubility was reached (monitored as in Fig. 2), i.e., after 35–50 min of stirring in the dark. O2-evolution activity of PSII was then probed by illumination with Xe flashes: At m/z = 32 the MIMS signal was indistinguishable from noise, at m/z = 34 excitation with 30 flashes produced a discernable signal (see Fig. S2, Inset), and at m/z = 36 a large signal well above the low background was observed due to the high enrichment of the suspension with H218O (see Fig. 2, Inset).
Fig. 3 shows the  -evolving activity of dark-adapted PSII-core particles in response to a series of 200 Xe flashes (2 Hz) for three different conditions in the gas phase, namely under air (1 bar, 0.21 bar O2), O2 (21.7 bars), and N2 (20 bars). Under all three conditions light-induced
-evolving activity of dark-adapted PSII-core particles in response to a series of 200 Xe flashes (2 Hz) for three different conditions in the gas phase, namely under air (1 bar, 0.21 bar O2), O2 (21.7 bars), and N2 (20 bars). Under all three conditions light-induced  evolution was observed, but the extent of the m/z = 36 signals at high pressure was by 20–25% smaller than at atmospheric pressure. Because this holds true for both O2 and N2, this was attributed to an effect of pressure on the sensitivity of the MIMS apparatus rather than to a chemical effect of O2 on the OEC. This assignment is in full agreement with the observed lower sensitivity of our MIMS setup toward
evolution was observed, but the extent of the m/z = 36 signals at high pressure was by 20–25% smaller than at atmospheric pressure. Because this holds true for both O2 and N2, this was attributed to an effect of pressure on the sensitivity of the MIMS apparatus rather than to a chemical effect of O2 on the OEC. This assignment is in full agreement with the observed lower sensitivity of our MIMS setup toward  at high applied pressures (see Materials and Methods and Fig. S1). The relative magnitudes of the
at high applied pressures (see Materials and Methods and Fig. S1). The relative magnitudes of the  release as documented in Fig. 3, i.e., 100% (air), 79% (O2, 20 bars), and 75% (N2, 20 bars), were reproducible with a standard deviation of 5% (n = 4). As a control in all cases m/z = 40 (Ar) traces were recorded, and one example is shown in Fig. 3. This documents the absence of changes in the gas permeability of the MIMS inlet during the measurements.
release as documented in Fig. 3, i.e., 100% (air), 79% (O2, 20 bars), and 75% (N2, 20 bars), were reproducible with a standard deviation of 5% (n = 4). As a control in all cases m/z = 40 (Ar) traces were recorded, and one example is shown in Fig. 3. This documents the absence of changes in the gas permeability of the MIMS inlet during the measurements.
Fig. 3.

 evolution of PSII-cc from Syn. sp. induced by a series of 200 saturating Xe flashes (2 Hz; open arrow) at 1 bar air, 21.7 bars O2, or 20 bars N2. The time dependence of the Ar level (m/z = 40) is presented by a gray line. Other conditions: see Fig. 2, but ∼30% H218O and [Chl] = 50 μM.
evolution of PSII-cc from Syn. sp. induced by a series of 200 saturating Xe flashes (2 Hz; open arrow) at 1 bar air, 21.7 bars O2, or 20 bars N2. The time dependence of the Ar level (m/z = 40) is presented by a gray line. Other conditions: see Fig. 2, but ∼30% H218O and [Chl] = 50 μM.
It is therefore obvious that, in contrast to expectations based on the previous UV measurements, there is no major specific effect of O2 pressure (up to 20 bars) versus N2 pressure on the extent of photosynthetic O2 evolution under repetitive excitation at 2-Hz frequency.
The Influence of Flash Frequency and pH.
One difference between the above experiments (Figs. 2 and 3) and the original UV-absorption data (23) is the frequency of flash excitation (2 Hz in this work versus 10 Hz in the former). This raised the possibility that an O2-induced kinetic limitation may have caused the previously observed inhibition. In order to address this question, experiments were performed using 20 flashes at 50-Hz frequency that were followed by 30 flashes at 2 Hz. The normalization of the former to the latter MIMS signals eliminated the unspecific pressure effects (see above). The normalized signal levels of 50-Hz traces obtained at 20 bars O2 and 17 bars N2 (Fig. 4A) were 40.2 (0.5) and 40.8 (1.0), respectively, where the numbers in parentheses give the standard deviation (n = 2). It was noteworthy that also under these conditions no O2-specific inhibition was observed, which exceeded the kinetic limitations of the acceptor side electron transfer to 2,5-dichloro-p-benzoquinone (DCBQ) that caused the above signal levels to be below the theoretical value of about 67% (20/30).
Fig. 4.

Flash-induced  evolution of PSII-cc from Syn. sp. measured by MIMS at 20 bars O2, or 17 bars N2 and pH 6.7 (A) or pH 5.5 (B).
evolution of PSII-cc from Syn. sp. measured by MIMS at 20 bars O2, or 17 bars N2 and pH 6.7 (A) or pH 5.5 (B).  evolution was induced by a series of 20 Xe flashes (arrows) given at 50 Hz. Other conditions: see Fig. 2. All data normalized to a consecutive series of 30 flashes at 2 Hz.
evolution was induced by a series of 20 Xe flashes (arrows) given at 50 Hz. Other conditions: see Fig. 2. All data normalized to a consecutive series of 30 flashes at 2 Hz.
In previous UV experiments it was observed that lowering the pH from 6.7 to 5.5 mimics the effect of elevated O2 pressure on the OEC (30). This is plausible because a pH jump by one unit can exert a similar back pressure on the S4 → S0 transition as a tenfold increased O2 pressure, assuming one proton is released during the S4 → S0 step (reaction 1). In order to maximize the back pressure, we explored by MIMS the combined effect of O2 pressure and reduced pH. Again, no O2-specific inhibition was observed at 50 Hz (Fig. 4B) and at 2 Hz (Fig. S3) that exceeded the reduction of ∼35% caused by the lower pH alone.
Flash-Induced Oxygen-Evolution Patterns (FIOPs).
If PSII is dark-adapted under ambient air pressure, the OEC is predominantly in the S1 state, such that the release of O2 occurs after absorption of the third flash in a row, but negligibly after the second. Based on time-resolved X-ray absorption measurements, Haumann et al. proposed that exposure to elevated O2 pressure (11–13 bars) causes either a shift of the Si state distribution in the dark (30–40% from S1 toward S2) or an increase of photophysical double hits from normally 0–3% to about 15% (22). The double hit factor gives the percentage of centers that have been excited twice in a single flash (for details see ref. 3). Both effects imply that a considerable amount of O2 would be produced already by the second flash of light.
Because a drastic change in the dark Si state distribution or of the miss or double hit parameters could in principle also explain the previous UV observations, we scrutinized this possibility by MIMS. We first tried obtaining a flash pattern of PSII-cc. This is complicated, because due to the slow diffusion of O2 through the MIMS membrane, dark times between the flashes of 25 s have to be employed in order to resolve the individual flash yields. Although the obtained flash pattern under 1 bar air, 20 bars O2, and 17 bars N2 all looked identical, they were highly damped, most likely due to fast back reactions during the long dark periods (see Fig. S4). We therefore repeated these experiments with PSII-mf. Fig. 5A shows that the normalized FIOPs at 20 bars N2 and 20 bars O2 are practically identical. We further tested if the absence of an increased second flash O2 yield holds true also for PSII-cc by measuring the sum of the O2 yields obtained after excitation of dark-adapted samples with either two or three flashes of light given at 10 Hz (Fig. 5B). There was negligible  production in the former and considerable in the latter case. This holds true both for equilibration with 1 bar air (0.21 bar O2) and with 20 bars O2. In other words there was no evidence, by MIMS experiments, for the postulated Si state shift (22) by elevated
production in the former and considerable in the latter case. This holds true both for equilibration with 1 bar air (0.21 bar O2) and with 20 bars O2. In other words there was no evidence, by MIMS experiments, for the postulated Si state shift (22) by elevated  pressure of the flash pattern of
pressure of the flash pattern of  release. In addition, no evidence for effects of elevated O2 pressure on the miss and double hit parameters was found.
release. In addition, no evidence for effects of elevated O2 pressure on the miss and double hit parameters was found.
Fig. 5.

 evolution measured by MIMS at elevated (and normal) O2-(N2-)back pressure at m/z = 36 (A) FIOPs measured with PSII-mf from spinach at 20.4 bars O2 or 20.2 bars N2.
evolution measured by MIMS at elevated (and normal) O2-(N2-)back pressure at m/z = 36 (A) FIOPs measured with PSII-mf from spinach at 20.4 bars O2 or 20.2 bars N2.  evolution was induced by a series of saturating laser flashes separated by dark times of 25 s. Other conditions: ∼40% H218O; [Chl] = 50 μM; 1 mM K3[Fe(CN)6]; pH 6.5; 20 °C. (B)
evolution was induced by a series of saturating laser flashes separated by dark times of 25 s. Other conditions: ∼40% H218O; [Chl] = 50 μM; 1 mM K3[Fe(CN)6]; pH 6.5; 20 °C. (B)  evolution induced by two (2f) or three (3f) Xe flashes (10 Hz) measured by MIMS in PSII-cc from Syn. sp. at 0.2 bar or at 20.3 bars O2. Other conditions: see Fig. 2, but ∼40% H218O and [Chl] = 50 μM. Data in both panels normalized as in Fig. 4.
evolution induced by two (2f) or three (3f) Xe flashes (10 Hz) measured by MIMS in PSII-cc from Syn. sp. at 0.2 bar or at 20.3 bars O2. Other conditions: see Fig. 2, but ∼40% H218O and [Chl] = 50 μM. Data in both panels normalized as in Fig. 4.
Discussion
In the MIMS experiments of this work the transient O2 evolution and the concentration of dissolved O2 in the PSII suspensions were directly detected and studied as a function of the O2 pressure in the adjacent gas phase. We found both the extent of the  -MIMS signals and its distribution over sequential flashes unimpaired by an at least 50-times increased O2 concentration in the suspension. Below we discuss these findings in relation to the conflicting results of previous studies on this topic. Such conflicts may have three reasons: (i) mismatch between experimental boundary conditions, (ii) misconception in the interpretation of either data, or (iii) necessity to revise the present understanding of the addressed mechanism.
-MIMS signals and its distribution over sequential flashes unimpaired by an at least 50-times increased O2 concentration in the suspension. Below we discuss these findings in relation to the conflicting results of previous studies on this topic. Such conflicts may have three reasons: (i) mismatch between experimental boundary conditions, (ii) misconception in the interpretation of either data, or (iii) necessity to revise the present understanding of the addressed mechanism.
(i). Equivalence of the Experimental Boundary Conditions?
The chemical boundary conditions in MIMS experiments were essentially the same as in the previous UV-spectroscopic work (23) (for a detailed comparison, see SI Text and Figs. S5–S7). Still we found (by MIMS) that O2 production by PSII was not suppressed by elevated O2 pressure as postulated in refs. 21 and 23, not even at acidic pH as in ref. 30. The MIMS results are thus in sharp contrast to the conclusions based on previous UV and DF studies (21, 23, 30), being in line with recent fluorescence (29) and TR-XAS experiments (22). The latter experiments showed at slightly different conditions (sample type, concentration, buffers, and acceptors; see Table S1) that the Si state transitions are not blocked and that the Mn reduction during the S4-S0 transition occurs with unchanged rate and amplitude. Our present MIMS data show in addition that neither the formation nor the unbinding of O2 from the Mn4OxCa cluster are impaired by up to 50-fold elevated O2 pressure.
However, there are also some minor discrepancies among the latter results. Whereas Haumann et al. (22), in their TR-XAS experiments, gave evidence for either an increased S2 state dark population or an increased double hit parameter under 11–13 bars O2 pressure, Kolling et al. (29), who monitored the pattern of variable fluorescence as a function of the flash number, found no changes in the oscillation patterns in thylakoids, under O2 pressures up to 20 bars. Whereas the latter was coincident with our results, the former was not. Our finding of an unperturbed pattern of O2 production (maxima upon flashes 3, 7, 11,…) together with the data by Kolling et al. (29) dismiss the notion that the UV-absorption study might have been compromised by an influence of elevated O2 pressure on the double hit factor or the miss parameter or other changes to the oscillation pattern. They rather support the possibility already considered by Haumann et al. (22) that the changed oscillation pattern in their work might be caused by reactive oxygen species such as singlet O2 formed by PSII during the illumination with strong laser flashes and X-rays at elevated O2 pressures.
(ii). Misconception in the Interpretation of the UV or the MIMS Data?
The flash-light-induced UV-difference spectra of PSII are composites of events at the donor and the acceptor sides of PSII. The relative spectral contributions of YZ and of Mn during the stepwise oxidation of Mn (plus ligand shell) during the transitions from state S0 via S1 and S2 to S3 have been characterized by several authors, and so was the spontaneous reduction of  and Mn, after S3 being oxidized to yield S4, and the decay in about 1 ms into state S0 (see ref. 18 and references therein). It has also been established that the wavelength of 360 nm is particularly well suited to monitor Mn transitions because the extent of the reduced-oxidized difference of YZ is very small at this wavelength (18, 31). The parallel time course of O2-release and UV-absorption transients (360 nm) with a half-time of about 1 ms in WT-PSII also holds in a mutant (D1-D61N) where the half-time is tenfold longer (13 ms) (32). There has been no obvious reason from the literature to doubt (i) the attribution of the UV-millisecond phase to the reduction of Mn and (ii) its parallel time course with O2 release.
and Mn, after S3 being oxidized to yield S4, and the decay in about 1 ms into state S0 (see ref. 18 and references therein). It has also been established that the wavelength of 360 nm is particularly well suited to monitor Mn transitions because the extent of the reduced-oxidized difference of YZ is very small at this wavelength (18, 31). The parallel time course of O2-release and UV-absorption transients (360 nm) with a half-time of about 1 ms in WT-PSII also holds in a mutant (D1-D61N) where the half-time is tenfold longer (13 ms) (32). There has been no obvious reason from the literature to doubt (i) the attribution of the UV-millisecond phase to the reduction of Mn and (ii) its parallel time course with O2 release.
In contrast to all the other techniques, MIMS detects the production of  from H218O directly, the only caveat being that the MIMS signal in response to an O2 pulse rises rather slowly (instrument-limited half-rise ≈10 s), so that a possible slowing of O2 release may be unnoticed.
from H218O directly, the only caveat being that the MIMS signal in response to an O2 pulse rises rather slowly (instrument-limited half-rise ≈10 s), so that a possible slowing of O2 release may be unnoticed.
(iii). Revision of the Present Understanding?
In line with the above caveat we attempted to reconcile the MIMS results with the previous UV results (23, 30) by assuming that not the Mn reduction and O2 production, but the effective O2 release time was slowed down by elevated O2 pressure and that the driving force for O2 release comes partially from a conformational change between an initially formed  state to the more stable S0 state, a scenario that has been previously proposed by Vos et al. (33). Take the following simplified reaction scheme of the terminal cascade, wherein redox intermediates of the OEC (between states S4 and S0), the protonation state of bound water, proton release, and rebinding of substrate water (see reaction 1) are neglected for simplicity.
state to the more stable S0 state, a scenario that has been previously proposed by Vos et al. (33). Take the following simplified reaction scheme of the terminal cascade, wherein redox intermediates of the OEC (between states S4 and S0), the protonation state of bound water, proton release, and rebinding of substrate water (see reaction 1) are neglected for simplicity.
 |
[2] |
Herein  denotes a conformationally strained but fully reduced state of the OEC with oxygen already released into the medium, and S0 its fully relaxed equivalent. All reactions but the one back from
denotes a conformationally strained but fully reduced state of the OEC with oxygen already released into the medium, and S0 its fully relaxed equivalent. All reactions but the one back from  to S4 are of first order. Because of the abundance of O2, the latter can be considered as pseudofirst order, such that k-1—its magnitude being proportional to the O2 concentration—comes in s-1, like the other three rate constants. Let us consider the following arbitrary choice of parameters ki/s-1 for the situation at ambient O2 concentration (0.2 bar):
to S4 are of first order. Because of the abundance of O2, the latter can be considered as pseudofirst order, such that k-1—its magnitude being proportional to the O2 concentration—comes in s-1, like the other three rate constants. Let us consider the following arbitrary choice of parameters ki/s-1 for the situation at ambient O2 concentration (0.2 bar):![]()
Then the appearance of O2 in solution is kinetically limited by the first reaction. Broadly speaking, its exponential rise time is 1 ms and to a lesser extent 10 ms.  is stabilized by 60 meV compared to S4, and S0 is relative to
is stabilized by 60 meV compared to S4, and S0 is relative to  by 120 meV (34). The total stabilization of fully relaxed S0 is larger, namely 180 meV. Let us now increase the O2 pressure 100-fold from 0.2 to 20 bars. Only one parameter depends on the O2 concentration, namely k-1. Its value increases from 102 to 104:
by 120 meV (34). The total stabilization of fully relaxed S0 is larger, namely 180 meV. Let us now increase the O2 pressure 100-fold from 0.2 to 20 bars. Only one parameter depends on the O2 concentration, namely k-1. Its value increases from 102 to 104:![]()
The increase changes the situation dramatically, the rate of net O2 production is now governed by the second reaction, and it is furthermore delayed by the adverse equilibrium of the first reaction. The rise time of the pulse of O2 is now about 100 ms instead of 1 ms. In other words, the “millisecond phase” of S4 reduction is seemingly suppressed (truly slowed down). If the reaction takes place in H218O and with  in the gas and liquid phase, then first
in the gas and liquid phase, then first  appears with the rate k1 (i.e., in 1 ms) in the solution concomitant with
appears with the rate k1 (i.e., in 1 ms) in the solution concomitant with  uptake. The appearance of
uptake. The appearance of  is detected by MIMS, whereas the transient intake of
is detected by MIMS, whereas the transient intake of  (if present) is not detectable because of the high background signal at m/z = 32 under 20 bars of
(if present) is not detectable because of the high background signal at m/z = 32 under 20 bars of  . At 100-fold increased O2 pressure
. At 100-fold increased O2 pressure  is destabilized by 60 meV relative to S4. Because S0 is still by 120 meV more stable than
is destabilized by 60 meV relative to S4. Because S0 is still by 120 meV more stable than  —this is independent of the O2 pressure—the total driving force for proceeding from S4 to S0 is still 60 meV. This is sufficient to expel O2 into the solution. Rather than to be provided by the very O2-liberating step (
—this is independent of the O2 pressure—the total driving force for proceeding from S4 to S0 is still 60 meV. This is sufficient to expel O2 into the solution. Rather than to be provided by the very O2-liberating step (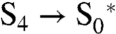 ), the forward drive is then provided by the conformational relaxation in the OEC (the transition from
), the forward drive is then provided by the conformational relaxation in the OEC (the transition from  to S0). The published UV transients (23) (see also Fig. S8) have revealed that the hypothetical
to S0). The published UV transients (23) (see also Fig. S8) have revealed that the hypothetical  transition would have to last 100 ms or longer.
transition would have to last 100 ms or longer.
The above kinetic model predicts that elevated O2 pressure can suppress the ms phase of Mn reduction (as detected by UV), whereas the appearance of O2 in solution, detected by MIMS, is seemingly unimpaired. This concept for reconciling the conflicting data on kinetic grounds was tested by the experiments documented in Fig. 4A where a series of 20 light flashes was fired to a dark-adapted sample. The spacing of flashes, 20 ms, was chosen tight enough as to reveal a possible kinetic limitation of the  transition. The extent of
transition. The extent of  release was almost independent of exposing the PSII suspension to either 20 bars O2 or 17 bars N2. This result rules out the above discussed kinetic limitation, in agreement with the TR-XAS finding that the S3 → S4 → S0 kinetic is basically unaffected by elevated O2 pressure (22).
release was almost independent of exposing the PSII suspension to either 20 bars O2 or 17 bars N2. This result rules out the above discussed kinetic limitation, in agreement with the TR-XAS finding that the S3 → S4 → S0 kinetic is basically unaffected by elevated O2 pressure (22).
Consequences for the Energetics of Water Oxidation.
MIMS experiments revealed unconstrained O2 production up to at least 10 bars, i.e., 50-fold elevated partial O2 pressure, without any indication for the transient retention of O2 in PSII. O2 polarography with a bare platinum electrode and other techniques have revealed a half-rise time of 1 ms (see Introduction), and rapid exchange between freshly produced O2 and O2 in the medium is probable. As a consequence, the dilution of O2 into the suspending medium adds an entropic term to the free energy of the product side. If the driving force of the O2-liberating step was −100 meV under ambient conditions (0.21 bars O2, pH 6.5), then the 50-times increased O2 pressure was expected to raise the backward driving force entropically by 100 meV, such that one expects half of the previous O2 release into the medium. If the driving force was greater than before, say 160 meV, still 90% of the extent found in air is to be expected. The MIMS data at pH 6.5 and with at least 10 bars dissolved O2 therefore imply that the terminal step of water oxidation has a driving force greater than 160 meV. Under the likely assumption that the S4 → S0 transition is coupled to the release of at least one proton (see description of reaction 1), the MIMS data under 20 bars O2 in the gas phase and at pH 5.5 (see Fig. 4B) raise the lower limit for the driving force even to 220 meV. How does this estimate compare with the current ones for the free energies in PSII? The midpoint potential of pheophytin a (Pheo a) in Thermosynechococcus elongatus has been recently determined to be  (35), i.e., 100 mV higher than previous estimates (summarized in ref. 36). Based thereupon the midpoint potentials of P680 and YZ became also more positive, Em(P680/P680•+) = +1.210 V and
(35), i.e., 100 mV higher than previous estimates (summarized in ref. 36). Based thereupon the midpoint potentials of P680 and YZ became also more positive, Em(P680/P680•+) = +1.210 V and  (35–37). Given that the mean midpoint potential for the oxidation of water under ambient conditions is +0.8 V, it appears that there is indeed leeway for such a high driving force for the terminal reaction cascade.
(35–37). Given that the mean midpoint potential for the oxidation of water under ambient conditions is +0.8 V, it appears that there is indeed leeway for such a high driving force for the terminal reaction cascade.
Conclusion
By MIMS we monitored oxygen ( ) production from water (H218O) under variation of the oxygen (
) production from water (H218O) under variation of the oxygen ( ) pressure around PSII. There was no indication for any inhibition by up to 50-fold increased back pressure of
) pressure around PSII. There was no indication for any inhibition by up to 50-fold increased back pressure of  . This result qualifies the previous conclusion to the contrary that has been based on UV-absorption transients (23, 30) and delayed fluorescence (21), and it corroborates results by X-ray absorption transients (22) and visible fluorescence (29). Despite the well-established relation between the millisecond phase of UV-absorption transients (at 360 nm) and the reduction of manganese (see Introduction), the MIMS versus UV discrepancy calls for a reevaluation of the difference spectrum of UV-absorption transients as a function of flash number, and in response to low and high oxygen pressure.
. This result qualifies the previous conclusion to the contrary that has been based on UV-absorption transients (23, 30) and delayed fluorescence (21), and it corroborates results by X-ray absorption transients (22) and visible fluorescence (29). Despite the well-established relation between the millisecond phase of UV-absorption transients (at 360 nm) and the reduction of manganese (see Introduction), the MIMS versus UV discrepancy calls for a reevaluation of the difference spectrum of UV-absorption transients as a function of flash number, and in response to low and high oxygen pressure.
It is an important result of the present work that the driving force of the terminal reaction cascade of photosynthetic water oxidation is rather strong, namely at least 160 meV, but taking into account a probable proton release during the S4 → S0 transition, it is even > 220 meV (at pH 6.5 and atmospheric conditions). It is obvious that the construction of PSII, by itself, does not impose a narrow constraint to the oxygen content of the atmosphere that can be raised, in principle, by orders of magnitude.
Materials and Methods
Samples and Media.
O2-evolving PSII-cc were isolated from modified WT cells of Synechocystis sp. PCC6803 as described earlier (38). Typical rates of O2 evolution under continuous illumination were 2,000–3,000 μmol (O2) × mg(Chl)-1 × h-1 (38) (see also SI Text). Prior to MIMS measurements PSII-cc were thawed in the dark on ice and diluted to the desired chlorophyll concentrations (5 or 50 μM) with standard medium (100 mM sucrose, 25 mM CaCl2, 10 mM NaCl, 1 M glycine betaine, and 50 mM MES at pH 6.7 or 5.5) containing H216O/H218O mixtures as described below. In some experiments PSII-mf were employed that were prepared from spinach as described previously (39). For the measurements, they were suspended in the following buffer: 150 mM sucrose, 35 mM NaCl, and 40 mM MES at pH 6.5. The rates of O2 evolution for such samples were 400–500 μmol (O2) × mg(Chl)-1 × h-1.
MIMS Setup for Experiments at Elevated O2/N2 Pressure.
To perform MIMS measurements (40) of O2 produced under elevated O2 pressure, a MIMS reaction cell resisting pressures up to 20 bars was developed (Fig. 1) and connected to the vacuum line of our magnetic sector field isotope-ratio MS (ThermoFinniganPlus XP) via a cooling trap (dry ice/C2H5OH or liquid N2). The nominal sample space of the chamber was 1 mL, but a volume of 600 μL was used to increase the interface between gas phase and liquid. Gas diffusion into the liquid was further increased by rapid stirring of the suspension. The suspension was separated from the high vacuum (3 × 10-8 mbar) of the MS by a gas-permeable ∼150-μm-thick metallic mesh silicon membrane (Franatech GmbH) resting on a porous Teflon support (∅ 3 mm) (Small Parts Inc.). For monitoring the pressure in a gas phase above the samples, the cover of the reaction chamber was connected to a pressure detector (Huba Control). The MIMS cell was thermostated to 20 °C.
The response of our MIMS setup is linear from ambient pressure up to about 8 bars O2 (about 99.5%  ), but then deviates slightly resulting in a dissolved O2 signal of about 60–80% of what would be expected based on lower pressures (Fig. S1). Possible reasons for this deviation, for example, are (i) pressure effects on the membrane permeability and (ii) a decrease in sensitivity of the mass spectrometer caused by a reduced internal vacuum at high applied pressures. Calibration of the O2 signals at ambient pressure results in a value of 100 mV ≈ 30 nM
), but then deviates slightly resulting in a dissolved O2 signal of about 60–80% of what would be expected based on lower pressures (Fig. S1). Possible reasons for this deviation, for example, are (i) pressure effects on the membrane permeability and (ii) a decrease in sensitivity of the mass spectrometer caused by a reduced internal vacuum at high applied pressures. Calibration of the O2 signals at ambient pressure results in a value of 100 mV ≈ 30 nM  (m/z = 36 peak) (for details, see SI Text).
(m/z = 36 peak) (for details, see SI Text).
Protocol for the detection of O2 evolution Under Elevated Pressure.
H218O (97%; Larodan Fine Chemicals AB) was used to enrich the aqueous sample suspensions with 30–50% of H218O. For this, 18O-labeled H2O was added to the reaction mixture that contained either dark-adapted PSII-cc and 2,5-DCBQ as electron acceptor (250-μM final concentration; added from 20 mM stock solution in ethanol) or PSII-mf and ferricyanide (1 mM). The MIMS chamber was then flushed with the desired gas, sealed, and exposed to elevated O2/N2 pressure. The following rise of the dissolved  or
or  concentrations in the PSII suspension was monitored at m/z = 32 and m/z = 28, respectively (Fig. 2 and Fig. S2). During this equilibration period (typically about 35–50 min), the reaction mixture was stirred in darkness.
concentrations in the PSII suspension was monitored at m/z = 32 and m/z = 28, respectively (Fig. 2 and Fig. S2). During this equilibration period (typically about 35–50 min), the reaction mixture was stirred in darkness.
PSII-cc were excited via the sapphire window of the MIMS cell (Fig. 1) with a group of short saturating Xe flashes provided by one of the following Xe flash lamps: Perkin Elmer, LS-1130-4 (max. 50 Hz; ∼5-μs half-width) or EG&G, model PS 302, light pack FY-604 (max. 10 Hz; ∼15-μs half-width). The experiments on spinach PSII-mf (Fig. 5 A andB) were performed employing a frequency doubled Nd-YAG laser (Continuum Inlite II-20; 532 nm).
O2 produced by the PSII samples was detected online as 18O-double-labeled O2 at m/z = 36 (see Fig. 2, Inset). In case of N2-pressure experiments the increase of the N2 level in the sample solution was monitored at m/z = 28 until the maximum of its solubility was reached, and then the Faraday cups of the MS were switched for the detection of O2 species (see Fig. S2 and SI Text). For monitoring the membrane permeability the concentration of Ar was simultaneously recorded at m/z = 40 during all O2 measurements.
Supplementary Material
Acknowledgments.
J.C. and W.J. are very grateful to Hella Kenneweg for skillful and engaged technical assistance. W.J. and J.M. enjoyed fruitful discussions with Fabrice Rappaport. We thank Gennady Ananyev, Charles Dismukes, and the referees for helpful comments on the manuscript. W.J. acknowledges financial support by the Deutsche Forschungsgemeinschaft, the MWK Niedersachsen, and the Fonds der Chemie. J.M. and D.S. gratefully acknowledge the support by the Wallenberg Stiftelse, Vetenskapsrådet, and the Max-Planck Gesellschaft.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1014249108/-/DCSupplemental.
References
- 1.Kok B, Forbush B, McGloin M. Cooperation of charges in photosynthetic O2 evolution. Photochem Photobiol. 1970;11:457–476. doi: 10.1111/j.1751-1097.1970.tb06017.x. [DOI] [PubMed] [Google Scholar]
- 2.Joliot P, Barbieri G, Chabaud R. Un nouveau modele des centres photochimiques du systeme II. Photochem Photobiol. 1969;10:309–329. [Google Scholar]
- 3.Messinger J, Renger G. In: Primary Processes of Photosynthesis, Part 2 Principles and Apparatus. Renger G, editor. Cambridge, UK: RSC Publishing; 2008. pp. 291–351. [Google Scholar]
- 4.Siegbahn PEM. Structures and energetics for O2 formation in photosystem II. Acc Chem Res. 2009;42:1871–1880. doi: 10.1021/ar900117k. [DOI] [PubMed] [Google Scholar]
- 5.Dau H, Haumann M. The manganese complex of photosystem II in its reaction cycle—Basic framework and possible realization at the atomic level. Coord Chem Rev. 2008;252:273–295. [Google Scholar]
- 6.Junge W, Haumann M, Ahlbrink R, Mulkidjanian A, Clausen J. Electrostatics and proton transfer in photosynthetic water oxidation. Philos T R Soc Lon B. 2002;357:1407–1417. doi: 10.1098/rstb.2002.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rappaport F, Blanchard-Desce M, Lavergne J. Kinetics of electron transfer and electrochromic change during the redox transitions of the photosynthetic oxygen evolving complex. Biochim Biophys Acta. 1994;1184:178–192. [Google Scholar]
- 8.Haumann M, et al. Photosynthetic O2 formation tracked by time-resolved X-ray experiments. Science. 2005;310:1019–1021. doi: 10.1126/science.1117551. [DOI] [PubMed] [Google Scholar]
- 9.Etienne AL. Etude de l’etape thermique de l’emission photosynthetique d’oxygene par une methode d’ecoulement. Biochim Biophys Acta. 1968;153:895–897. doi: 10.1016/0005-2728(68)90018-2. [DOI] [PubMed] [Google Scholar]
- 10.Razeghifard MR, Pace RJ. EPR kinetic studies of oxygen release in thylakoids in PSII membranes: a kinetic intermediate in the S3 to S0 transition. Biochemistry. 1999;38:1252–1257. doi: 10.1021/bi9811765. [DOI] [PubMed] [Google Scholar]
- 11.Strzalka K, Walczak T, Sarna T, Swartz HM. Measurement of time-resolved oxygen concentration changes in photosynthetic systems by nitroxide-based EPR oximetry. Arch Biochem Biophys. 1990;281:312–318. doi: 10.1016/0003-9861(90)90449-9. [DOI] [PubMed] [Google Scholar]
- 12.Clausen J, Debus RJ, Junge W. Time-resolved oxygen production by PSII: Chasing chemical intermediates. Biochim Biophys Acta. 2004;1655:184–194. doi: 10.1016/j.bbabio.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 13.Jursinic PA, Dennenberg RJ. Oxygen release time in leaf disks and thylakoids of peas and photosystem II membrane fragments of spinach. Biochim Biophys Acta. 1990;1020:195–206. [Google Scholar]
- 14.Meunier PC, Popovic R. The time for oxygen release in photosynthesis: reconciliation of flash polarography with other measurement techniques. Photosynth Res. 1991;28:33–39. doi: 10.1007/BF00027174. [DOI] [PubMed] [Google Scholar]
- 15.Lavergne J. Mitochondrial responses to intracellular pulses of photosynthetic oxygen. Proc Natl Acad Sci USA. 1989;86:8768–8772. doi: 10.1073/pnas.86.22.8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babcock GT, Blankenship RE, Sauer K. Reaction kinetics for positive charge accumulation on the water side of chloroplast photosystem II. FEBS Lett. 1976;61:286–289. doi: 10.1016/0014-5793(76)81058-7. [DOI] [PubMed] [Google Scholar]
- 17.Razeghifard MR, Klughammer C, Pace RJ. Electron paramagnetic resonance kinetic studies of the S states in spinach thylakoids. Biochemistry. 1997;36:86–92. doi: 10.1021/bi9614287. [DOI] [PubMed] [Google Scholar]
- 18.Lavergne J. Improved UV-visible spectra of S-state transitions in the photosynthetic oxygen evolving system. Biochim Biophys Acta. 1991;1060:175–188. [Google Scholar]
- 19.Renger G, Hanssum B. Studies on the reaction coordinates of the water oxidase in PSII membrane-fragments from spinach. FEBS Lett. 1992;299:28–32. doi: 10.1016/0014-5793(92)80092-u. [DOI] [PubMed] [Google Scholar]
- 20.Karge M, Irrgang K-D, Renger G. Analysis of the reaction coordinate of photosynthetic water oxidation by kinetic measurements of 355 nm absorption changes at different temperatures in photosystem II preparations suspended in either H2O or D2O. Biochemistry. 1997;36:8904–8913. doi: 10.1021/bi962342g. [DOI] [PubMed] [Google Scholar]
- 21.Clausen J, Junge W, Dau H, Haumann M. Photosynthetic water oxidation at high O2 backpressure monitored by delayed chlorophyll fluorescence. Biochemistry. 2005;44:12775–12779. doi: 10.1021/bi051183a. [DOI] [PubMed] [Google Scholar]
- 22.Haumann M, Grundmeier A, Zaharieva I, Dau H. Photosynthetic water oxidation at elevated dioxygen partial pressure monitored by time-resolved X-ray absorption measurements. Proc Natl Acad Sci USA. 2008;105:17384–17389. doi: 10.1073/pnas.0802596105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clausen J, Junge W. Detection of an intermediate of photosynthetic water oxidation. Nature. 2004;430:480–483. doi: 10.1038/nature02676. [DOI] [PubMed] [Google Scholar]
- 24.Dau H, Haumann M. Photosynthetic oxygen production—Response. Science. 2006;312:1471–1472. [Google Scholar]
- 25.Junge W, Clausen J. Photosynthetic oxygen production. Science. 2006;312:1470–1470. doi: 10.1126/science.312.5779.1470c. [DOI] [PubMed] [Google Scholar]
- 26.Penner-Hahn JE, Yocum CF. Photosynthetic oxygen production—Response. Science. 2006;312:1470–1471. doi: 10.1126/science.312.5779.1470c. [DOI] [PubMed] [Google Scholar]
- 27.Penner-Hahn JE, Yocum CF. Biochemistry—The photosynthesis “oxygen clock” gets a new number. Science. 2005;310:982–983. doi: 10.1126/science.1120919. [DOI] [PubMed] [Google Scholar]
- 28.Raven JA, Larkum AWD. Are there ecological implications for the proposed energetic restrictions on photosynthetic oxygen evolution at high oxygen concentrations? Photosynth Res. 2007;94:31–42. doi: 10.1007/s11120-007-9211-z. [DOI] [PubMed] [Google Scholar]
- 29.Kolling DRJ, Brown TS, Ananyev G, Dismukes GC. Photosynthetic oxygen evolution is not reversed at high oxygen pressures: Mechanistic consequences for the water-oxidizing complex. Biochemistry. 2009;48:1381–1389. doi: 10.1021/bi801774f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clausen J, Junge W. The inhibitory effects of acidification and augmented oxygen pressure on water oxidation. Photosynth Res. 2008;98:229–233. doi: 10.1007/s11120-008-9321-2. [DOI] [PubMed] [Google Scholar]
- 31.Dekker JP, van Gorkom HJ, Wensink J, Ouwehand L. Absorbance difference spectra of the successive redox states of the oxygen-evolving apparatus of photosynthesis. Biochim Biophys Acta. 1984;767:1–9. [Google Scholar]
- 32.Hundelt M, Hays A-MA, Debus RJ, Junge W. Oxygenic photosystem II: The mutation D1-D61N in Synechocystis sp PCC 6803 retards S-state transitions without affecting electron transfer from YZ to P680+ Biochemistry. 1998;37:14450–14456. doi: 10.1021/bi981164j. [DOI] [PubMed] [Google Scholar]
- 33.Vos MH, van Gorkom HJ, van Leeuwen PJ. An electroluminescence study of stabilization reactions in the oxygen-evolving complex of photosystem II. Biochim Biophys Acta. 1991;1056:27–39. [Google Scholar]
- 34.Renger G. A model for the molecular mechanism of photosynthetic oxygen evolution. FEBS Lett. 1977;81:223–228. [Google Scholar]
- 35.Kato Y, Sugiura M, Oda A, Watanabe T. Spectroelectrochemical determination of the redox potential of pheophytin a, the primary electron acceptor in photosystem II. Proc Natl Acad Sci USA. 2009;106:17365–17370. doi: 10.1073/pnas.0905388106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rappaport F, Guergova-Kuras M, Nixon PJ, Diner BA, Lavergne J. Kinetics and pathways of charge recombination in photosystem II. Biochemistry. 2002;41:8518–8527. doi: 10.1021/bi025725p. [DOI] [PubMed] [Google Scholar]
- 37.Rappaport F, Diner BA. Primary photochemistry and energetics leading to the oxidation of the (Mn)4Ca cluster and to the evolution of molecular oxygen in Photosystem II. Coord Chem Rev. 2008;252:259–272. [Google Scholar]
- 38.Clausen J, et al. Photosynthetic water oxidation in Synechocystis sp PCC6803: mutations D1-E189K, R and Q are without influence on electron transfer at the donor side of photosystem II. Biochim Biophys Acta. 2001;1506:224–235. doi: 10.1016/s0005-2728(01)00217-1. [DOI] [PubMed] [Google Scholar]
- 39.Berthold DA, Babcock GT, Yocum CF. A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes. FEBS Lett. 1981;134:231–234. [Google Scholar]
- 40.Beckmann K, Messinger J, Badger MR, Wydrzynski T, Hillier W. On-line mass spectrometry: membrane inlet sampling. Photosynth Res. 2009;102:511–522. doi: 10.1007/s11120-009-9474-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.