

Abstract
Using adoptive transfer models we determined that an adeno-associated viral vector of serotype 2 (AAV2) induces in mice proliferation of CD8+ T cells that recognize an epitope within the viral capsid. Proliferation to an endogenous epitope within viral protein (VP)3 could be observed for at least 3 weeks while a foreign epitope placed at multiple copies within VP2 elicited CD8+ T cell expansion for at least 10 weeks. These data show that capsid antigens of AAV2 degrade slowly over a period of weeks and during this period provide targets to CD8+ T cells.
Introduction
A clinical gene transfer trial for treatment of severe hemophilia B by hepatic infusion of adeno-associated viral vectors derived from human serotype 2 (AAV2) expressing human factor IX (AAV2-hF.IX) under a hepatocyte-specific promoter resulted in subclinical transient transaminitis with concomitant loss of circulating F.IX.1 Results were suggestive of immune-mediated destruction of AAV-transduced hepatocytes preventing sustained expression of the therapeutic transgene product. As this result contrasted with numerous preclinical trials with AAV vectors, which consistently had shown sustained transgene product expression upon application of AAV vectors,2,3,4,5 we hypothesized that humans unlike experimental animals have pre-existing immunity to antigens of AAV due to natural infections and that the AAV gene transfer vehicle had elicited a recall response of AAV capsid-specific CD8+ T cells, which in turn lysed AAV-transduced cells. Subsequent clinical trials confirmed that AAV vectors given at high doses for gene transfer augment frequencies of AAV capsid-specific CD8+ T cells circulating in the recipients' blood.6,7,8 Nevertheless, a causative link between increases in circulating CD8+ T cells to AAV capsid following AAV-mediated gene transfer and loss of the therapeutic transgene products has not yet been formally established and de facto our hypothesis remains under debate as so far animal models that attempted to mimic pre-existing cellular immunity in humans failed to faithfully recapitulate loss of AAV vectors or their transgene products.9,10,11
Within AAV gene transfer vectors, the genes encoding antigens of AAV are deleted and replaced with the transgene's expression cassette. Targets for CD8+ T cell-mediated destruction of AAV-transduced cells could thus only be present transiently while the virions' capsid antigens are being degraded and resulting epitopic peptides are being displayed on the cell surface upon their binding to major histocompatibility complex molecules. To assist in the design of clinical trials, we conducted a series of mouse experiments to address the following two issues. First, the argument has been made that the amount of epitopes derived from AAV capsid antigens and displayed on transfected cells does not suffice to allow for recognition by CD8+ T cells and that hence the increase in AAV-specific CD8+ T cell frequencies observed in patients that had received AAV gene transfer was caused by contamination of vector preparations with those that encapsidated parts of the AAVcap gene (Recombinant DNA Advisory Committe meeting, June 2007). Second, in some of the ongoing clinical trials AAV gene transfer recipients are being treated with transient immunosuppression (IS) to prevent reactivation of CD8+ T cells by AAV capsid.12 The longevity of AAV capsid antigens in vivo and kinetics of their degradation remain unknown thus making it difficult to render informed decisions on the duration of IS.
In mouse studies described in this manuscript, we took two approaches to determine how long CD8+ T cells can recognize AAV capsid antigens delivered by intravenously infused AAV2 vector. The basic method employed for either approach was to inject mice with AAV2 vectors and then transfer, at different times there after, splenocytes from Thy1 congenic mice that contained CD8+ T cells directed to an epitope displayed by the AAV capsid. The use of congenic mice allows one to distinguish host from donor cells with simple staining procedures followed by flow cytometry. In the first set of experiments mice were injected with our standard AAV2 vectors and received lymphocytes from AAV capsid-immune mice. In the second set of experiments mice were injected with AAV2 vectors that contained multiple copies of SIINFEKL, a potent CD8+ T cell epitope from ovalbumin, within viral protein (VP)2 of the capsid. These mice received splenocytes from OT-1 mice, which are transgenic for the SIINFEKL-specific CD8+ T cell receptor (TcR). Proliferation of AAV2 capsid- or SIINFEKL-specific donor CD8+ T cells was then assessed as a measure for the presence of the T cells' targets from the degrading AAV2 capsid. Results show that AAV2 capsid degrades rather slowly in vivo and that in the more sensitive system based on the SIINFEKL-modified vector, T cell proliferation could be triggered for at least 10 weeks following AAV2 gene transfer. Our results also show that early proliferation of antigen-inexperienced CD8+ T cells is primarily mediated by dendritic cells rather than by transduced hepatocytes. These results indicate that, provided AAV2 capsid decay rates are similar in humans, a 3–4 month course of IS may be needed to prevent T cell-mediated destruction of AAV-transduced cells.
Results
Expression of multiple copies of SIINFEKL by the AAV2(MOVA)-hF.IX capsid
An AAV2 vector that carries five copies of SIINFEKL within VP2 and expresses human factor IX as its transgene, termed AAV2(MOVA)-hF.IX, was constructed using the two plasmids shown in Figure 1a. To ensure that AAV2(MOVA)-hF.IX vector carries the inserted sequence within VP2, the vector was boiled and then probed with an anti-AAV VP1, VP2, and VP3 antibody. AAV2-hF.IX vector was used as a control. The results showed that AAV2(MOVA)-hF.IX vector and AAV2-hF.IX vectors contained comparable levels of the AAV2 VPs. The higher molecular weight of VP2 in AAV2(MOVA)-hF.IX vector compared to that in AAV2-hF.IX vector suggests the presence of the inserted SIINFEKL sequences (Figure 1b).
Figure 1.

Adeno-associated viral vector of serotype 2 (MOVA) expressing hF.IX under a hepatocyte-specific promoter [AAV2(MOVA)- hF.IX]. (a) AAV-helper plasmids expressing AAV2(MOVA) capsid consist of one plasmid expressing the AAV2 viral protein 1 (VP1) and VP3 protein with a mutated VP2 start codon and another expressing the AAV2 VP2 protein with mutated VP1 and VP3 start codons. Five copies of SIINFEKLAA sequences were inserted into the VP2-expressing plasmid right after the VP2 start codon. (b) The expression of VP1, VP2, and VP3 proteins of the AAV2(MOVA)-hF.IX vector and the AAV2-hF.IX vector was tested for by western blot. (c) C57BL/6 mice were intramuscularly injected with 2 × 1011 vector genomes of AAV2(MOVA)-hF.IX or 1 × 109 virus particles of AdC68-NP.OVA.GFP. After 14 and 10 days respectively, peripheral blood mononuclear cells were isolated and tested by intracellular cytokine staining for interferon-γ (IFN-γ).
SIINFEKL-specific CD8+ T cell responses to the AAV2(MOVA)-hF.IX
To determine whether SIINFEKL as presented by the capsid protein VP2 of AAV2(MOVA)-hF.IX can induce specific CD8+ T cell responses, C57BL/6 mice were injected intramuscularly with 2 × 1011 vector genomes of AAV2(MOVA)-hF.IX or 1 × 109 virus particles of AdC68-NP.OVA.GFP, an E1-deleted adenovirus (Ad) vector of chimpanzee serotype 68 that expresses SIINFEKL as a fusion protein with the nucleoprotein of influenza virus and green fluorescent protein as a positive control. Peripheral blood mononuclear cells were isolated 10–14 days later. Ovalbumin (OVA)-specific CD8+ T cell responses were tested upon restimulation of cells with SIINFEKL followed by intracellular cytokine staining for interferon-γ (IFN-γ). Mice injected with AdC68-NP.OVA.GFP generated a strong SIINFEKL-specific CD8+ T cell response. Mice injected with AAV2(MOVA)-hF.IX only showed marginal frequencies of SIINFEKL-specific CD8+ T cells, which failed to exceed our cutoff point for positive T cell responses tested for by intracellular cytokine staining in mice (Figure 1c). These data suggest that at the given vector dose the AAV2(MOVA)-hF.IX vector failed to stimulate detectable primary SIINFEKL-specific IFN-γ-producing CD8+ T cell responses. Comparable levels of SIINFEKL-specific CD8+ T cell responses were also detected when mice were boosted intramuscularly with 1 × 109 virus particles of AdC68-NP.OVA.GFP 6 weeks after injection of the AAV2(MOVA)-hF.IX vector or phosphate-buffered saline (PBS) (data not shown) again confirming that the AAV2(MOVA)-hF.IX vector had failed to prime a capsid-specific T cell response.
The proliferation of AAV2 capsid-specific memory CD8+ T cells in AAV2-hF.IX transduced mice
We hypothesized that AAV2 capsid-specific CD8+ T cell responses that were observed in human AAV2 gene transfer recipients7 reflected recall responses due to reactivation of AAV capsid-specific CD8+ T cells that had been induced upon natural infections and that expanded upon injection of large doses of the vector. To test whether AAV2 capsid-specific memory CD8+ T cells became reactivated upon AAV2 gene transfer, we first performed an experiment to test whether AAV2 capsid-specific memory CD8+ T cells proliferate in mice that had been injected with AAV2-hF.IX given intravenously (i.v.), a route that favors AAV2-mediated transduction of hepatocytes. The experiment was also designed to assess duration of CD8+ T cell recognition of AAV2 capsid components. Thy1.1 BALB/c mice first received the AAV2-hF.IX vector. Then at different times thereafter, AAV2-hF.IX-injected mice were adoptively transferred with carboxyfluorescein succinimidyl ester (CFSE)-labeled splenocytes from congenic Thy1.2 BALB/c mice that had been immunized with 1011 virus particles of the Ad-AAV2 capsid vectors at least 4 months earlier. Transferring in parallel donor lymphocytes into BALB/c mice that had not been injected with AAV2-hF.IX controlled the experiments. Proliferation of CFSE-labeled AAV2 capsid-specific memory CD8+ T cells of donor origin was tested 10 days later by staining splenocytes with fluorochrome-labeled antibodies to Thy1.2, CD8, and the AAV2 capsid-specific tetramer13. Donor-derived AAV2 capsid-specific memory CD8+ T cells showed marked proliferation if they were transferred into mice 2 days and 3 weeks after the AAV2-hF.IX gene transfer compared to their proliferation in naive control mice (Figure 2a,b) suggesting that AAV2 capsid-specific memory CD8+ T cells can be reactivated and proliferated upon AAV2 gene transfer. However, no proliferation was observed if the AAV2 capsid-specific memory CD8+ T cells were transferred 6 weeks after AAV2 gene transfer (Figure 2a,b), which suggests that AAV2 capsid is degraded by 6 weeks after AAV2 gene transfer to no longer supply sufficient amounts of the endogenous epitope for CD8+ T cell recognition. The gating method for the proliferation assay and the AAV2 capsid-specific tetramer staining are showed in Supplementary Figure S1.
Figure 2.

Proliferation of adeno-associated viral vector of serotype 2 (AAV2) capsid-specific memory CD8+ T cells in AAV2 vectors expressing hF.IX under a hepatocyte-specific promoter (AAV2-hF.IX) transduced mice. Thy1.1 BALB/c mice were intravenously injected with 2 × 1011 vector genomes of AAV2-hF.IX. Mice then received 5 × 107 carboxyfluorescein succinimidyl ester-labeled splenocytes from the Thy1.2 BALB/c mice, which had been immunized with 1 × 1011 virus particles of adenovirus human serotype 5 vectors expressing capsid antigens of AAV2 under a cytomegalovirus promoter (Ad-AAV2cap) at least 4 months earlier, at day 2, week 3 and week 6 following AAV2 transfer. (a) The proliferation of Thy1.2+CD8+AAV2-tet+ cells was tested 10 days later from the spleens of the recipient mice. Control mice did not receive AAV2-hF.IX before the adoptive transfer of splenocytes. Each group contains two to three control mice, results for one of those are shown. Similar results were obtained with the other control mice. (b) The average number of division cycle (proliferation index) and the percentage of cells of the original sample which divided (% divided) were calculated. Error bars represent standard deviation (SD) for 2–3 mice per group.
Proliferation of OT-1 CD8+ T cells in AAV2(MOVA) hF.IX-transduced mice
To enhance the stringency of the analyses, we incorporated SIINFEKL, a very immunodominant epitope of ovalbumin, into AAV capsid. Our initial attempts to incorporate a foreign epitope into VP3, the most abundant of the capsid proteins and the target of most CD8+ T cells, failed to result in a vector that expressed its transgene product upon intravenous transfer into C57BL/6 mice, and we therefore inserted five copies of SIINFEKLAA (SIINFEKL plus a 2-alanine linker) into VP2. The new vector, termed AAV2(MOVA)-hF.IX was used to perform a similar series of experiments as described above. Specifically, Thy1.1 C57BL/6 mice first received 5 × 1011 vector genomes of AAV2(MOVA)-hF.IX by tail vein injection. Control mice were injected with PBS. At day 2, week 2, 10, or 16, mice were injected i.v. with CFSE-labeled OT-1 mouse splenocytes, which were Thy1.2+ and contained SIINFEKL-specific CD8+ T cells that express the TcR Vα2 chain.14 Proliferation of CD8+ OT-1 splenocytes was tested 10 days later by staining for the donor Thy1.2 phenotype, CD8 and the Vα2 TcR. Strong proliferation of transgenic SIINFEKL-specific CD8+ T cells was detected if cells were injected 2 days after AAV transfer. SIINFEKL-specific CD8+ T cell proliferation gradually decreased at later time points and finally ceased by week 16 (Figure 3a,b). At week 10 time point, weak proliferation of T cells was still detected within samples from spleen and liver; at this time point the difference in T cell proliferation was only significant for cells isolated from liver. These results suggest that OT-1 splenocytes can be activated by the SIINFEKL epitope presented in the AAV2 capsid VP2 protein. They also demonstrate that an epitope within the AAV2 capsid can trigger expansion of antigen-inexperienced CD8+ T cells. The data further suggest that in vivo degradation of AAV2 capsid proceeds over an extended period of time, providing targets to CD8+ T cells directed against a potent epitope for at least 10 weeks.
Figure 3.

Proliferation of splenic CD8+ T cells from OT-1 mice in adeno-associated viral vector of serotype 2 (MOVA) vectors expressing hF.IX under a hepatocyte-specific promoter [AAV2(MOVA)-hF.IX]transduced mice. Thy1.1 C57BL/6 mice were intravenously injected with 5 × 1011 vector genomes of AAV2(MOVA)-hF.IX. Mice then received 5 × 106 CFSE-labeled splenocytes from OT-1 mice at day 2, week 2, week 10 and week 16 following AAV2 transfer. (a) The proliferation of Thy1.2+CD8+Vα2+ cells in spleens of individual mice and pooled lymphocytes isolated from livers was tested 10 days later. Control mice did not receive AAV2(MOVA)-hF.IX before splenocytes were adoptively transferred. Results are shown for one of the three control mice; similar results were obtained with the other control mice. (b) The average number of division cycle (Proliferation Index) and the percentage of cells of the original sample which divided (% divided) were calculated. Error bars represent SD for two to three mice per group. The proliferation of cells from liver was tested with pooled samples; thus p values were not calculated for the liver samples.
Stability of AAV2(MOVA)-hF.IX and AAV2-hF.IX vectors
The native AAV2 capsid did not induce proliferation of specific CD8+ T cells if splenocytes from AAV2-immune mice were injected 6 weeks after gene transfer while the SIINFEKL-modified capsid induced proliferation of OT-1 CD8+ T cells for at least 10 weeks. It is feasible that this reflects differences in the sensitivity of the two assay systems or alternatively that insertion of SIINFEKL had altered overall stability of the capsid. This was tested by assessing proliferation of memory CD8+ T cells directed to an endogenous epitope within the VP3 loop of AAV2 capsid in BALB/c mice injected with the AAV2(MOVA)-hF.IX vector. Thy1.1+ BALB/c mice were injected i.v. with AAV2(MOVA)-hF.IX or PBS as control. Two days later, CFSE-labeled splenocytes from Thy1.2+ BALB/c mice, which had been immunized with Ad-AAV2cap vector 4 months earlier were adoptively transferred into the immunized Thy1.1+ BALB/c mice. Lymphocytes were harvested 10 days later and proliferation of Thy1.2+CD8+ AAV2 tetramer+ cells was tested by staining of lymphocytes followed by flow cytometry. We detected similar levels of proliferation in mice receiving AAV2(MOVA)-hF.IX (Figure 4a) and AAV2-hF.IX (Figure 2a, 2 days). There was no significant difference in proliferation index and the percentages of cells that had divided between these two groups (Figure 4b), which suggests that the extended and enhanced proliferation of OT-1 splenocytes in mice injected with AAV2(MOVA)-hF.IX was unlikely to be a reflection of differences in AAV2 capsid stability.
Figure 4.

The stability of adeno-associated viral vector of serotype 2 (MOVA) vectors expressing hF.IX under a hepatocyte-specific promoter [AAV2(MOVA)-hF.IX] vector. Thy1.1 BALB/c mice were intravenously injected with 2 × 1011 vector genomes of AAV2(MOVA)-hF.IX or AAV2-hF.IX. On day 2, mice received 5 × 107 CFSE-labeled splenocytes from Thy1.2 BALB/c mice, which had been immunized with 1 × 1011 virus particles of adenovirus human serotype five vectors expressing capsid antigens of AAV2 under a cytomegalovirus promoter (Ad-AAV2cap) at least 4 months earlier. (a) Proliferation of Thy1.2+CD8+AAV2-tet+ cells was tested 10 days later using lymphocytes isolated from spleens. The control mice were intravenously injected with phosphate-buffered saline before splenocytes were adoptively transferred. One control mouse is shown here and similar results were obtained in other control mice. (b) The average number of division cycle (proliferation index) and the percentage of cells of the original sample which divided (% divided) were calculated. Error bars represent SD for two to three mice per group.
Presentation of AAV capsid by dendritic cells
To further explore the cell type that presents epitopes within AAV capsid to CD8+ T cells, we used B6.FBV.TG mice.15 These mice express the receptor of diphtheria toxin (DT) under the control of the CD11c promoter. Mice lack this receptor and are thus resistant to DT. Expression of the DT receptor in cells that express CD11c such as myeloid dendritic cells, the primary subset of cells able to present antigen to naive CD8+ T cells, causes transient depletion of these cells upon injection of mice with DT. Pre-experiments were conducted to ensure that DT treatment depleted CD11c+ cells from B6.FBV.TG mice (Figure 5a). For the proliferation assay, two groups of B6.FBV.TG mice were then injected i.v. with AAV2(MOVA)-hF.IX. Two days later one of the groups was treated with 4 ng/g of DT. Mice of the other group were injected with PBS. CFSE-labeled OT1-Thy1.1+ mouse splenocytes were adoptively transferred into AAV2(MOVA)-hF.IX-immunized B6.FBV.TG mice 5 hours after DT or PBS injection. The experiment was further controlled by injecting CFSE-labeled OT1-Thy1.1+ splenocytes into naive C57BL/6 recipients. Proliferation of Thy1.1+CD8+Vα2+ cells was tested 3 days later. This early time point was chosen as DT-mediated depletion of dendritic cells is very transient in B6.FBV.TG; by 5 days after drug treatment dendritic cells differentiate from bone marrow precursors and repopulate the periphery. For the same reason, cells were transferred early on day 2 after AAV2 injection; testing for cell proliferation after 3 rather than 10 days reduces the sensitivity of the assay such that by 2 weeks after AAV2 transfer proliferation could no longer be detected (data not shown).
Figure 5.

Proliferation of splenic CD8+ T cells from OT-1 mice in dendritic cell depleted adeno-associated viral vector of serotype 2 (MOVA) vectors expressing hF.IX under a hepatocyte-specific promoter [AAV2(MOVA)-hF.IX]-injected recipient mice. (a) B6.FVB.TG mice were injected with DT and splenocytes were isolated the next day. CD11c+ dendritic cells were tested from spleens of mice that were or were not treated with diphtheria toxin (DT). (b) B6.FVB.TG mice were injected with 5 × 1011 vector genomes of AAV2(MOVA)-hF.IX. Two days later, some mice were injected with 4 ng/g DT to deplete dendritic cells. 4–5 hours after DT treatment, mice received 5 × 106 CFSE-labeled splenocytes from OT1-Thy1.1 mice. Proliferation of Thy1.1+CD8+Vα2+ cells in mouse spleens was measured 3 days later. (c) The percentage of cells of the original sample which divided (% divided) and the average number of division cycle (proliferation index) were calculated. Error bars represent SD for two to three mice per group. Negative control mice were injected with splenocytes from OT1-Thy1.1 mice but did not receive AAV2(MOVA)-hF.IX.
Depletion of dendritic cells strongly reduced proliferation of SIINFEKL-specific CD8+ T cell in mice that had been injected with AAV2(MOVA)-hF.IX 2 days earlier (Figure 5b). The percentage of cells of the original sample which divided (% divided) and the average number of division cycle (proliferation index) were calculated and both parameters were decreased by about 2/3 after dendritic cell depletion (Figure 5c). These results show that dendritic cells elicit the initial proliferative response of CD8+ T cells directed against an epitope within the AAV2 capsid.
Presentation of AAV capsid by hepatocytes
To determine whether AAV2-transduced hepatocytes can elicit proliferation of AAV capsid-specific CD8+ T cells, we first transferred CFSE-labeled OT-1 mouse splenocytes into Thy1.1+ C57BL/6 mice. The following day mice were injected through the tail vein with 107 hepatocytes harvested from Thy1.2+ C57BL/6 mice that had been injected with AAV2-hF.IX or AAV2(MOVA)-hF.IX 1 week earlier. To further control the experiment, some mice were injected with hepatocytes harvested from Thy1.2+ C57BL/6 mice immunized with the AdC68-NP.OVA.GFP vector or an AdC68 vector expressing an irrelevant protein, i.e., the glycoprotein of rabies virus (rab.gp). Lymphocytes were isolated 10 days later from spleens and liver of the recipient mice and proliferation of Thy1.2+CD8+Vα2+ cells was determined. SIINFEKL-specific CD8+ T cells had proliferated in mice that had received hepatocytes from AdC68-NP.OVA.GFP-immunized mice (Figure 6b), whereas no proliferation above background was observed in recipients of hepatocytes from AdC68-rab.gp- or AAV2-hF.IX-injected mice (Figure 6a,c). SIINFEKL-specific CD8+ T cells from only one of the three mice, which had been transplanted with hepatocytes from AAV2(MOVA)-hF.IX-injected mice showed modest proliferation (Figure 6f). However, there was no statistically significant difference in the proliferation index and % divided between the mice which were transplanted with hepatocytes from AAV2(MOVA)-hF.IX or AAV2-hF.IX- or AdC68-rab.gp-injected mice (Figure 6g,h). These data indicate that hepatocytes can readily trigger T cell proliferation upon transduction with an Ad vector that encodes the T cells' cognate antigen, but appear to be only marginally involved in driving proliferation of antigen-inexperienced CD8+ T cells that recognize an epitope within AAV capsid.
Figure 6.

Proliferation of splenic CD8+ T cells from OT-1 mice in mice transplanted with hepatocytes from AAV2(MOVA)-hF.IX transduced mice. Thy1.1 C57BL/6 mice were injected with 5 × 106 CFSE-labeled splenocytes from OT-1 mice. The following day, mice then received 1 × 107 hepatocytes from Thy1.2 C57BL/6 mice, which had been injected with either 5 × 1011 vector genomes of AAV2(MOVA)-hF.IX or AAV-hF.IX, or 1 × 1011 virus particles of AdC68-NP.OVA.GFP or AdC68-rab.gp 1 week earlier. Proliferation of Thy1.2+CD8+Vα2+ cells was tested 10 days later. (a–c) The pooled results from two to three mice. (d–f) The results for three individual mice, which received hepatocytes from mice transduced with AAV2(MOVA)-hF.IX. (g) The average number of division cycle (proliferation index) and (h) the percentage of cells of the original sample which divided (% divided) were calculated. Error bars represent SD for two to three mice per group.
Discussion
Experiments described here were designed to test whether and how long epitopes derived from the capsid of an AAV2 vector could trigger expansion of specific CD8+ T cells. Although clinical trials using high doses of AAV2 for gene replacement showed expansion of T cells directed to AAV capsid antigens in blood, which ultimately may have caused rejection of AAV-transduced cells,2,7 this problem was not encountered in preclinical animals, such as hemophilic dogs, which upon AAV-mediated gene transfer achieved sustained expression of the AAV vectors' transgene.2 We argued that humans unlike most species used for preclinical testing of AAV gene transfer vectors are naturally infected with AAV together with a helper virus during childhood and thus carry memory T cells, which are more readily expanded by low doses of antigen compared to antigen-inexperienced T cells.7,16 Nevertheless, attempts to substantiate the hypothesis that rejection of high doses of AAV vectors is mediated by AAV capsid-specific memory CD8+ T cells using relevant mouse models have failed thus far. We and two other groups showed that mice with pre-existing CD8+ T cells to AAV capsid do not eliminate AAV-transduced cells, although they kill AAV-peptide pulsed cells in vivo.10,11,17 The group of Samulski et al.18 used a system similar to ours by incorporating SIINFEKL into VP3 of an AAV2 vector. This vector induced a weak CD8+ T cell response to SIINFEKL, which appeared to reduce rather than eliminate transgene expression with a time course distinct from that observed in human subjects. We conducted a similar series of experiments but our results differed. For our studies, we used AAV2 vectors that expressed five copies of SIINFEKL within VP2 of the capsid. AAV capsid epitopes identified to date in mice or humans are clustered in the sequence shared with VP319,20 the most abundant of the three capsid proteins, which is present at tenfold higher copy numbers compared to VP1 or VP2. To at least partially compensate for the lower copy number of an epitope within VP2 we incorporated five copies of SIINFEKLAA into the capsid of the AAV2(MOVA) vectors. This vector, even if used at high doses, did not induce a SIINFEKL-specific CD8+ T cell response detectable by intracellular cytokine staining for IFN-γ upon in vitro stimulation of lymphocytes. In additional experiments (data not shown), we established that the AAV2(MOVA) vector also failed to elicit a response in mice that had been transferred with CD8+ T cells from OT-1 mice nor did the vector prime for a SIINFEKL-specific CD8+ T cell response, i.e., naive mice injected with this vector failed to develop an enhanced response upon a booster immunization with an Ad vector expressing SIINFEKL (data not shown). These results are in agreement with one of our previous publications10 which showed that AAV2 vectors only induce capsid-specific CD8+ T cell responses if they encoded an immunogenic transgene product, which was furthermore linked to an agent that enhances costimulation by blockade of an immunoinhibitory pathway.21 With the caveat that we only tested for CD8+ T cells producing IFN-γ, one of the most common cytokines secreted by virus-specific CD8+ T cells and may have missed responses of T cells producing other cytokines or chemokines, the data suggest that AAV2 vectors expressing poorly immunogenic transgene products, such as hF.IX, do not induce in mice a pronounced CD8+ T cell response to their capsid antigens.
It was shown that in mice alternate reading frames of hF.IX contain an epitope that when expressed by a peptide and pulsed onto dendritic cells elicited a CD8+ T cell response, which in turn was boosted upon injection of an AAV2-hF.IX vector.22 In turn mice with pre-existing CD8+ T cells to this epitope developed lower levels of circulating hF.IX compared to control mice leading the authors to conclude that lack of sustained hF.IX expression upon AAV2-mediated gene transfer into humans was caused by CD8+ T cell to epitopes created by alternative reading frames of the transgene. Although we cannot exclude that some alternative reading frames within F.IX as expressed by AAV vectors may indeed evoke CD8+ T cell responses in patients with F.IX mutations that could interfere with sustained gene transfer, we would like to point out that the same open reading frames should be present within the human genome unless the mutation deleted an alternative start codon. Patients enrolled in the AAV2-hF.IX trial1 had defects in clotting factors due to point mutations within their F.IX gene rather than deletions and one would thus expect that their T cells should have been tolerant to F.IX and any polypeptides derived from alternative start codons.
Although epitopes within the AAV capsid failed to elicit a specific CD8+ T cell response that could be detected by functional assays for IFN-γ, they were nevertheless able to drive expansion of AAV capsid-specific memory CD8+ T cells thus providing a tool to study the in vivo kinetics of AAV capsid degradation. It should be pointed out that T cells are exquisitely sensitive to antigen. They can respond to a few copies of their cognate antigen, i.e., a peptide displayed on an major histocompatibility complex class I molecule.23 Each AAV particle contains the three VP at ratios of 1:1:10 with VP3, the most common protein being present at ~60 copies, AAV2 vectors thus contain 60 copies of each native epitope within VP3 and AAV2(MOVA) vectors contain 30 copies of SIINFEKL within VP2. Major histocompatibility complex class I peptide complexes are stable with half-lives that range from hours to several days.24 Doses of 1011 vector genomes of AAV vector as given here or above 1013 vector genomes as used in clinical trials in humans that commonly recognize more than one epitope within AAV2 capsid1,19, would thus be expected to provide ample opportunities for CD8+ T cell recognition.
To avoid destruction of AAV-transduced cells by AAV capsid-specific CD8+ T cells, clinical trials are underway that are using transient IS following AAV gene transfer.25 To limit the time of IS it is crucial to know how long the capsid proteins of AAV vectors can be recognized by T cells as this will drive the duration of IS regimens, which should be continued for the minimum time needed to achieve the therapeutic goal.
Experiments conducted here with a single-stranded AAV2 vector show that the endogenous epitope within VP3 induces proliferation of AAV capsid-specific CD8+ T cells for at least 3 weeks while proliferation ceases after 6 weeks. The system based on recognition of SIINFEKL within AAV capsid by TcR-transgenic OT-1-origin CD8+ T cells shows prolonged proliferation for at least 10 weeks, which then ceases after 16 weeks. One could argue that degradation of the capsid, which contained five copies of SIINFEKL within VP2, was delayed. We view this as unlikely as the proliferative CD8+ T cells response to the endogenous AAV2 epitope within VP3 was similar to that of the same epitope within an unmodified AAV2 capsid. Instead, we assume that the combination of a very high affinity epitope, i.e., SIINFEKL and high precursor frequencies of SIINFEKL-specific CD8+ T cells upon OT-1 cell transfer contributed to the prolonged CD8+ T cell response to the AAV2(MOVA) vector. How humans will respond to transfer of an AAV vector is expected to vary depending on the trial design, i.e., type of vector, vector dose and route of application, as well as pertinent features of the patients such as frequencies and stimulation history of their AAV capsid-specific CD8+ T cells26 and their human leukocyte antigen type, which dictates numbers and avidities of epitopes present within the AAV capsid. Provided that AAV2 degrades at a similar rate in mice and human subjects, our data indicate that even under the worst-case scenario a 4-month IS regimen should suffice to avoid immune-mediated rejection of AAV2-transduced cells upon hepatic transfer. The caveat should be added that AAV2 vectors delivered to other tissues such as the central nervous system, muscle, or cancers may show different rates of degradation as one would expect that levels and types of proteolytic enzymes that ultimately destroy the capsid antigens will vary between different types of cells. In the same token AAV2 pseudotyped with capsids from other serotypes may each show distinct rates of decay. These assumptions remain to be tested experimentally.
We attempted to define the cell substrate that drives proliferation of AAV capsid-specific CD8+ T cells in mice that received an i.v. injection of an AAV2 vector. Early after AAV2 transfer CD8+ T cell proliferation was mainly induced by dendritic cells, which most likely were transduced by the vector or acquired vector by phagocytosis. Purified hepatocytes isolated from mice that received AAV2 vectors 1 week earlier at best induced marginal proliferation of capsid epitope-specific CD8+ T cells. These data are in agreement with earlier findings that AAV capsid-specific CD8+ T cells in mice fail to eliminate AAV2 transduced hepatocytes. As the immune system of mice does not always mirror that of humans27 these results do not necessarily preclude that in humans AAV-transduced hepatocytes provide targets to AAV capsid-specific CD8+ T cells.
Other investigators are exploring alternative28 or artificial29 serotypes of AAV or double-stranded AAV vectors,30 which potentially could either evade pre-existing immune responses or could be used at lower doses, which would be assumed to also lower the likelihood of reactivation of immunological memory. For example, it has been proposed that mutations of surface-exposed tyrosine residues on AAV2 capsid reduces ubiquitination and proteasome-mediated degradation of the vector particle, which in turn results in higher efficiency of gene transfer thus allowing for a reduction of the vector dose.31 Whether changes in proteolytic degradation will reduce memory T cell responses to AAV capsid remains to be investigated in more depth. Alternative serotype or double-stranded AAV vectors may have different degradation rates, which may vary depending on the inflammatory potential of a vector, which for example may differ for single versus double-stranded AAV vectors. Accelerated degradation caused by upregulation of proteolytic enzymes under inflammatory conditions32 would be expected to exacerbate AAV capsid-specific CD8+ T cell responses by providing large doses of epitopes concurrently rather than spaced over a prolonged time. The assay systems described here may provide preclinical guidance on the likelihood of different AAV vector platforms to elicit AAV capsid-specific CD8+ T cell responses in clinical trials.
Materials and Methods
Mice. Male 6–8-week-old BALB/c mice (Thy1.1 and Thy1.2) and C57BL/6 mice (Thy1.1 and Thy1.2) were purchased from the National Cancer Institute (Bethesda, DC) or the Jackson Laboratory (Bar Harbor, ME). B6.FVB.TG mice were purchased from the Jackson Laboratory and bred at the Wistar Institute (Philadelphia, PA). Male OT-1 mice and female OT1-Thy1.1 mice were bred at the Wistar Institute and male F1 mice were used at 6–10 weeks of age. Mice were housed at the Animal Facility of the Wistar Institute and treated according to the institutional rules for animal welfare.
Vectors. The E1- and E3-deleted Ad human serotype 5 vectors expressing capsid antigens of AAV2 under a cytomegalovirus promoter (Ad-AAV2cap) and the AdC68-expressing NP.OVA.GFP or rab.gp were prepared as described before.6 The preparation of AAV2-hF.IX has also been described previously.6
The plasmids and methods used to propagate and purify AAV2(MOVA)-hF.IX were similar to those of AAV2-hF.IX,6 except that the plasmids encoding the AAV2 capsid genes differed. Two plasmids encoding the AAV2 capsid gene and the sequence of SIINFEKL within VP2 were produced to generate AAV2 vectors expressing SIINFEKL on the viral capsid. The first plasmid has a mutated VP2 start codon and only expresses AAV2 VP1 and VP3 proteins. The second plasmid has mutated VP1 and VP3 start codons and only expresses VP2 protein, in which five SIINFEKL-encoding sequences (TCGATCATCAATTTTGAGAAACTG) were inserted right after the ACG start codon into the BglII site (AGATCT). Each SIINFEKL-encoding sequence was linked by 2-alanine-encoding sequences (GCAGCT) (Figure 1a). To ensure that AAV2 vectors were not contaminated with plasmid vectors, they were treated with endonucleases and tested further by bacterial transformation assays. To ensure that AAV particles did not inadvertently encapsidate AAV rep/cap sequences vectors were designed to minimize the chance of packaging of AAV capsid sequences.33
Immunization of mice. Ad-AAV2cap vectors were diluted in sterile PBS to a total volume of 100 µl and intramuscularly injected into the hindlegs of mice. AAV2-hF.IX and AAV2(MOVA)-hF.IX vectors were diluted in 200 µl of sterile PBS and injected i.v. into the tail vein of mice.
Intracellular cytokine staining. To examine SIINFEKL-specific CD8+ T cell responses, peripheral blood mononuclear cells were isolated and incubated with the SIINFEKL peptide (AnaSpec, Fremont, CA; purity >95%) for 5 hours at 37 °C in a 5% CO2 incubator. Control cells were stimulated with an irrelevant peptide derived from Gag of HIV-1 (AMQMLKETI). Cells were surface stained with an anti-CD8 antibody conjugated to fluorescein isothiocyanate, then fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences, San Jose, CA) and stained with an anti-IFN-γ antibody conjugated to phycoerythrin. Both of the antibodies were purchased from BD Biosciences. Frequencies of cytokine-producing CD8+ T cells over all CD8+ T cells from cultures stimulated with the irrelevant peptide were subtracted from frequencies of cytokine-producing cells from cultures stimulated with the AAV or SIINFEKL peptide. Responses were considered positive if frequencies were three times or more higher than those of controls.
Western blotting of AAV2(MOVA)-hF.IX. AAV2(MOVA)-hF.IX and AAV2-hF.IX viral vectors were boiled for 10 minutes in Laemmli sample buffer (Bio-Rad, Hercules, CA). Western blot was performed and the membrane was blotted with a monoclonal antibody to AAV VP1, VP2, and VP3 (Fitzgerald Industries, Concord, MA).
In vivo proliferation assay. Lymphocytes were isolated from spleens of donor mice (Thy1.2), and red blood cells were lysed with 1× RBC Lysing Buffer (eBioscience, San Diego, CA). Cells were then labeled with 7 µmol/l CFSE using the CellTrace CFSE Cell Proliferation Kit (Invitrogen, Carlsbad, CA) and transferred to recipient mice (Thy1.1) in 200 µl PBS by tail vein injection. At 3 or 10 days after transfer, splenocytes or liver lymphocytes of recipient mice were harvested; stained with antibodies to CD8, Thy1.2, Vα2, which is the V region used by the SIINFEKL-specific TcR of OT-1 mice or the AAV2-tetramer (tet). They were then analyzed by flow cytometry. Gates were set to identify antigen-specific CD8+ T cells of donor origin. These cells were then analyzed for decreases in CFSE expression, with a decrease being indicative for cell proliferation. The percentage of cells that proliferated was calculated as follows. Cells were gated according to CFSE intensity into different populations that had undergone 1-N cycles. Numbers of cells present in each generation were determined using FlowJo (Ashland, OR). The percentage of cells of the original sample which divided (% divided) was calculated as follows:
ni is the cell number of the i-th generation (i = 1, 2,…, N).
1. The % divided (%D) is defined as
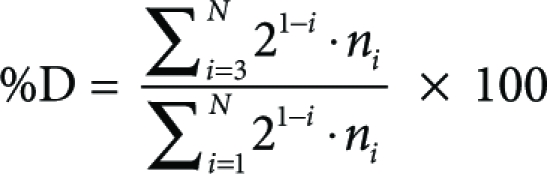 |
The average number of divisions that those cells, which didded, underwent (prolifration index, PI) was calculated using the following formula. 2. The proliferation index (PI) is defined as
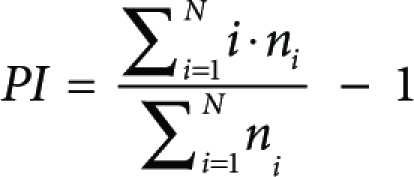 |
Dendritic cells depletion in mice. B6.FVB.TG mice, which carry the receptor for DT under control of the CD11c promoter so that treatment with DT results in transient depletion of CD11c+ dendritic cells15 were intraperitonealy injected with 4 ng/g of DT (Sigma, St Louis, MO) diluted in 200 µl PBS. To confirm depletion, mice were killed the day following DT treatment and splenocytes were isolated and stained with PE-labeled antibody to CD11c (BD Biosciences, San Jose, CA).
Hepatocytes isolation and transplantation. Mouse livers were first perfused in situ with liver perfusion medium (Invitrogen, Grand Island, NY) and then digested with liver digest medium (Invitrogen, Grand Island, NY) through a catheter inserted into the inferior vena cava with the portal vein severed. The liver was transferred into a Petri dish with hepatocyte wash medium (Invitrogen, Grand Island, NY) and cut into small pieces to release the hepatocytes. Cells were spun at 50g for 5 minutes at 4 °C and gently resuspended in 50% Percoll diluted in hepatocyte wash medium. Hepatocytes were then spun at 120g for 5 minutes at 4 °C. Live hepatocytes at the bottom were collected and washed two more times with hepatocyte wash medium and one time with PBS. On average 107 live hepatocytes were isolated from each mouse. Hepatocytes were resuspended in PBS and injected at 107 cells in 1 ml of PBS into each recipient mouse by intraperitoneal injection.
Statistics. Significance was determined by one-tailed Student's t-tests comparing results obtained with individual mice from one group to those obtained with individual mice from the other group. Significance was set at P ≤ 0.05.
SUPPLEMENTARY MATERIAL Figure S1. Gating for the proliferation of CFSE stained AAV2-specific memory CD8+ T cells.
Acknowledgments
This work was funded by P01 HL78810 from NIH. We thank Ms C. Cole for help in preparation of the manuscript, the Vector Core Facilities at Wistar, and Children's Hospital of Philadelphia for preparation of vectors.
Supplementary Material
Gating for the proliferation of CFSE stained AAV2-specific memory CD8+ T cells.
REFERENCES
- Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- Arruda VR, Schuettrumpf J, Herzog RW, Nichols TC, Robinson N, Lotfi Y, et al. Safety and efficacy of factor IX gene transfer to skeletal muscle in murine and canine hemophilia B models by adeno-associated viral vector serotype 1. Blood. 2004;103:85–92. doi: 10.1182/blood-2003-05-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog RW, Yang EY, Couto LB, Hagstrom JN, Elwell D, Fields PA, et al. Long-term correction of canine hemophilia B by gene transfer of blood coagulation factor IX mediated by adeno-associated viral vector. Nat Med. 1999;5:56–63. doi: 10.1038/4743. [DOI] [PubMed] [Google Scholar]
- Mount JD, Herzog RW, Tillson DM, Goodman SA, Robinson N, McCleland ML, et al. Sustained phenotypic correction of hemophilia B dogs with a factor IX null mutation by liver-directed gene therapy. Blood. 2002;99:2670–2676. doi: 10.1182/blood.v99.8.2670. [DOI] [PubMed] [Google Scholar]
- Scallan CD, Lillicrap D, Jiang H, Qian X, Patarroyo-White SL, Parker AE, et al. Sustained phenotypic correction of canine hemophilia A using an adeno-associated viral vector. Blood. 2003;102:2031–2037. doi: 10.1182/blood-2003-01-0292. [DOI] [PubMed] [Google Scholar]
- Brantly ML, Chulay JD, Wang L, Mueller C, Humphries M, Spencer LT, et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc Natl Acad Sci USA. 2009;106:16363–16368. doi: 10.1073/pnas.0904514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE, et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, Meulenberg JJ, Hui DJ, Basner-Tschakarjan E, Hasbrouck NC, Edmonson SA, et al. AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood. 2009;114:2077–2086. doi: 10.1182/blood-2008-07-167510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lin SW, Giles-Davis W, Li Y, Zhou D, Xiang ZQ, et al. A preclinical animal model to assess the effect of pre-existing immunity on AAV-mediated gene transfer. Mol Ther. 2009;17:1215–1224. doi: 10.1038/mt.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Murphy SL, Giles-Davis W, Edmonson S, Xiang Z, Li Y, et al. Pre-existing AAV capsid-specific CD8+ T cells are unable to eliminate AAV-transduced hepatocytes. Mol Ther. 2007;15:792–800. doi: 10.1038/sj.mt.6300090. [DOI] [PubMed] [Google Scholar]
- Wang L, Figueredo J, Calcedo R, Lin J., and, Wilson JM. Cross-presentation of adeno-associated virus serotype 2 capsids activates cytotoxic T cells but does not render hepatocytes effective cytolytic targets. Hum Gene Ther. 2007;18:185–194. doi: 10.1089/hum.2007.001. [DOI] [PubMed] [Google Scholar]
- Jiang H, Couto LB, Patarroyo-White S, Liu T, Nagy D, Vargas JA, et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood. 2006;108:3321–3328. doi: 10.1182/blood-2006-04-017913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SW, Hensley SE, Tatsis N, Lasaro MO., and, Ertl HC. Recombinant adeno-associated virus vectors induce functionally impaired transgene product-specific CD8+ T cells in mice. J Clin Invest. 2007;117:3958–3970. doi: 10.1172/JCI33138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JM, Sterry SJ, Cose S, Turner SJ, Fecondo J, Rodda S, et al. Identification of conserved T cell receptor CDR3 residues contacting known exposed peptide side chains from a major histocompatibility complex class I-bound determinant. Eur J Immunol. 1993;23:3318–3326. doi: 10.1002/eji.1830231239. [DOI] [PubMed] [Google Scholar]
- Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertl HC. Challenges of immune responses in gene replacement therapy. IDrugs. 2005;8:736–738. [PubMed] [Google Scholar]
- Li C, Hirsch M, Asokan A, Zeithaml B, Ma H, Kafri T, et al. Adeno-associated virus type 2 (AAV2) capsid-specific cytotoxic T lymphocytes eliminate only vector-transduced cells coexpressing the AAV2 capsid in vivo. J Virol. 2007;81:7540–7547. doi: 10.1128/JVI.00529-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Hirsch M, DiPrimio N, Asokan A, Goudy K, Tisch R, et al. Cytotoxic-T-lymphocyte-mediated elimination of target cells transduced with engineered adeno-associated virus type 2 vector in vivo. J Virol. 2009;83:6817–6824. doi: 10.1128/JVI.00278-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen D, Cantwell ER, O'Brien T, Johnson PA., and, Mahon BP. Adeno-associated virus serotype 2 induces cell-mediated immune responses directed against multiple epitopes of the capsid protein VP1. J Gen Virol. 2009;90 Pt 11:2622–2633. doi: 10.1099/vir.0.014175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino DE, Mingozzi F, Hui DJ, Chen H, Colosi P, Ertl HC, et al. Identification of mouse AAV capsid-specific CD8+ T cell epitopes. Mol Ther. 2005;12:1023–1033. doi: 10.1016/j.ymthe.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Li C, Goudy K, Hirsch M, Asokan A, Fan Y, Alexander J, et al. Cellular immune response to cryptic epitopes during therapeutic gene transfer. Proc Natl Acad Sci USA. 2009;106:10770–10774. doi: 10.1073/pnas.0902269106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasaro MO, Tatsis N, Hensley SE, Whitbeck JC, Lin SW, Rux JJ, et al. Targeting of antigen to the herpesvirus entry mediator augments primary adaptive immune responses. Nat Med. 2008;14:205–212. doi: 10.1038/nm1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzavecchia A., and, Sallusto F. Antigen decoding by T lymphocytes: from synapses to fate determination. Nat Immunol. 2001;2:487–492. doi: 10.1038/88678. [DOI] [PubMed] [Google Scholar]
- Hamilos DL. Antigen presenting cells. Immunol Res. 1989;8:98–117. doi: 10.1007/BF02919073. [DOI] [PubMed] [Google Scholar]
- Mingozzi F., and, High KA. Immune responses to AAV in clinical trials. Curr Gene Ther. 2007;7:316–324. doi: 10.2174/156652307782151425. [DOI] [PubMed] [Google Scholar]
- Masopust D, Ha SJ, Vezys V., and, Ahmed R. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol. 2006;177:831–839. doi: 10.4049/jimmunol.177.2.831. [DOI] [PubMed] [Google Scholar]
- Davis MM. A prescription for human immunology. Immunity. 2008;29:835–838. doi: 10.1016/j.immuni.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Vandenberghe LH., and, Wilson JM. New recombinant serotypes of AAV vectors. Curr Gene Ther. 2005;5:285–297. doi: 10.2174/1566523054065057. [DOI] [PubMed] [Google Scholar]
- Li W, Asokan A, Wu Z, Van Dyke T, DiPrimio N, Johnson JS, et al. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol Ther. 2008;16:1252–1260. doi: 10.1038/mt.2008.100. [DOI] [PubMed] [Google Scholar]
- McCarty DM. Self-complementary AAV vectors; advances and applications. Mol Ther. 2008;16:1648–1656. doi: 10.1038/mt.2008.171. [DOI] [PubMed] [Google Scholar]
- Zhong L, Li B, Mah CS, Govindasamy L, Agbandje-McKenna M, Cooper M, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci USA. 2008;105:7827–7832. doi: 10.1073/pnas.0802866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloetzel PM. The proteasome and MHC class I antigen processing. Biochim Biophys Acta. 2004;1695:225–233. doi: 10.1016/j.bbamcr.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Hauck B, Murphy SL, Smith PH, Qu G, Liu X, Zelenaia O, et al. Undetectable transcription of cap in a clinical AAV vector: implications for preformed capsid in immune responses. Mol Ther. 2009;17:144–152. doi: 10.1038/mt.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gating for the proliferation of CFSE stained AAV2-specific memory CD8+ T cells.