

Abstract
PA4608 is a single PilZ domain protein from Pseudomonas aeruginosa that binds to cyclic dimeric guanosine monophosphate (c-di-GMP). Although the monomeric structure of unbound PA4608 has been studied in detail, the molecular details of c-di-GMP binding to this protein are still uncharacterized. Hence, we determined the solution structure of c-di-GMP bound PA4608. We found that PA4608 undergoes conformational changes to expose the c-di-GMP binding site by ejection of the C-terminal 310 helix. A dislocation of the C-terminal tail in the presence of c-di-GMP implies that this region acts as a lid that alternately covers and exposes the hydrophobic surface of the binding site. In addition, mutagenesis and NOE data for PA4608 revealed that conserved residues are in contact with the c-di-GMP molecule. The unique structural characteristics of PA4608, including its monomeric state and its ligand binding characteristics, yield insight into its function as a c-di-GMP receptor.
Keywords: c-di-GMP, PA4608, PilZ, NMR, alanine scanning mutagenesis
Introduction
Bis-(3′-5′)-cyclic dimeric guanosine monophosphate (c-di-GMP) is a recently discovered bacterial second messenger. c-di-GMP is involved in various cellular functions that promote cell to cell communication and biofilm formation, inhibit cellular motility, and repress bacterial virulence.1–3 The synthesis of c-di-GMP is regulated by diguanyl cyclase and its degradation by phosphodiesterase.4 Diguanyl cyclase contains the GGDEF domain, whereas phosphodiesterase contains the EAL domain.5 Although the role of GGDEF and EAL domains was discovered several years ago, there is little information on c-di-GMP-mediated signaling pathways. Amikam and Galperin identified the PilZ domain in the sequence of bacterial cellulose synthase through bioinformatics analysis and suggested that c-di-GMP binds to PilZ domains.6 However, the molecular mechanism of c-di-GMP signaling remains to be determined.
Recently, two crystal structures of PilZ domain proteins have been solved. The structures of Vibrio cholera VCA0042 and Pseudomonas putida PP4397 illustrate the molecular details for complexes of c-di-GMP and PilZ domains. Both VCA0042 and PP4397 are composed of two domains, a YcgR-N domain at the N-terminus and a PilZ domain at the C-termius. The YcgR-N domain in these proteins has weak sequence homology with the N-terminal domain of YcgR, and its tertiary structure is similar to that of the PilZ domain of YcgR. The crystal structures of both holo-VCA0042 and holo-PP4397 showed that c-di-GMP binding triggers a change in the interdomain orientation, inducing the PilZ domain to move close to the YcgR-N domain. However, the binding stoichiometry and domain orientations are substantially different for each protein: PP4397 undergoes a dimer-to-monomer transition upon binding of two c-di-GMP molecules to one PP4397 molecule, whereas VCA0042 remains a dimer during binding of one c-di-GMP molecule to one VCA0042 molecule.7 These differences in binding stoichiometry and oligomeric state are expected to play a role in generating diverse forms of c-di-GMP-mediated regulation.8
This manuscript describes the molecular characterization of c-di-GMP binding to Pseudomonas aeruginosa PA4608. PA4608 is a single PilZ domain protein whose NMR structure (PDB ID, 1YWU) was determined previously by Ramelot.9 Here, we studied the conformational differences between the unbound and c-di-GMP bound forms of PA4608 using NMR spectroscopy. The conformation of c-di-GMP in complex with PA4608 was analyzed with 15N-filtered NOESY, and the role of phylogenetically conserved residues was investigated by mutational studies. Using these multiple approaches, our data demonstrate that the detachment of the 310 helix at its C-terminal region exposes the binding site for c-di-GMP, and suggest that PA4608 is a single-domain c-di-GMP receptor.
Results and Discussion
Overall structure of c-di-GMP bound PA4608
1H, 13C, and 15N resonances were assigned for all residues of PA4608 (1–125) except Ile69 and Glu109-Glu113. Amide resonances for these residues were not observed on the NMR timescale. The 20 final structures of c-di-GMP bound PA4608 were calculated from 1584 NOE restraints, 44 hydrogen bond restraints, 116 backbone dihedral angle restraints, and 55 RDC restraints (Table I). The restraints of c-di-GMP were not included in the calculation due to the ambiguity of c-di-GMP peak assignments. The quality of the structures was validated using PROCHECK.10
Table I.
NMR and Refinement Statistics for Protein Structures
| NMR distance and dihedral constraints | ||
|---|---|---|
| Distance constraints | ||
| Total NOE | 1567 | |
| Intra-residue | 418 | |
| Inter-residue | ||
| Sequential (|i−j| = 1) | 471 | |
| Medium-range (|i−j| < 4) | 203 | |
| Long-range (|i−j > 5) | 475 | |
| Hydrogen bonds | 44 | |
| Total dihedral angle restraints | 178 | |
| Phi | 89 | |
| Psi | 89 | |
| Total RDCs(NH)a | 55 | |
| Q-factor | 0.068 ± 0.003 | |
| Structure statistics | ||
| Violations (mean and s.d.) | ||
| Distance violation (>0.3Å) | 0 | |
| Dihedral angle violation (5°) | 0 | |
| Energies | ||
| Total energy (kcal/mol) | −4925.76 ± 58.30 | |
| Average pairwise r.m.s.d.b (Å) | ||
| Heavy | 1.31 ± 0.12 | |
| Backbone | 0.63 ± 0.09 | |
| Ramachandran statistics | All residues | Ordered regionc |
| Most favored regions | 74.3 | 90.4 |
| Additional allowed regions | 22.1 | 9.4 |
| Generously allowed regions | 2.6 | 0.2 |
| Disallowed regions | 0.9 | 0.0 |
Tensor axiality (Da): -3.688 ± 0.009e-4, Tensor rhombicity(Dr): -1.517 ± 0.023e-4.
Pairwise r.m.s.d. was calculated among 20 structures with residues 14–104.
Ordered regions: 14–24, 27–37, 40–44, 57–61, 69–79, 82–87, 92–104, and 116–120.
The structure of c-di-GMP bound PA4608 includes a six-stranded anti-parallel β-barrel, which is commonly found in hydrophobic ligand binding proteins, as well as one long α-helix and one short 310 helix at the C-terminus (Fig. 1). The secondary structures of c-di-GMP bound PA4608 are arranged β-β-β-β-β-β-α-310, which is similar to the order of unbound PA4608 except that one short 310 helix (109–113) is missing from the C-terminal region. The structure of residues 109–113 was not converged because the amide resonances on residues 109–113 are not observed on 1H-15N-HSQC. The c-di-GMP binding site, which was previously predicted by chemical shift perturbation,11 is the surface that is surrounded by the RxxxR motif (residues 10–13) and the DxSxxG motif (residues 35–40 in the β2–β3 turn).
Figure. 1.
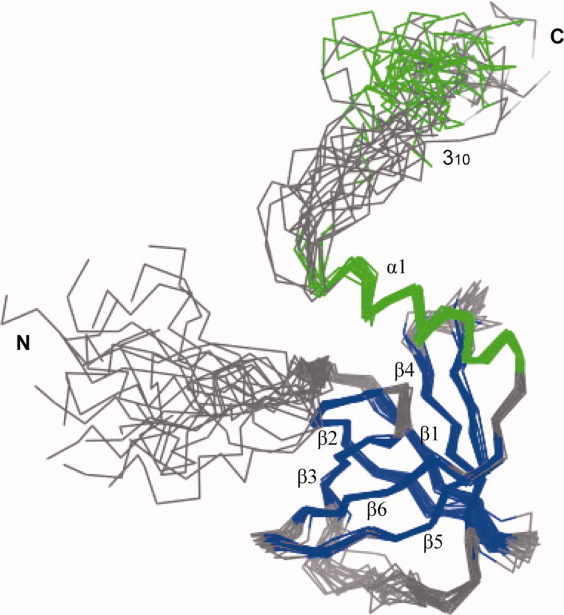
Structure of c-di-GMP bound PA4608. Superposition of the final 20 energy-minimized structures. The structures are superimposed for minimal mutual deviation of the backbone atoms. The secondary structures are color coded as follows: helices (green) and β sheets (blue).
Conformational changes upon c-di-GMP binding
Although the β-barrel structures of unbound9 and c-di-GMP bound PA4608 are very similar, a comparison between the unbound and c-di-GMP bound PA4608 structures reveals conformational changes induced by c-di-GMP binding (Fig. 2). The 310 helices in the C-terminal region undergo the most marked conformational changes. Although we do not have structural information about the first 310 helix (residues 109–113) in the c-di-GMP bound PA4608 structure (because 15N-based resonances of residues 109–113 were not observed), a large dislocation of the second 310 helix (residues 117–120) is observed between the unbound and c-di-GMP bound PA4608 structures. Opening of this 310 “lid” substantially alters the shape of the c-di-GMP binding site. In the free form, the binding site was not exposed to solvent, but became fully exposed by the 310 lid opening. The binding pocket has enough space to accommodate the two mutually intercalated c-di-GMP molecules.
Figure. 2.
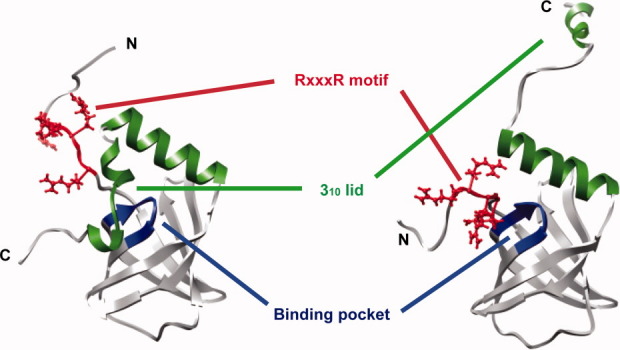
Conformational changes of PA4608 upon c-di-GMP binding. Ribbon representation of unbound PA4608 (left) and c-di-GMP bound PA4608 (right). Residues are colored as follows: RxxxR motif (red), DxSxxG motif/binding pocket (blue), and 310 helix (green).
The RxxxR motif in the N-terminal region of PA4608 also undergoes a structural change upon c-di-GMP binding. This motif moves to the binding surface that is exposed by the 310 lid opening in the c-di-GMP bound structure. In unbound PA4608, the surface of the binding site is blocked by two 310 helices (residues 109–113 and 117–120), but in c-di-GMP bound PA4608, the N- and C-terminal tails switch positions, allowing c-di-GMP to enter the binding site (Fig. 2).
NMR relaxation data (Fig. 3) show the changes in internal mobility upon c-di-GMP binding. The average 15N-NOE value of residues 11–16, including the Arg-rich motif, increased from 0.70 to 0.84, but R2 values were similar upon c-di-GMP binding, indicating that the internal mobility of residues 11–16 is decreased without a noticeable conformational fluctuation. Although residues 8–13 are not converged in Figure 1, because c-di-GMP bound PA4608 structures were calculated using only intramolecular NOEs of protein, intermolecular NOEs at guanidine group of Arg8 and Arg10 (Supporting Information Fig. 2) suggest that residues 8–13 are fixed by c-di-GMP molecule. Significant changes are observed in the 310 lid (residues 109–120), which covers the c-di-GMP binding site. Although the backbone amide resonances of residues 109–113 were not be observed, R2 values around 108–116 were increased upon c-di-GMP binding, suggesting that this stretch of residues, which includes the first 310 helix, undergoes conformational fluctuation induced by c-di-GMP binding. The other change was observed at residues 117–120, whose NOE values decreased noticeably, but whose R2 values were similar upon c-di-GMP binding. Thus, the internal mobilities of residues 117–120, which includes the second 310 helix, are increased upon c-di-GMP binding, but their tumbling is still significantly restricted by the rest of protein.
Figure. 3.
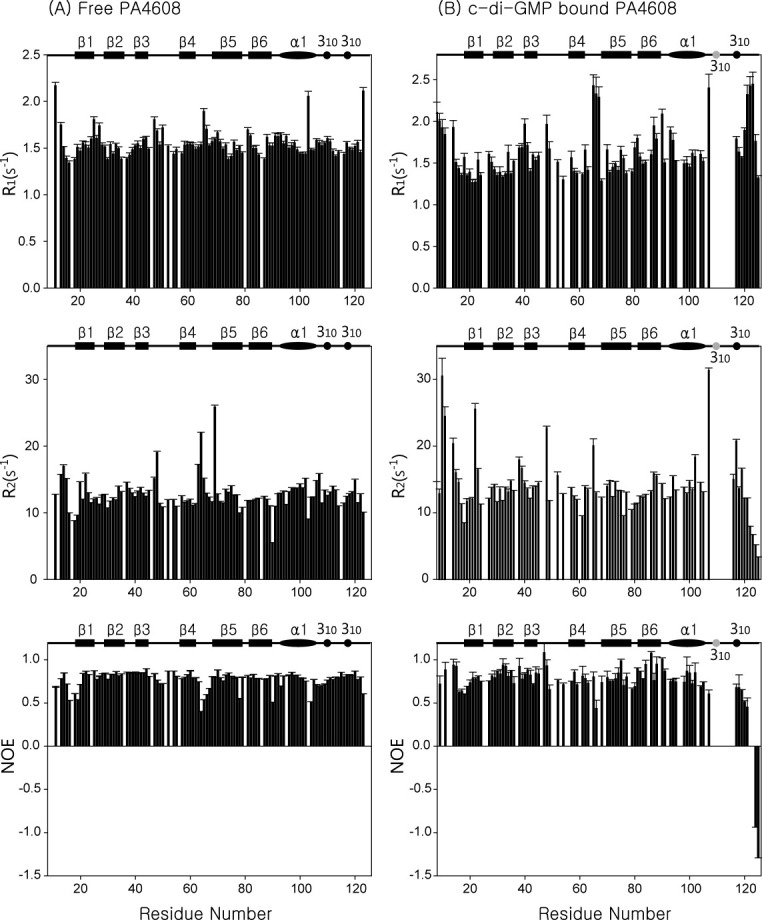
NMR relaxation data of PA4608. 15N relaxation parameters are shown for (A) unbound PA4608 and (B) c-di-GMP bound PA4608. In both panels, R1 results are in the top graph, R2 results in the middle, and 1H-15N NOE results in the bottom graph.
To confirm the participation of the 310 lid in c-di-GMP binding, we prepared the deletion mutant Δ116–125. Shorter constructs of PA4608 (Δ113–125, Δ109–125) were very unstable or not expressed at all. The imino peaks on the 1D spectrum of the complex, indicative of the interaction between deletion mutant Δ116–125 and c-di-GMP, were similar to those of the native complex (Supporting Information Fig. 3). This result indicates that the detached 310 helix (117–120) is not necessary for c-di-GMP binding.
Two mutually intercalated c-di-GMP molecules bind to PA4608
To investigate the binding ratio between PA4608 and c-di-GMP, we monitored the titration of PA4608 with c-di-GMP using 15N-HSQC spectra (results not shown). The cross peaks of PA4608 were observed to be in slow exchange until the ratio of PA4608 to c-di-GMP was 1:2 and showed no change at higher ratios. Titration results confirmed that the dissociation constant between PA4608 and c-di-GMP was less than 50 nM, as previously reported,11 and showed that the binding ratio of PA4608:c-di-GMP was 1:2.
To investigate the structure of c-di-GMP when bound to PA4608, we conducted a 1D NMR titration of c-di-GMP with PA4608 protein. Four imino peaks were observed on the 1D proton spectra upon complex formation between c-di-GMP and PA4608, and some cross peaks with sugar ring protons on the 2D 15N-filtered NOESY were observed upon binding of unlabeled c-di-GMP to 15N, 13C-labeled PA4608. The cross peaks were seen on the three imino peak resonances, but not on the peak at 13.3 ppm (Supporting Information Fig. 4). These cross peaks imply that two c-di-GMP molecules bind to PA4608 with a mutually intercalated conformation similar to that of holo-PP4397.8 Thus, based on this information and the chemical shift perturbation experiment,11 a model structure for PA4608 in complex with c-di-GMP was generated (Fig. 4). This model structure suggests that c-di-GMP is located in the binding pocket.
Figure. 4.
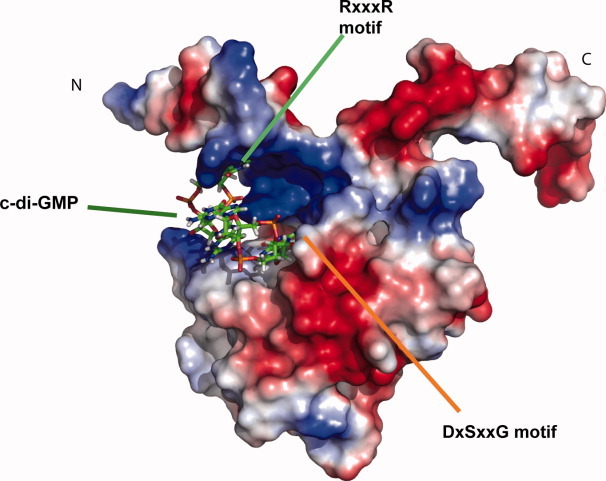
Model of PA4608: c-di-GMP complex. Surface representation of the model structure of PA4608 complexed with c-di-GMP. The c-di-GMP molecules are shown using a stick representation. The surface of PA4608 is rendered with electrostatic surface potentials using PyMOL (DeLano Scientific).
Mutagenesis studies reveal that conserved residues are critical to c-di-GMP binding
To determine the PA4608 residues that are critical for c-di-GMP binding, we prepared alanine substituted mutant proteins for conserved residues Arg9, Arg13, Asp35, Ser37, and Gly40 (Supporting Information Fig. 5), which were previously investigated by PSI-BLAST studies.6 There were problems preparing NMR samples of the D35A and S37A mutants because of difficulties with overexpression and concentration. 15N-HSQC spectra of the R9A, R13A, and G40A mutants were obtained, and their peak dispersion and pattern were similar to those of native PA4608 (Fig. 5). This result demonstrates that alanine substitution of these conserved residues does not disturb the tertiary structure of the protein. No chemical shift perturbations were observed during titration of any of the alanine mutants with c-di-GMP, suggesting that the guanidinium groups of arginines 9 and 13 are important for c-di-GMP binding. In the case of the G40A mutant, the lack of perturbation indicates that the additional CH3 group of the alanine residue prohibits c-di-GMP from binding. These titration results agreed with intermolecular NOEs from the NOESY-HSQC of the PA4608:c-di-GMP complex (Supporting Information Fig. 2). Several intermolecular NOEs of the Arg8 and Arg9 sidechain amide atoms indicate that the guanidine groups are directly involved in c-di-GMP binding. The intermolecular NOEs with moderate intensity near Gly40 support that the pocket for c-di-GMP binding is defined by this residue. The mutagenesis and intermolecular NOE results agree with the results of the previously reported chemical shift perturbation experiment of PA4608 with c-di-GMP.11
Figure. 5.

1H-15N HSQC spectra of PA4608 alanine mutants. (A) Overlapping 1H-15N spectrum of unbound native PA4608 before (red) and after (blue) addition of c-di-GMP. (B) PA4608 R13A mutant before (red) and after (green) addition of c-di-GMP. (C) PA4608 G40A mutant before (red) and after (green) addition of c-di-GMP.
Structure comparison among PilZ domains
The second messenger c-di-GMP, which has many cellular functions, binds to effector proteins and allosterically changes their structure. PilZ domain proteins are c-di-GMP receptors that seem to be altered by c-di-GMP binding. They presumably function with downstream partner proteins that remain to be identified. The PilZ domain is usually detected in multidomain proteins such as Pseudomonas putida protein PP4397 and Vibrio cholera protein VCA0042, which have the combination of a YcgR-N domain in their N-terminal region and a PilZ domain in their C-terminal region. The tertiary structures of the PilZ domains of apo-PA4608 (PDB code 1YWU),9 apo-VCA0042 (PDB code 1YLN: Zhang, R., Zhoi, M., Moy, S., Collart, F., Joachimiak, A.), and apo-PP4397 (PDB code 2GJG: Joint Center for Structural Genomics) exhibit very similar six-stranded β-barrel core structures with one α-helix. However, the C-terminal regions after the α-helix have no sequence similarity (Supporting Information Fig. 5) and no common tertiary structure (Fig. 6). Apo-PA4608 has two 310 helices in the C-terminal region, which is located at one face of the β-barrel [Fig. 6(A)]. Unlike apo-PA4608, apo-PP4397 has no additional secondary structures at its C-terminus, but does include a very extended α-helix after the β-barrel [Fig. 6(B)]. Apo-VCA0042 has an additional β-turn that is located on top of the β-barrel [Fig. 6(C)].
Figure. 6.

Structural comparison between PilZ domain proteins PA4608 and PP4397. (A) Ribbon representation of apo PA4608 (1YWU). Residues are colored as follows: RxxxR motif (red), DxSxxG motif (blue), and 310 helices (green). (B) Ribbon representation of apo PP4397 (2GJG). Residues are colored as follows: RxxxR motif (red), DxSxxG motif (blue), and long α helix in the C-terminal region (green). (C) Ribbon representation of apo VCA0042 (1YLN). Residues are colored as follows: RxxxR motif (red), DxSxxG motif (blue), and β-turn (green).
In the case of PP4397, the dimeric structure of the apo form shows that the c-di-GMP binding surface is protected by dimerization at the PilZ domain. Binding of two c-di-GMP molecules induces a dimer-to-monomer transition, and the monomeric structure of holo-PP4397 shows that the YcgR-N domain covers the c-di-GMP binding site in the monomeric state.8 In contrast to apo-PP4397, the dimeric interface of apo-VCA0042 is located in the YcgR-N domain; VCA0042 retains its dimeric state before and after binding of a single c-di-GMP molecule. The binding surface of the VCA0042 PilZ domain is not covered by the YcgR-N domain in the absence of c-di-GMP, whereas it is tightly covered as a result of the inter-domain reorientation between the YcgR-N and PilZ domains in the presence of c-di-GMP.7 The β-turn of the PilZ domain seems to stabilize the altered inter-domain orientation by inter-subunit hydrophobic interactions.
The two different conformational changes exhibited by the PP4397 and VCA0042 PilZ domains hint at an explanation of the conformational changes in PA4608 upon c-di-GMP binding. Although PA4608 retains its monomeric state upon c-di-GMP binding,11 the conformational change of PA4608 that occurs upon c-di-GMP binding is similar to that of PP4397. In PP4397, dimerization covers the c-di-GMP binding surface in the unbound form, whereas in PA4608, the 310 lid covers the c-di-GMP binding surface in the unbound form. When complexed with c-di-GMP, the N-terminal region of PA4608 covers that surface, just as the YcgR-N domain of PP4397 does. This similarity of conformational change implies that the c-di-GMP binding mode of PA4608 is similar to that of PP4397. Also, the stabilization of the altered holo-VCA002 inter-domain orientation by a β-turn interaction suggests that the hydrophobic 310 lid, which is detached from the c-di-GMP binding site upon c-di-GMP binding, would be a possible interface for interaction with a downstream protein partner. Although we found that the 310 lid of PA4608 does not participate in c-di-GMP binding in vitro, frameshift mutations in the C-terminal region of DgrA, a PA4608 homolog, induce motile dgrA loss-of-function,11 indicating that the C-terminal region is likely to be involved in interactions with downstream partners in vivo.
In conclusion, our biochemical and structural studies of PA4608 identified the binding site for c-di-GMP, which includes conserved residues. We also identified a role for the 310 lid in the C-terminal tail in covering the c-di-GMP binding site in the unbound form. The findings suggest that c-di-GMP binding to the single PilZ domain protein PA4608 is similar to that of PP4397. Furthermore, the 310 lid is a potential binding surface for unknown downstream proteins.
Materials and Methods
Expression and purification of PA4608
PA4608 (125 a.a) was cloned into the pET-15b expression vector, which encodes an additional 18 amino acids, including a six-histidine affinity tag (His-tag) at the N-terminus of the inserted sequence. The recombinant protein was expressed in BL21 (DE3) pLysS cells. The recombinant PA4608-expressing bacteria were grown in minimal M9 medium containing 15NH4Cl (to obtain 15N-labeled protein) or 13C-glucose/15NH4Cl (to obtain 15N, 13C-labeled protein) at 37°C until the optical density reached 0.6–0.8 at 600 nm. Protein expression was induced with 0.4 mM IPTG for 3 h at 30°C. The recombinant protein was purified using Ni-NTA affinity chromatography. The His-tag was removed by thrombin digestion, and the protein was purified by ion-exchange chromatography and gel filtration chromatography. Three times molar excess of c-di-GMP was added to PA4608; residual c-di-GMP was removed by gel filtration chromatography. The PA4608 concentration in the final NMR sample was about 1.2 mM in a buffer of 20 mM sodium phosphate (pH 7.0), 10 mM magnesium chloride, and 10 mM DTT in 90%/10% H2O/D2O.
Structure calculation of c-di-GMP bound PA4608
All NMR data were obtained at 298 K using Varian INOVA 600 MHz or Bruker Avance 900 MHz spectrometers. 1H-15N HSQC, HNCACB, CBCA(CO)NH, HNHA, HNCO, HN(CA)CO, (H)CC(CO)NH, H(CC) (CO)NH, HCCH-COSY, HCCH-TOCSY, CCH-TOCSY, 2D 15N-filtered NOESY (τm = 150 msec), 3D 15N-edited NOESY-HSQC (τm = 100 msec), and 3D 13C-edited NOESY-HSQC (τm = 100 msec) spectra were acquired for the structure analysis. All FIDs were processed by NMRPipe.12 All spectra and assignments were manually analyzed with Sparky.13 Backbone dihedral angle restraints were derived from chemical shifts of the backbone atoms using TALOS+.14 Hydrogen bond restraints were determined by recording 1H-15N HSQCs with H-D exchange experiments. Residual dipolar coupling (RDC) restraints were measured with 1H-15N IPAP HSQCs using PF1 phage. Structure calculations were performed using CYANA 2.1.15 To improve the qualities of structures, RDC and explicit water refinement16,17 were achieved using Xplor-NIH 2.9.6.18 RDC quality was checked using the program PALES19 and plotted in Supporting Information Figure 1. The final 20 energy-minimized structures with the lowest target function energy were selected for further analysis. The restraints used for the final structure refinement are listed in Table I.
Molecular modeling of PA4608 complex with c-di-GMP
A structural model of the PA4608/c-di-GMP complex was generated using the software Discovery Studio 2.1 (Accelrys). The structure of c-di-GMP from the c-di-GMP/holo-PP4397 complex (PDB code 3KYF) was used as the initial structure of the ligand. Based on the contact points from previous chemical shift perturbation data,11 this c-di-GMP structure was manually docked into the binding site of our solution structure of c-di-GMP bound PA4608. To eliminate steric clashes, energy minimization of the modeled structure was performed until 0.01 kcal/mol/Å. The CHARMM forcefield was employed for energy minimization of the complex. Figures were prepared with the software Chimera20 and Pymol.21
NMR dynamics
15N relaxation measurements were made using two-dimensional, proton-detected heteronuclear NMR experiments in Varian Biopack. T1 and T2 relaxation were recorded with spectral widths of 9000 Hz sampled over 1024 complex points in the w2 (1H) dimension, and 1700 Hz over 128 complex points in the w1 (15N) dimension, with 12 scans for each increment in the indirect dimension. The relaxation delay for T1 and T2 measurements was 2 sec. T1 values were measured in a series of spectra with relaxation delays of 50, 100, 150, 200, 250, 300, 400, 500, 700, and 900 msec. T2 measurements were made with relaxation delays of 10, 30, 50, 70, 90, 110, 130, and 150 msec. The heteronuclear steady-state NOE spectra were acquired with a spectral width of 9000 Hz over 1024 complex points in the w2 (1H) dimension and 1700 Hz over 128 complex points in the w1 (15N) dimension. The proton saturation period was 5 sec. All the spectra were processed and analyzed with NMRPipe and Sparky.
NMR titration experiments with PA4608 mutants
PA4608 alanine substituted mutants (R9A, R13A, D35A, S37A, and G40A) were prepared using the Quick-Change protocol (Stratagene). Like the wild-type protein, the PA4608 alanine mutant proteins were expressed in BL21 (DE3) pLysS cells. Strains were cultured, protein expression was induced, and proteins were purified as described above. The protein concentration was 0.2 mM in 500 μL of a buffer containing 90% H2O/10% D2O, 20 mM sodium phosphate (pH 7.0), 100 mM sodium chloride, and 1 mM DTT. Titration samples were prepared with protein:c-di-GMP ratios of 1:0, 1:1, and 1:2. All 1H-15N HSQC spectra were recorded at 25°C on Varian INOVA 600 MHz spectrometers equipped with triple-resonance (1H/13C/15N) probes. A PA4608 C-terminal deletion mutant (Δ116–125) was amplified from the original PA4608 expression vector using appropriate primers, cloned into the pET-15b expression vector, and expressed and purified as described above. The protein concentration was 0.1 mM in 500 μL of a buffer containing 90% H2O/10% D2O, 20 mM sodium phosphate (pH 7.0), 100 mM sodium chloride and 1 mM DTT. 1D spectra with water suppression by echo-antiecho pulse were acquired for the PA4608 C-terminal deletion mutant by adding c-di-GMP with protein:c-di-GMP ratios of 1:0, 1:1, and 1:2.
Acknowledgments
This work was supported by the KAIST High Risk High Return Project (HRHRP) and grant from High Field NMR Research Program of Korea Basic Science Institute, which is supported by the Bio NMR Research Program of the Korean Ministry of Science and Technology (E28070). We thank Melissa Stauffer, PhD, of Scientific Editing Solutions, for editing the manuscript.
Supplementary material
References
- 1.Lim B, Beyhan S, Meir J, Yildiz FH. Cyclic-diGMP signal transduction systems in Vibrio cholerae: modulation of rugosity and biofilm formation. Mol Microbiol. 2006;60:331–348. doi: 10.1111/j.1365-2958.2006.05106.x. [DOI] [PubMed] [Google Scholar]
- 2.Cotter PA, Stibitz S. c-di-GMP-mediated regulation of virulence and biofilm formation. Curr Opin Microbiol. 2007;10:17–23. doi: 10.1016/j.mib.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Jenal U, Malone J. Mechanisms of cyclic-di-GMP signaling in bacteria. Annu Rev Genet. 2006;40:385–407. doi: 10.1146/annurev.genet.40.110405.090423. [DOI] [PubMed] [Google Scholar]
- 4.Christen B, Christen M, Paul R, Schmid F, Folcher M, Jenoe P, Meuwly M, Jenal U. Allosteric control of cyclic di-GMP signaling. J Biol Chem. 2006;281:32015–32024. doi: 10.1074/jbc.M603589200. [DOI] [PubMed] [Google Scholar]
- 5.Ryjenkov DA, Simm R, Romling U, Gomelsky M. The PilZ domain is a receptor for the second messenger c-di-GMP: the PilZ domain protein YcgR controls motility in enterobacteria. J Biol Chem. 2006;281:30310–30314. doi: 10.1074/jbc.C600179200. [DOI] [PubMed] [Google Scholar]
- 6.Amikam D, Galperin MY. PilZ domain is part of the bacterial c-di-GMP binding protein. Bioinformatics. 2006;22:3–6. doi: 10.1093/bioinformatics/bti739. [DOI] [PubMed] [Google Scholar]
- 7.Benach J, Swaminathan SS, Tamayo R, Handelman SK, Folta-Stogniew E, Ramos JE, Forouhar F, Neely H, Seetharaman J, Camilli A, Hunt JF. The structural basis of cyclic diguanylate signal transduction by PilZ domains. EMBO J. 2007;26:5153–5166. doi: 10.1038/sj.emboj.7601918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ko J, Ryu KS, Kim H, Shin JS, Lee JO, Cheong C, Choi BS. Structure of PP4397 reveals the molecular basis for different c-di-GMP binding modes by Pilz domain proteins. J Mol Biol. 2010;398:97–110. doi: 10.1016/j.jmb.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Ramelot TA, Yee A, Cort JR, Semesi A, Arrowsmith CH, Kennedy MA. NMR structure and binding studies confirm that PA4608 from Pseudomonas aeruginosa is a PilZ domain and a c-di-GMP binding protein. Proteins. 2007;66:266–271. doi: 10.1002/prot.21199. [DOI] [PubMed] [Google Scholar]
- 10.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 11.Christen M, Christen B, Allan MG, Folcher M, Jeno P, Grzesiek S, Jenal U. DgrA is a member of a new family of cyclic diguanosine monophosphate receptors and controls flagellar motor function in Caulobacter crescentus. Proc Natl Acad Sci USA. 2007;104:4112–4117. doi: 10.1073/pnas.0607738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 13.Goddad TDKD. SPARKY 3. San Francisco: University of California; 2004. [Google Scholar]
- 14.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 16.Nabuurs SB, Nederveen AJ, Vranken W, Doreleijers JF, Bonvin AM, Vuister GW, Vriend G, Spronk CA. DRESS: a database of Refined solution NMR structures. Proteins. 2004;55:483–486. doi: 10.1002/prot.20118. [DOI] [PubMed] [Google Scholar]
- 17.Linge JP, Williams MA, Spronk CA, Bonvin AM, Nilges M. Refinement of protein structures in explicit solvent. Proteins. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]
- 18.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 19.Zweckstetter M, Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]
- 20.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 21.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific; 2002. http://www.pymol.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.