

Abstract
Mitochondrial DNA (mtDNA) has been reported to contain 5-methylcytosine (5mC) at CpG dinucleotides, as in the nuclear genome, but neither the mechanism generating mtDNA methylation nor its functional significance is known. We now report the presence of 5-hydroxymethylcytosine (5hmC) as well as 5mC in mammalian mtDNA, suggesting that previous studies underestimated the level of cytosine modification in this genome. DNA methyltransferase 1 (DNMT1) translocates to the mitochondria, driven by a mitochondrial targeting sequence located immediately upstream of the commonly accepted translational start site. This targeting sequence is conserved across mammals, and the encoded peptide directs a heterologous protein to the mitochondria. DNMT1 is the only member of the three known catalytically active DNA methyltransferases targeted to the mitochondrion. Mitochondrial DNMT1 (mtDNMT1) binds to mtDNA, proving the presence of mtDNMT1 in the mitochondrial matrix. mtDNMT1 expression is up-regulated by NRF1 and PGC1α, transcription factors that activate expression of nuclear-encoded mitochondrial genes in response to hypoxia, and by loss of p53, a tumor suppressor known to regulate mitochondrial metabolism. Altered mtDNMT1 expression asymmetrically affects expression of transcripts from the heavy and light strands of mtDNA. Hence, mtDNMT1 appears to be responsible for mtDNA cytosine methylation, from which 5hmC is presumed to be derived, and its expression is controlled by factors that regulate mitochondrial function.
Keywords: mitochondrial epigenetics, epigenetics, 5-hydroxymethylation
In the nucleus, cytosine methylation cooperates with N-terminal histone modifications to establish a silenced chromatin structure (1), thus regulating nuclear gene expression. Methylation patterns are established in the developing embryo by two de novo DNA methyltransferases, DNMT3a and -3b (2). Maintenance of this pattern in somatic cells is believed to be the predominant function of DNMT1, with functional cooperation evident between the two groups of enzymes (3). Cytosine methylation is essential for normal development, and deletion of DNMT1 results in embryonic lethality in mice and mitotic catastrophe in cultured cells (4). Recently, the presence of significant levels of 5-hydroxymethylcytosine (5hmC) was demonstrated in DNA from neurons, brain (5), and embryonic stem cells (6). 5hmC is derived from 5-methylcytosine (5mC) oxidation catalyzed by the TET family of methylcytosine oxygenases, and its functional significance is under intense investigation. This modification is likely to have an impact on local chromatin structure, and it has been proposed that 5hmC acts as an intermediate in active or passive demethylation (7).
Cytosine methylation of mitochondrial DNA (mtDNA) has been controversial and, remarkably, infrequently studied. The earliest study, conducted over three decades ago, reported that there was no methylation of mtDNA (8). Subsequently, low levels of methylation restricted to CpG dinucleotides were reported in mitochondria of several species, using methylation-sensitive restriction endonuclease cleavage and nearest-neighbor analysis (9–11). Mammalian mtDNA shows a similar level of CpG suppression to that of nuclear DNA (12), suggesting that 5mC is susceptible to mutation in mtDNA. To date, 5mC is the only modified base described in mtDNA, but the mechanisms establishing and maintaining mtDNA methylation, and the functional significance of this modification in mtDNA, are not known.
Mammalian mtDNA is a 16.5-kb double-stranded, circular molecule, present in multiple copies per mitochondrion (13). The mitochondrial genome encodes 13 of the proteins present in the respiratory chain complexes of mammalian mitochondria, as well as two ribosomal RNAs and 22 transfer RNAs specific to this organelle. All other mitochondrial proteins, including those required for mtDNA replication and transcription, are encoded in the nucleus and translocated to the mitochondria using specialized import systems which often involve N-terminal mitochondrial targeting sequences (MTSs) (14). In contrast to the nuclear genome, mtDNA is not complexed with histones. However, mtDNA is present in protein-containing complexes called nucleoids, each containing multiple copies of mtDNA bound to a complex mixture of proteins (15).
Transcription of the mitochondrial genome is thought to be coregulated with nuclear components of the respiratory chain complexes (16). In mammals, oxidative stress results in stabilization of peroxisome proliferator-activated receptor γ-coactivator 1α (PGC1α), which activates the transcription of several nuclear-encoded transcription factors, including nuclear respiratory factor 1 (NRF1). PGC1α and NRF1 form a complex that in turn up-regulates transcription of transcription factor of activated mitochondria (TFAM) and multiple members of mitochondrial respiratory chain complexes (17). Several nuclear-encoded genes involved in mitochondrial function, including PGC1α (18), are regulated by DNA methylation. Conversely, it has been suggested that mitochondria are able to influence cytosine methylation levels in the nucleus by modulating the flux of one-carbon units for the generation of S-adenosylmethionine, the methyl donor in DNA methylation (19). Thus, epigenetic regulation of nuclear gene expression appears to have a mitochondrial component. The presence of cytosine methylation in mtDNA led us to question whether this epigenetic modification might play a role in the coordinated regulation of mitochondrial gene expression from both nuclear and mitochondrial genomes.
Results
Human and Mouse DNMT1 Encode Mitochondrial Targeting Sequences.
Early reports of DNA methylation in the mitochondrial genome (9–11) led us to ask whether one or more of the catalytically active mammalian DNA methyltransferases might be targeted to mitochondria. Examination of the 5′ UTR and 5′ flanking genomic DNA upstream of the published transcription start sites (20) for both human and mouse DNMT1 revealed that sequence equivalent to 101 codons in human and 63 codons in mouse DNMT1 was in-frame with the highly conserved amino acid sequence of DNMT1, starting with the ATG reported (20) to be the primary translational start codon (Fig. 1 A and B). This upstream sequence includes two additional in-frame codons for methionine, each in a moderate context for ribosome binding (21); the upstream ATG codons are denoted ATG1 and ATG2, whereas the published translation start is shown as ATG3. RT-PCR using sense primers located over ATG1 or ATG2 and antisense primers crossing the exon 1-2 boundary by 4 nucleotides detected transcripts capable of encoding these N-terminal extensions in human and mouse cells. Transcripts initiating upstream of ATG1 in mouse and ATG2 (but not ATG1) in human mRNA were easily detected (Fig. 1C), suggesting the utilization of an upstream transcription start site encoding an N-terminal extension.
Fig. 1.

Mouse and human DNMT1 loci have an in-frame sequence upstream of the published start codon. (A) Sequence encoding an additional 101 in-frame amino acids, including two potential translation start ATG codons, is present upstream of the published human DNMT1 start ATG (ATG3). (B) In mouse, 63 in-frame amino acids are encoded upstream of the published translational start site, with two additional potential translation start ATGs. Overlapping binding sites for p53 (underlined) and NRF1 (gray box) are located across one of the two potential start ATG codons in both species. Published transcription start sites are indicated by horizontal arrows. (C) Mature transcripts capable of encoding these upstream sequences are detected in both mouse and human cells. No RT, no reverse transcriptase control.
Mouse or human DNMT1 isoforms containing this additional N-terminal sequence were predicted by MitoProt II (http://ihg.gsf.de/ihg/mitoprot.html) (22) to be targeted to the mitochondria with very high probability, compared with proteins beginning at the published start codon, ATG3 (Table S1). The genome databases also contain upstream sequences for chimpanzee, rat, and cow DNMT1; in each species, one or more in-frame potential start codons encode a peptide with a strong probability (>90%) of mitochondrial localization (Table S1). All are predicted to form amphiphilic α-helices, although sequence conservation between them is low, as is often the case for mitochondrial leader peptides across species.
Immunoblots of purified mitochondria from mouse embryonic fibroblasts (MEFs) and HCT116 human colon carcinoma cells showed the presence of DNMT1 (Fig. 2A) but not DNMT3a or DNMT3b in this organelle (Fig. 2B). Full-length DNMT1 and a smaller peptide are detected by an N-terminal DNMT1 antibody, suggesting that proteolytic processing occurs upon entry into the mitochondria. Absence of the nuclear marker H3K4me3 in the mitochondrial fraction indicated purity from contamination by nuclear material, the primary site of localization of DNA methyltransferases.
Fig. 2.

Mouse and human DNMT1 are associated with the mitochondrial compartment. (A) DNMT1 antibody directed against an N-terminal epitope (amino acids 1–10) detects two protein species in the mitochondrial fraction, full-length DNMT1 (185 kDa; arrow) and a 60-kDa peptide. (B) DNMT3a or DNMT3b de novo methyltransferases are not detected in mitochondria. Antibodies against compartment-specific markers voltage-dependent anion carrier (VDAC) (mitochondrial), tubulin (cytosolic), and H3K4me3 (nuclear) demonstrate purity of each subcellular fraction. C, cytosol; M, mitochondrial lysate; W, whole-cell lysate. For DNMT3b, the mitochondrial fraction was overloaded twofold to exclude the possibility of low levels of DNMT3b in the mitochondria. (C) Mouse and human leader peptides direct GFP to the mitochondria. Transiently transfected NIH/3T3 cells show distinct colocalization of GFP-tagged targeting sequences with MitoTracker Red stain in the mitochondria. CAT-GFP does not localize to the mitochondria.
We cloned the mouse and human leader sequences, from ATG1 to upstream of ATG3, in-frame with the C-terminal GFP tag of pcDNA6.2/GFP, and transfected the plasmids into NIH/3T3 fibroblasts. Confocal microscopy showed that both human and mouse leader sequences targeted GFP to the mitochondria, indicated by colocalization of MitoTracker Red with green fluorescence (Fig. 2C). Mitochondria in untransfected cells within the same visual field remained red in the merged photomicrographs, serving as negative controls for colocalization, whereas a chloramphenicol acetyl transferase (CAT)-GFP control plasmid remained cytosolic. We also transfected these constructs into HCT116 human colon carcinoma cells for immunoblot analysis of purified mitochondria using anti-GFP antibody (Fig. S1). HCT116 mitochondria clearly accumulated GFP. Thus, human and mouse leader peptides represent bona fide MTSs capable of tracking heterologous proteins to this organelle. Each MTS is able to operate across species, indicating functional conservation.
mtDNMT1 Expression Is Regulated by Factors That Respond to Oxidative Stress.
MatInspector (http://www.genomatix.de/en/index.html) predicted a binding site for NRF1 in both human and mouse DNMT1. This consensus sequence was located over one of the upstream in-frame start codons and was conserved in all other mammalian species studied (Table S1; Fig. 3A). Under conditions of oxidative stress, the coactivator PGC1α activates and interacts with NRF1 to up-regulate multiple nuclear-encoded mitochondrial genes (17). Accordingly, we transiently transfected NRF1, PGC1α, or both together into HCT116 cells and analyzed the levels of mitochondrial DNMT1 (mtDNMT1) by immunoblot (Fig. 3B). A small increase in mtDNMT1 was seen in cells transfected with NRF1 or PGC1α alone, whereas cotransfection with both PGC1α and NRF1 resulted in an approximately fivefold increase in mtDNMT1 relative to control. Thus, this locus is sensitive to regulation by activators that respond to oxidative stress.
Fig. 3.

Regulation of mtDNMT1 expression. (A) NRF1 consensus binding sites are conserved over one of the potential mitochondrial start ATG codons in mammalian DNMT1. (B) NRF1 and PGC1α up-regulate mtDNMT1. (C) Loss of p53 selectively up-regulates mtDNMT1 expression. Values represent mean ± SD for two independent experiments. (D) Immunoblot analysis of WT and p53−/− MEFs shows increased mtDNMT1 expression in p53-null cells. (E) Gene-specific changes in mitochondrial transcription relative to 18S rRNA. To test for differences in expression of four mitochondrial genes across the two cell lines, we applied an ANOVA using least-squares means. ND1 and ND6 showed significant differences in expression (P < 0.05). In p53−/− MEFs, ND1 mRNA expression was significantly higher (mean = 1.73, SE = 0.18, 95% C.I. = 1.37, 2.09) than in WT MEFs (mean = 0.95, SE = 0.09, 95% C.I. = 0.76, 1.15). ND6 mRNA expression in p53−/− MEFs was significantly lower (mean = 0.75, SE = 0.07, 95% C.I. = 0.59, 0.90) than in WT MEFs (mean = 1.14, SE = 0.11, 95% C.I. = 0.91, 1.38).
The NRF1 binding site is coincident with a p53 consensus binding site (Fig. 1 A and B), which we previously demonstrated to repress DNMT1 transcription (23). Our earlier study showed a three- to sixfold increase in DNMT1 transcription following either activation or genetic deletion of p53 in HCT116 cells and MEFs. Because p53 is known to regulate mitochondrial respiration (24), we asked whether this tumor suppressor protein also affected mtDNMT1 mRNA expression. We used RT-quantitative (q)PCR with primers that distinguish the mitochondrial transcript from the total DNMT1 transcript (Fig. 1C); the mitochondrial transcript comprised 1–2% of the total DNMT1 synthesized in log-phase MEFs or HCT116 cells. The relative abundance of mtDNMT1 transcript increased sixfold in p53−/− MEFs compared with WT MEFs, whereas total DNMT1 mRNA increased threefold (Fig. 3C), suggesting a preferential up-regulation of the mitochondrial transcript in cells lacking p53. Immunoblot analysis of these isogenic cells showed a striking increase in mtDNMT1 protein with loss of p53 (Fig. 3D).
Gene-Specific Changes in Mitochondrial Transcription.
We asked whether this mtDNMT1 overexpression was reflected in an alteration in transcription of the mitochondrial genome (Fig. 3E). NADH dehydrogenase subunit 6 (ND6), the only protein-coding gene on the light (L) strand, was significantly underexpressed in response to increased mtDNMT1, suggesting a role for mtDNA methylation in repression of L-strand transcription. On the heavy (H) strand, ATPase subunit 6 (ATP6) and cytochrome c oxidase subunit 1 (COX1) were unaltered in their expression levels. However, NADH dehydrogenase subunit 1 (ND1), the first H-strand protein-coding region following the ribosomal RNA genes, was significantly increased in response to elevated mtDNMT1. These data support a gene-specific effect on mitochondrial gene transcription, as discussed below.
Mitochondrial DNMT1 Is Bound to mtDNA.
We created an HCT116 cell line (25) in which one endogenous allele of DNMT1 carries a C-terminal tandem-affinity purification (TAP) tag (26). TAP-tagged DNMT1 translocated efficiently to mitochondria (Fig. 4B). We therefore used these cells to ask whether mtDNMT1 interacted directly with mtDNA. Formaldehyde-crosslinked mitochondrial lysates were immunoprecipitated with IgG beads (26), and qPCR with primers specific for mtDNA (Table S2) was used to quantitate the interaction between mtDNMT1 and mtDNA. Immunoprecipitates from TAP-tagged cells were substantially enriched for mtDNA in comparison with immunoprecipitates from untagged cells, except for an amplicon containing no CpG dinucleotides, which gave equally low signal from both cell lines (Fig. 4C). These data suggest CpG-dependent interaction of mtDNMT1 with the mitochondrial genome and confirm the localization of this protein to the mitochondrial matrix. Interaction was evident in the D-loop control region, which carries the mitochondrial origin of replication and promoters, as well as in rRNA and protein-coding regions. The level of enrichment was dependent on the target amplicon; five of the six regions probed showed a three- to fivefold enrichment of mtDNA sequences. However, qPCR of the region covering the junction between 12S and 16S rRNA genes (primer 2) showed only twofold enrichment in binding of mtDNMT1-TAP. The density of CpG dinucleotides in this amplicon is <50% that in all other amplicons analyzed, suggesting that interaction of mtDNMT1 with mtDNA is proportional to CpG density, and supporting a functional role for mtDNMT1 in establishment and maintenance of mtDNA methylation.
Fig. 4.

mtDNMT1 specifically binds to mitochondrial DNA. (A) Organization of the human mitochondrial genome. tRNA genes (black bars, single-letter code) intersperse the protein-coding and rRNA genes on both the H strand (dark gray) and L strand (light gray). Regions amplified in mtIP are shown with broken white lines. (B) Endogenous DNMT1 carrying a TAP tag translocates efficiently into the mitochondria. (C) mtDNMT1-TAP interacts specifically with mtDNA. Nonspecific background was assessed using mitochondrial lysates from nontagged HCT116 cells. Values shown are the mean ± SD of six replicate samples from two independent experiments.
5-Hydroxymethylcytosine Is Present in mtDNA.
We immunoprecipitated randomly sheared mtDNA with an antibody to 5mC or 5hmC and probed the precipitated DNA by qPCR to determine the presence and relative abundance of these two modified bases in mtDNA (Fig. 5 A and B). Immunoprecipitated samples were enriched 10- to 20-fold for 5mC relative to IgG control for all regions tested. mtDNA immunoprecipitated using anti-5hmC was highly enriched (85- to 580-fold) relative to IgG controls, except across the D loop (primer 27), which was enriched 38-fold. The specificity of each antibody for its respective modification was confirmed using control DNA in which every cytosine was converted to either 5mC or 5hmC (Fig. S2). The presence of both cytosine modifications in mtDNA suggests that earlier studies underestimated the degree of epigenetic modification of the mitochondrial genome.
Fig. 5.

mtDNA contains both 5mC and 5hmC. (A) mtDNA immunoprecipitation using antibody directed against 5mC shows 10- to 20-fold enrichment compared with IgG controls. (B) mtDNA immunoprecipitation using antibody directed against 5hmC demonstrates substantial levels of this modified base in mtDNA. (C) Site-specific detection of 5hmC in mtDNA. DNA was incubated in the presence or absence of β-gt, fully methylated with M.Sss1 and M.CviP1, and cleaved with Gla1. The degree of protection from cleavage was assessed using qPCR with mtDNA-specific primers. Data shown represent mean ± SD of six replicates from two independent experiments, normalized to input DNA.
We used phage T4 5hmC-β-glucosyltransferase (β-gt) (27) to determine the presence of 5hmC at Gla1 restriction endonuclease cleavage sites (28). Control experiments using defined DNA sequences containing cytosine, 5mC, or 5hmC confirmed that Gla1 cleaved only sites modified by methylation or hydroxymethylation, but not sites containing glucosylated 5hmC (Fig. S3A). Protection of mtDNA from cleavage by 5hmC glucosylation was assessed by endpoint (Fig. S3 B and C) and qPCR (Fig. 5C). 5hmC was present in three different amplicons from human mtDNA and two amplicons from mouse mtDNA or genomic DNA. Amplicons containing two Gla1 restriction sites each (amplicons ATP6, 12S, and 16S-3) showed 50% protection in comparison with amplicons with a single Gla1 site (amplicons 2 and 16S-2), suggesting a similar level of 5hmC at all restriction sites tested. A mouse amplicon devoid of Gla1 sites (ATP6/COX3) was protected from cleavage irrespective of 5hmC glucosylation (Fig. S3C).
Discussion
Cytosine methylation of the mitochondrial genome has remained largely overlooked, in part because early reports using nearest-neighbor analysis indicated that this modification was present at only 2–5% of CpG dinucleotides (11), well below the level of methylation seen in the nucleus. The data presented here show a 10- to 20-fold enrichment of mtDNA sequences in immunoprecipitates using 5mC antibody, somewhat lower than that usually obtained from genomic DNA (∼100-fold for CpG islands). This likely reflects the CpG-sparse nature of the mitochondrial genome, which does not contain CpG islands. We demonstrate here the presence of 5hmC in mtDNA using two independent assays. Thus, epigenetic modification of cytosines in the mitochondrial genome is likely much more frequent than previously believed. In the nucleus, 5hmC is generated from 5mC by the action of the TET family of methylcytosine oxygenases (6). There is not yet evidence regarding the presence or absence of these enzymes in mitochondria, and the TET family proteins or loci do not contain recognizable mitochondrial targeting sequences (14). We therefore cannot rule out the possibility of a different mechanism for the generation of 5hmC, including covalent addition of 5-hydroxymethyl groups directly to DNA cytosine residues by mtDNMT1 (29) using formaldehyde generated from mitochondrial mixed-function oxidases. The apparently lower enrichment for 5hmC in the D-loop control region most likely reflects the less efficient amplification of a longer fragment (833 bp compared with 112–238 bp) from mtDNA sheared to an average size of 300–400 bp. However, the D loop exists as a stable triple-helical structure containing an RNA primer required for initiation of mtDNA replication (13), and we have found this region to be resistant to in vitro methylation by M.Sss1 cytosine methyltransferase. It is therefore possible that the kinetics of epigenetic modification in this region of the mitochondrial genome might be different from those in coding regions.
The function of 5hmC in the nuclear genome is not yet clear. It has been proposed that 5hmC is an intermediate metabolite in active demethylation of the genome by repair enzymes (30), in passive demethylation as a result of lack of recognition by enzymes involved in maintenance methylation (31), or that it alters local chromatin structure because 5hmC is not recognized by 5-methylcytosine-binding proteins (7). The role of 5hmC in the mitochondrial genome likely involves one or more of these processes. Although quantitative measurements of the relative abundance of 5hmC and 5mC can be achieved using methylated DNA immunoprecipitation (Me-DIP), 5hMe-DIP, HPLC, or enzymatic methods, mapping the location and distribution of 5hmC in either the nuclear or mitochondrial genome is not yet technically feasible, because this modified base is indistinguishable from 5mC by bisulfite modification (7).
This study reports a mitochondrial isoform of DNA methyltransferase 1, which is the only member of the catalytically active mammalian DNA methyltransferase family found in this organelle. The conservation of an ORF encoding a mitochondrial targeting sequence upstream of the commonly accepted translational start codon across multiple mammalian species suggests an important role for this enzyme in mitochondrial function.
Although DNMT1 is generally considered to be the maintenance DNA methyltransferase, it is able to methylate completely unmethylated DNA in vitro with an efficiency that exceeds that of the de novo methyltransferases DNMT3a and -3b (32). Thus, DNMT1 appears to be capable of both initiating and maintaining cytosine methylation in the nucleus, and the lack of de novo methyltransferases in mitochondria implicates mtDNMT1 in both processes in this organelle.
We show that mtDNMT1 binds to the mitochondrial genome in a manner proportional to the density of CpG dinucleotides. Of particular relevance is the binding of mtDNMT1 to the D-loop control region, which carries the promoters driving transcription initiation of both heavy and light strands, supporting a role for mtDNMT1 in regulation of mitochondrial gene expression. The asymmetric, gene-specific alteration in mitochondrial transcription patterns shown here suggests diverse roles for mtDNMT1 and cytosine modification in this organelle. Decreased expression of ND6 on the L strand implies that cytosine methylation in mtDNA represses gene expression from the light-strand promoter, as it does in the nucleus. However, increased transcription of ND1 with no change in transcription of ATP6 or COX1 raises the possibility of a different mode of action on the H strand. A binding site for mitochondrial terminator factor 1 (MTERF1) is located between the end of the 16S rRNA gene and the translation start of ND1 (33). MTERF1 binds to both H-strand promoter 1 (HSP1) and the terminator binding site (Fig. 4A), forming a transcription loop that maintains high-level production of rRNA. Transcripts initiating at HSP2 produce polycistronic messages encoding the entire H strand (13). Our data raise the possibility that mtDNMT1, either through modification of CpG dinucleotides or by direct protein–protein interaction, interferes with MTERF-dependent transcription termination, allowing read-through from HSP1 to the next transcriptional unit (ND1) without impacting polycistronic mRNA synthesis from HSP2.
We show here that DNMT1 is present in the mitochondrial matrix, bound to mtDNA, and modifies transcription of the mitochondrial genome in what appears to be a gene-specific fashion. We report the presence of both 5hmC and 5mC in mtDNA , suggesting that earlier studies may have underestimated the proportion of modified cytosines in this genome. Hence, mtDNMT1 appears to be responsible for the establishment and maintenance of cytosine methylation in mtDNA, from which 5hmC is presumably derived. Our data support a role for epigenetic modification of the mitochondrial genome in regulation of mitochondrial transcription.
Materials and Methods
Cell Lines.
HCT116p53+/+ and HCT116p53−/− were obtained from Bert Vogelstein, Johns Hopkins University (Baltimore, MD). Primary MEFs were prepared from E12.5–E13.5 embryos.
Plasmids and Transfections.
Primers used are listed in Table S2. Mitochondrial targeting sequences were amplified from random-primed human and mouse cDNAs. Murine NRF1 cDNA was obtained from the American Type Culture Collection and recloned into pDEST26/C-FLAG. PGC1α plasmid was a gift from Gregorio Gil, Virginia Commonwealth University. Cells were transfected using Polyjet liposomes (ProSci) (HCT116) or nucleofection (Amaxa) (MEF and NIH/3T3) according to the manufacturers’ specifications, and were harvested 48 h after transfection.
Mitochondrial Purification and Immunoblot Analysis.
Mitochondria were purified by dounce homogenization and differential centrifugation in the presence of complete protease inhibitors (Roche) (34). Proteins were resolved on 4–15% gradient SDS/PAGE gels. Antibodies used were anti-DNMT1 amino acids 1–10 (Abcam), anti-tubulin and anti–voltage-dependent anion carrier (VDAC) (Pierce), anti-H3K4me3 (Upstate Biotechnology), anti-DNMT3a and anti-DNMT3b (Imgenex), anti-GFP (Invitrogen), and anti-TAP (Open Biosystems). Protein was loaded onto SDS/PAGE gels to approximate equal cell equivalents, so that an equal signal for each compartment-specific antibody was obtained (whole-cell lysate, 75 μg; cytosol, 25 μg; mitochondria, 18 μg).
Confocal Microscopy.
NIH/3T3 cells were plated onto poly-l-lysine-coated coverslips and fixed with 4% paraformaldehyde 48 h after transfection. Cells were stained with 1 nM MitoTracker Red (Molecular Probes) for 15 min, washed three times with PBS, and mounted onto glass slides with ProLong Gold antifade reagent with DAPI (Invitrogen). Microsopy was carried out using a Leica TCS-SP2 AOBS confocal scanning microscope.
Gene Expression.
Total RNA was isolated using TRIzol (Invitrogen), DNase I-treated, and reverse-transcribed with SuperScript III (Invitrogen) and random hexamers. Gene expression was determined using qPCR with Quantitect SYBR Green PCR Mastermix (Qiagen). Values were normalized to 18S rRNA for mitochondrial transcription or to β-actin for mtDNMT1 quantitation.
Statistics.
Statistical analyses of differential mitochondrial gene expression profiles were performed using a random-effects ANOVA using the statistical package JMP version 7.0 (SAS). The least-squares mean for each gene for WT and p53−/− MEFs was obtained along with standard errors, and each dataset was normalized to 18S rRNA expression. Three independent sets of biological samples were analyzed for each gene, with triplicate technical replicates in each sample. The ANOVA model included technical replicates as nested effects and biological replicates as random effects. The corresponding 95% confidence intervals (C.I.) were obtained using statistical methods for transformations (Delta method). All other qPCR experiments include multiple replicates from two or more independent experiments, normalized to relevant controls or to input DNA. SDs were computed using the formula
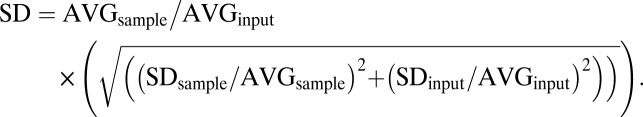 |
Mitochondrial Immunoprecipitation.
Purified mitochondria from DNMT1-TAP and nontagged HCT116 cells were formaldehyde-cross-linked, lysed, and immunoprecipitated as described (35). Mitochondrial DNA was sheared using a Diagenode Bioruptor water bath sonicator to an average length of 400 bp. We used 750 μg mitochondrial extract in each mitochondrial immunoprecipitation (mtIP). IgG beads were equilibrated in lysis buffer and added to mitochondrial lysates for overnight incubation at 4 °C to isolate DNMT1-TAP/DNA complexes. Immunoprecipitated samples were processed as described (35) and purified DNA was analyzed by qPCR with 1 μL mtIP DNA and Quantitect SYBR Green Mastermix. The abundance of mtDNA was determined from a standard curve of purified mtDNA and is expressed as ng mtDNA immunoprecipitated. Values obtained for non-TAP-tagged HCT116 cells represent nonspecific background. Primers used were from Lu et al. (35) (fragments of 800–900 bp, primers 1, 2, 3, and 27) or as listed in Table S2 (fragments < 200 bp).
Mitochondrial 5mC and 5hmC Immunoprecipitation.
Purified mtDNA (4 μg) was sheared to an average length of 400 bp and rotated overnight at 4 °C with 2 μg IgG, anti-5mC, or anti-5hmC (Active Motif). The specificity of both antibodies for their respective cytosine modifications in DNA was verified using defined DNA substrates synthesized in the presence of dCTP, 5m-dCTP, or 5hm-dCTP (Active Motif). Precleared protein-G beads (Amersham) were used as previously described (23) to immunoprecipitate the antibody/DNA complexes, and DNA was purified from immunoprecipitates using proteinase K, organic extraction, and precipitation (23). The abundance of mtDNA was determined from a standard curve of purified mtDNA and is expressed as ng mtDNA immunoprecipitated relative to input values.
Sequence-Specific Detection of 5hmC.
The presence of 5hmC at Gla1 restriction sites was determined using a Quest 5hmC Detection Kit (Zymo Research) as described by the manufacturer using 80 ng mtDNA or total cellular DNA. Gla1 cleaves DNA only when restriction-site cytosines are methylated or hydroxymethylated; glucosylation of 5hmC residues results in protection from Gla1 cleavage. Control DNAs used to validate the assay were from Active Motif.
Supplementary Material
Acknowledgments
Microscopy was performed at the Virginia Commonwealth University Department of Anatomy and Neurobiology Microscopy Facility, supported, in part, by National Institutes of Health (NIH)-National Institute of Neurological Disorders and Stroke Center Core Grant 5P30NS047463-02. Plasmid cloning and preparation were carried out in the Massey Cancer Center Macromolecule Core Facility, and statistical analysis with assistance from Dr. R. V. Ramakrishnan in the Biostatistics Core Facility. These core facilities are supported, in part, by NIH-National Cancer Institute (NCI) Cancer Center Support Grant P30 CA16059. This work was supported by NIH-NCI Grant CA106630 (to S.M.T.) and by a pilot project from the Massey Cancer Center.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1012311108/-/DCSupplemental.
References
- 1.Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- 2.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 3.Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10:805–811. doi: 10.1038/nrg2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen T, et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 5.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang Y, et al. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 2010;5:e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dawid IB. 5-Methylcytidylic acid: Absence from mitochondrial DNA of frogs and HeLa cells. Science. 1974;184:80–81. doi: 10.1126/science.184.4132.80. [DOI] [PubMed] [Google Scholar]
- 9.Nass MM. Differential methylation of mitochondrial and nuclear DNA in cultured mouse, hamster and virus-transformed hamster cells. In vivo and in vitro methylation. J Mol Biol. 1973;80:155–175. doi: 10.1016/0022-2836(73)90239-8. [DOI] [PubMed] [Google Scholar]
- 10.Shmookler Reis RJ, Goldstein S. Mitochondrial DNA in mortal and immortal human cells. Genome number, integrity, and methylation. J Biol Chem. 1983;258:9078–9085. [PubMed] [Google Scholar]
- 11.Pollack Y, Kasir J, Shemer R, Metzger S, Szyf M. Methylation pattern of mouse mitochondrial DNA. Nucleic Acids Res. 1984;12:4811–4824. doi: 10.1093/nar/12.12.4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cardon LR, Burge C, Clayton DA, Karlin S. Pervasive CpG suppression in animal mitochondrial genomes. Proc Natl Acad Sci USA. 1994;91:3799–3803. doi: 10.1073/pnas.91.9.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falkenberg M, Larsson NG, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem. 2007;76:679–699. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- 14.Mokranjac D, Neupert W. Protein import into mitochondria. Biochem Soc Trans. 2005;33:1019–1023. doi: 10.1042/BST20051019. [DOI] [PubMed] [Google Scholar]
- 15.Garrido N, et al. Composition and dynamics of human mitochondrial nucleoids. Mol Biol Cell. 2003;14:1583–1596. doi: 10.1091/mbc.E02-07-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cannino G, Di Liegro CM, Rinaldi AM. Nuclear-mitochondrial interaction. Mitochondrion. 2007;7:359–366. doi: 10.1016/j.mito.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 17.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 18.Barrès R, et al. Non-CpG methylation of the PGC-1α promoter through DNMT3B controls mitochondrial density. Cell Metab. 2009;10:189–198. doi: 10.1016/j.cmet.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 19.Smiraglia DJ, Kulawiec M, Bistulfi GL, Gupta SG, Singh KK. A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol Ther. 2008;7:1182–1190. doi: 10.4161/cbt.7.8.6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoder JA, Yen RW, Vertino PM, Bestor TH, Baylin SB. New 5′ regions of the murine and human genes for DNA (cytosine-5)-methyltransferase. J Biol Chem. 1996;271:31092–31097. doi: 10.1074/jbc.271.49.31092. [DOI] [PubMed] [Google Scholar]
- 21.Kozak M. How do eucaryotic ribosomes select initiation regions in messenger RNA? Cell. 1978;15:1109–1123. doi: 10.1016/0092-8674(78)90039-9. [DOI] [PubMed] [Google Scholar]
- 22.Claros MG, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 23.Peterson EJ, Bögler O, Taylor SM. p53-mediated repression of DNA methyltransferase 1 expression by specific DNA binding. Cancer Res. 2003;63:6579–6582. [PubMed] [Google Scholar]
- 24.Matoba S, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 25.Kohli M, Rago C, Lengauer C, Kinzler KW, Vogelstein B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 2004;32:e3. doi: 10.1093/nar/gnh009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rigaut G, et al. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 27.Kornberg SR, Zimmerman SB, Kornberg A. Glucosylation of deoxyribonucleic acid by enzymes from bacteriophage-infected Escherichia coli. J Biol Chem. 1961;236:1487–1493. [PubMed] [Google Scholar]
- 28.Tarasova GV, Nayakshina TN, Degtyarev SKH. Substrate specificity of new methyl-directed DNA endonuclease GlaI. BMC Mol Biol. 2008;9:7. doi: 10.1186/1471-2199-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liutkeviciute Z, Lukinavicius G, Masevicius V, Daujotyte D, Klimasauskas S. Cytosine-5-methyltransferases add aldehydes to DNA. Nat Chem Biol. 2009;5:400–402. doi: 10.1038/nchembio.172. [DOI] [PubMed] [Google Scholar]
- 30.Rusmintratip V, Sowers LC. An unexpectedly high excision capacity for mispaired 5-hydroxymethyluracil in human cell extracts. Proc Natl Acad Sci USA. 2000;97:14183–14187. doi: 10.1073/pnas.97.26.14183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–950. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- 32.Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 33.Martin M, Cho J, Cesare AJ, Griffith JD, Attardi G. Termination factor-mediated DNA loop between termination and initiation sites drives mitochondrial rRNA synthesis. Cell. 2005;123:1227–1240. doi: 10.1016/j.cell.2005.09.040. [DOI] [PubMed] [Google Scholar]
- 34.Perchiniak E, et al. Probing the mechanism of the hamster mitochondrial folate transporter by mutagenesis and homology modeling. Biochemistry. 2007;46:1557–1567. doi: 10.1021/bi062191+. [DOI] [PubMed] [Google Scholar]
- 35.Lu B, et al. Roles for the human ATP-dependent Lon protease in mitochondrial DNA maintenance. J Biol Chem. 2007;282:17363–17374. doi: 10.1074/jbc.M611540200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.