

Abstract
Factor for inversion stimulation (FIS), a 98-residue homodimeric protein, does not contain tryptophan (Trp) residues but has four tyrosine (Tyr) residues located at positions 38, 51, 69, and 95. The equilibrium denaturation of a P61A mutant of FIS appears to occur via a three-state (N2 ⇆ I2 ⇆ 2U) process involving a dimeric intermediate (I2). Although it was suggested that this intermediate had a denatured C-terminus, direct evidence was lacking. Therefore, three FIS double mutants, P61A/Y38W, P61A/Y69W, and P61A/Y95W were made, and their denaturation was monitored by circular dichroism and Trp fluorescence. Surprisingly, the P61A/Y38W mutant best monitored the N2 ⇆ I2 transition, even though Trp38 is buried within the dimer removed from the C-terminus. In addition, although Trp69 is located on the protein surface, the P61A/Y69W FIS mutant exhibited clearly biphasic denaturation curves. In contrast, P61A/Y95W FIS was the least effective in decoupling the two transitions, exhibiting a monophasic fluorescence transition with modest concentration-dependence. When considering the local environment of the Trp residues and the effect of each mutation on protein stability, these results not only confirm that P61A FIS denatures via a dimeric intermediate involving a disrupted C-terminus but also suggest the occurrence of conformational changes near Tyr38. Thus, the P61A mutation appears to compromise the denaturation cooperativity of FIS by failing to propagate stability to those regions involved mostly in intramolecular interactions. Furthermore, our results highlight the challenge of anticipating the optimal location to engineer a Trp residue for investigating the denaturation mechanism of even small proteins.
Keywords: intermediates, protein folding, dimer, fluorescence, FIS
Introduction
The aromatic amino acid tryptophan (Trp) is the intrinsic fluorescent probe most used to monitor protein conformational change, dynamics, and intermolecular interactions due to its indole chromophore, which is very sensitive to its environment.1 An advantage offered by Trp is that it may be selectively excited to exclude the fluorescence contribution of tyrosine (Tyr) residues. Thus, Trp has been engineered into the structure of many proteins, usually by replacing a phenylalanine (Phe) or Tyr residues to preserve the hydrophobic interactions, residue size, and aromatic properties.2–9 In this study, Trp residues were used to probe the denaturation mechanism of the Escherichia coli factor for inversion stimulation (FIS), a predominantly α-helical homodimeric DNA-binding protein of 98 residues found in many bacteria.10–13 FIS is involved in a variety of functions, such as stimulation of certain site-specific DNA recombination events,14–19 modulation of DNA topology,20,21 and regulation of transcription of numerous genes.22–25 The C-terminus of FIS, which is comprised of the C- and D-helices, has a high content of positive charge and is involved in DNA-binding.26,27 The N-terminus of FIS, which is comprised of the A-helix and the preceding 26 residues, is involved in the stimulation of Hin-mediated DNA inversion.26 The dimer interface comprises most of the hydrophobic core and involves mainly the interaction of Helix B from one subunit with the A- and B-helices of another FIS subunit (Fig. 1). Conveniently for our studies, FIS lacks disulfide bonds or cofactors and unfolds reversibly.28
Figure. 1.
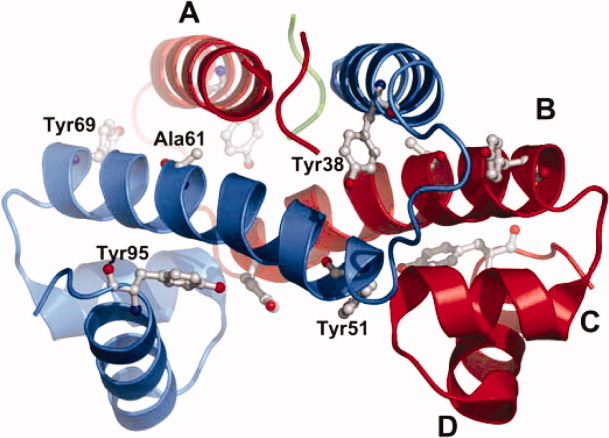
Ribbon model representation of the X-ray crystal structure of P61A FIS (PDB file: 1FIP). The first 25 residues of each monomer are flexible and do not resolve in the crystal structure. The tyrosine residues are highlighted and the four α-helices are labeled A through D. This figure was prepared using the software PyMol (The PyMol Molecular Graphics System, Version 1.3, Schrodinger, LLC.).
FIS has a proline (Pro) residue located at position 61 in the middle of Helix B that is fully conserved among 35 different bacteria examined.13 This Pro was thought to be largely responsible for the 20° kink in Helix B observed in the crystal structure, but the crystal structure of the mutant P61A FIS (Fig. 1) showed that its replacement with alanine reduced the angle of the kink by only 4°.29 Therefore, although Pro61 may support the kink of Helix B, the deformation of the helix is mainly caused by other structural factors. To investigate the role of the P61A mutation on the stability and equilibrium denaturation pathway of FIS, this mutant was studied by circular dichroism (CD) and fluorescence anisotropy.30 Initial equilibrium denaturation studies revealed that the unfolding curves for P61A FIS appeared consistent with a two-state (N2 ⇆ 2U) denaturation model.30 However, the slope (m-value) of the transition decreased as the concentration of P61A FIS increased, suggesting a more complex denaturation mechanism. Global analysis of all the unfolding curves revealed that the denaturation mechanism of P61A FIS was consistent with a three-state model involving a dimeric intermediate (N2 ⇆ I2 ⇆ 2U) and yielded a ΔGH2O for unfolding of 18.6 kcal/mol, 4 kcal/mol higher than that of WT FIS. Interestingly, the dimeric intermediate retained about two-thirds (12.4 kcal/mol) of the protein stability, which is consistent with most of the protein core being part of the monomer–monomer interface. Although the structure of the dimeric intermediate is not known, the crystal structure of native FIS shows that except for a few intermolecular interactions between the end of Helix C and the beginning of Helix B, the C-terminus of FIS is largely involved in intramolecular interactions (Fig. 1).
FIS does not contain Trp residues but has four Tyr residues located at positions 38, 51, 69, and 95 that are highly conserved in all known sequences of FIS.13 To obtain direct structural evidence about the denaturation mechanism of P61A FIS, and to further characterize the role of the C-terminus, each Tyr residue was individually mutated to Trp. The FIS crystal structure shows that the Tyr residues are distributed throughout the protein in very different environments, with each Tyr being involved in different intramolecular and intermolecular interactions (Fig. 1). Tyr38 is located near the end of Helix A and has the side chain hydroxyl group buried inside a hydrophobic pocket, with no hydrogen-bonding partner.13 Tyr51 is located near the beginning of Helix B, which is close to the C-terminus in three-dimensional space, and is involved in intermolecular hydrogen-bonding interactions with charged side chains of the opposite subunit.13 Tyr69 is highly solvent-exposed and is located near the end of Helix B where it is involved in an intermolecular ring-stacking interaction with Phe39.13 Tyr95 is positioned in the highly charged C-terminus, where it is partially buried and involved in intramolecular hydrogen bonds with lysine 94 and glutamic acid 59.13 This heterogenous environment of Tyr residues in FIS provides a convenient opportunity for engineering single Trp replacements at different positions that should serve as reporter fluorophores for the local conformational changes associated with the denaturation of P61A.
For this study, the equilibrium denaturation of three FIS double mutants (P61A/Y(38, 69, or 95)W) was studied by far ultraviolet (far-UV) CD and fluorescence spectroscopies. The mutant P61A/Y51W was omitted, because it could not be purified. Tyr51, which is located in Helix B and is involved in the electrostatic network involving the B- and D-helices, cannot be replaced with Trp without causing steric repulsion and severely destabilizing the protein. The denaturation studies with the remaining Trp mutants revealed that their ability to clearly detect the formation of the dimeric intermediate proceeds in sequential order: P61A/Y38W > P61A/Y69W > P61A/Y95W FIS. Although this trend was unexpected, it provided direct evidence for a dimeric intermediate in P61A FIS involving a disrupted C-terminus, and further suggested accompanying conformational changes near Trp 38 and 69. Of broader significance, these results provide insight on how the location of Trp residues might be better selected and exploited for probing conformational changes in proteins.
Results and Discussion
Structural environment of tryptophan 38, 69, and 95 in P61A FIS
To assess the suitability of the Trp replacements of Tyr38, 69, or 95 as fluorescent probes in our study, we compared the native and unfolded spectra of the three P61A/Y#W double mutants (# = 38, 69, or 95) and inspected the molecular environment of the engineered tryptophan residues in the crystal structure of P61A FIS.29
The fluorescence emission spectrum for P61A/Y38W FIS [Fig. 2(A)] show that under native conditions, the Trp residue exhibits much higher fluorescence than the corresponding unfolded state. In addition, the wavelength of maximum fluorescence (λmax) for native P61A/Y38W FIS is 329 nm, but upon unfolding undergoes a 24-nm Stokes shift to 353 nm, indicating that in the native state, Trp38 is buried in a nonpolar environment protected from solvent quenching [Fig. 3(A)].5 Because of its larger nonpolar side chain, Trp38 should reach deeper than Tyr into the surrounding hydrophobic pocket, consistent with its fluorescence emission behavior. Trp38 is expected to make intramolecular contacts with Phe39, Leu42, Val47, Leu50, and Leu53. The absence of contacts with the C-terminus and the large changes in fluorescence upon the denaturation of P61A/Y38W FIS suggest that Trp38 should be an excellent probe to monitor dimer dissociation.
Figure. 2.
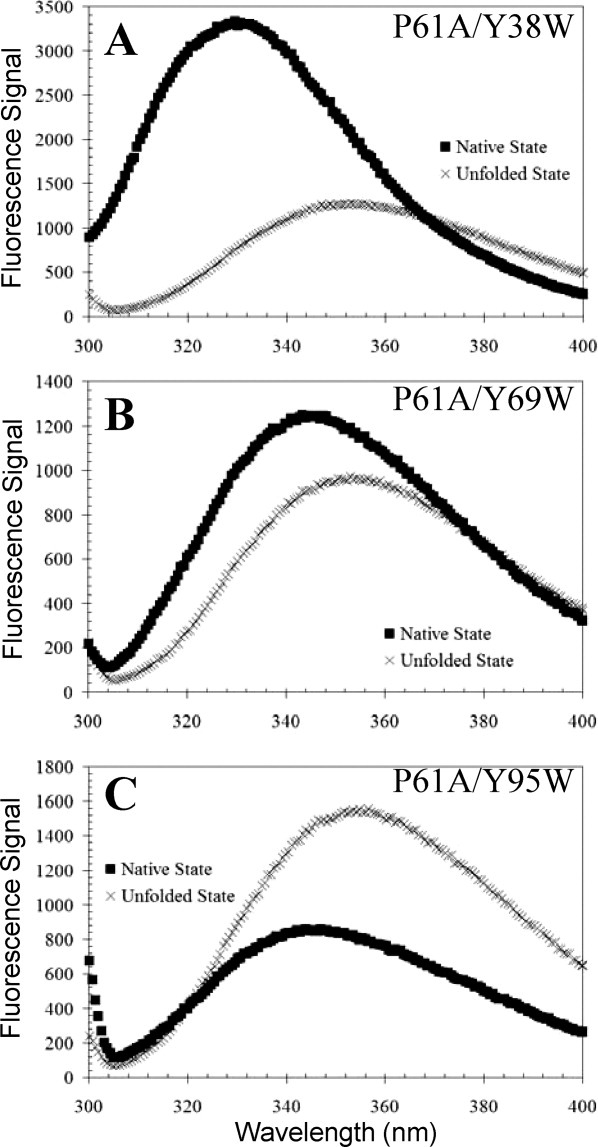
Fluorescence spectra of P61A/Y38W (A), P61A/Y69W (B), and P61A/Y95W (C) FIS were collected at 20°C under native and denaturing conditions. Emission spectra for all mutants were collected using an excitation wavelength of 295 nm in 10 mM PB at pH 7.4 with 0.1M NaCl for native conditions and 10 mM PB, 8M GuHCl at pH 7.4 with 0.1M NaCl for denaturing conditions. The concentration of the mutant proteins was 36 μM.
Figure. 3.
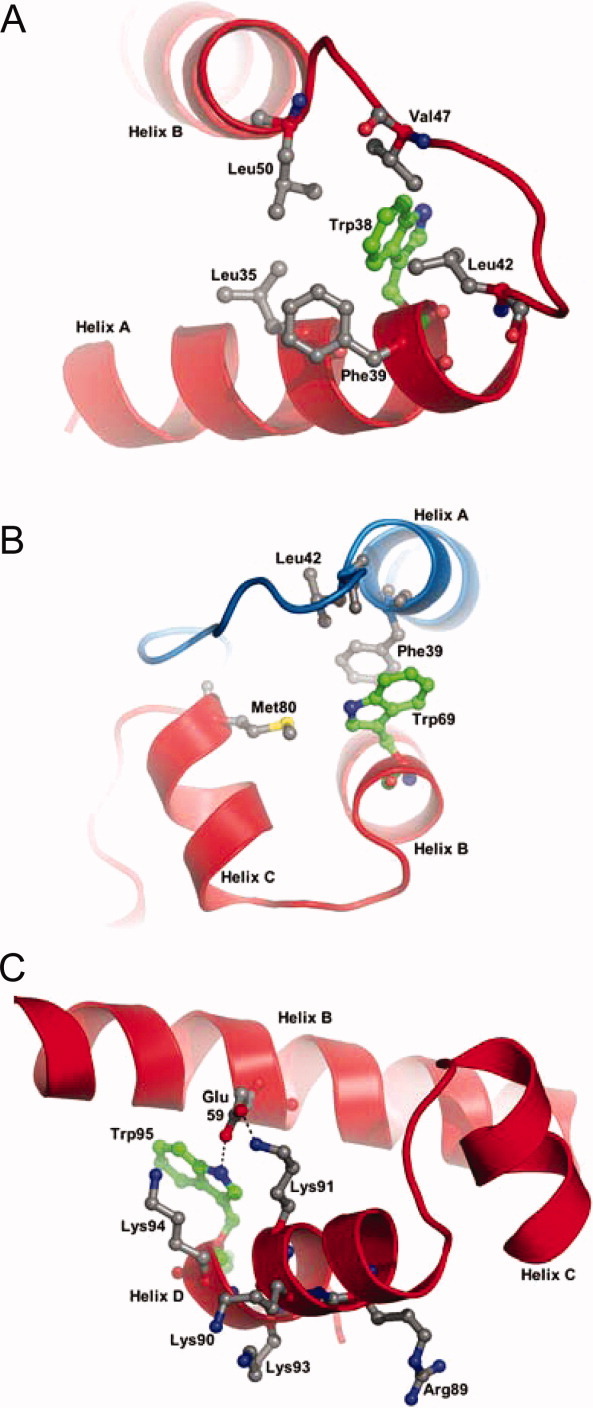
Structural environment around substituted tryptophan residues: Trp38 (A), Trp69 (B), and Trp95 (C) in P61A FIS. Replacements of tyrosines were made in silico in the structure of P61A FIS (1FIP) using the software PyMol. Residues that are proposed as interacting with tryptophan residues are labeled. Ribbon backbones of different subunits are colored differently. Hydrogen bonding in panel C is represented by a dotted line.
The fluorescence intensity of P61A/Y69W FIS is weaker than that of P61A/Y38W FIS, and therefore, decreases modestly upon unfolding [Fig. 2(B)]. In addition, the λmax shifts from 345 to 354 nm upon unfolding of P61A/Y69W, which is consistent with Trp69 being mostly solvent exposed in the native state. Inspection of an in silico replacement of Tyr69 with Trp in the structure of P61A FIS suggests that the intermolecular ring stacking interaction with Phe39 should be preserved [Fig. 3(B)]. Therefore, it appeared that Trp69 could serve as an adequate probe for dimer dissociation.
P61A/Y95W FIS displays lower fluorescence intensity in the native state than the unfolded state [Fig. 2(C)]. This suggests that the fluorescence of Trp95 is quenched in the native state, perhaps by nearby charged residues, such as Lys94 and Lys91 [Fig. 3(C)]. In addition, a shift of only 11 nm between the λmax of the native (345 nm) and unfolded (356 nm) states for P61A/Y95W FIS is consistent with Trp95 being in a polar environment largely exposed to the solvent. Analysis of an in silico Y95W mutation in the P61A structure suggests that Trp95 may not fit as well in the space designated for the native Tyr residue due to the bulkier side chain of Trp. In addition, Trp95 lacks the hydroxyl of Tyr and cannot form the hydrogen bonding interaction with Lys94 that is present in native FIS. This is consistent with a slight destabilizing effect (1.3 kcal/mol) caused by Y95W (Table I). Trp95 should still be able to form a hydrogen bond with Glu59 (on Helix B of the same chain) via its indole group [Fig. 3(C)]. Thus, the increase in P61A/Y95W fluorescence upon unfolding was expected to monitor the denaturation of the C-terminus.
Table I.
Global Analysis of Equilibrium Denaturation Data of WT, P61A, and P61A/Y(38, 69, or 95)W FIS Mutantsa
| Protein | Denaturant | ΔGN2 ⇆ I2 (kcal/mol) | mN2 ⇆ I2 [kcal/(mol M)] | ΔGI2 ⇆ 2U (kcal/mol) | mI2 ⇆ 2U [kcal/(mol M)] | ΔGN2 ⇆ 2U (kcal/mol) | mN2 ⇆ 2U [kcal/(mol M)] |
|---|---|---|---|---|---|---|---|
| WTb | GuHCl | 13.1 ± 0.2 | 4.7 ± 0.2 | ||||
| WTb | Urea | 14.7 ± 1.7 | 2.4 ± 0.1 | ||||
| P61Ac | GuHCl | 4.9 ± 0.2 | 1.9 ± 0.08 | 11.5 ± 0.2 | 1.9 ± 0.07 | 16.4 ± 0.3 | 3.8 ± 0.1 |
| P61Ab | Urea | 6.1 ± 0.3 | 1.03 ± 0.05 | 12.5 ± 0.4 | 1.10 ± 0.06 | 18.6 ± 0.5 | 2.13 ± 0.08 |
| P61A/Y38Wc | GuHCl | 6.5 ± 0.1 | 2.5 ± 0.05 | 15.8 ± 0.2 | 2.9 ± 0.05 | 22.3 ± 0.2 | 5.4 ± 0.07 |
| P61A/Y69W | GuHCl | 4.4 ± 0.1 | 2.3 ± 0.06 | 13.3 ± 0.1 | 2.1 ± 0.03 | 17.7 ± 0.1 | 4.4 ± 0.07 |
| P61A/Y95W | GuHCl | 4.0 ± 0.1 | 1.5 ± 0.03 | 11.1 ± 0.1 | 1.7 ± 0.3 | 15.1 ± 0.1 | 3.2 ± 0.3 |
Denaturation curves of WT and P61A were fit to a N2 ⇄ 2U denaturation mechanism. Denaturation curves of P61A and P61A/Y#W mutants were fit to a N2 ⇄ I2 ⇄ 2U denaturation mechanism. Most FIS variants were analyzed by CD and fluorescence spectroscopy, except P61A FIS, which was not amenable to fluorescence experiments due to its low Tyr fluorescence.
GuHCl-induced denaturation of P61A FIS monitored by far-UV CD
Previous studies of WT and P61A FIS were carried out using urea as the denaturant.28,30 As preliminary studies showed that urea could not fully denature P61A/Y38W FIS, we used the more powerful denaturant GuHCl. To facilitate comparisons between the P61A/Y#W mutants and P61A, the GuHCl-induced denaturation of P61A FIS was studied first. Tyr fluorescence was not a reliable method to monitor the unfolding of P61A FIS due to small changes in fluorescence and very steep pre transition and post-transition baselines. Therefore, the unfolding was monitored at different protein concentrations using far-UV CD (Fig. 4). As was previously demonstrated when using urea, the GuHCl-induced denaturation curves of P61A exhibited a decreasing slope with increasing protein concentration, indicating that the mechanism is more complex than two-state. Indeed, global fitting of the data shown in Figure 4 was consistent with the same three-state model (N2 ⇆ I2 ⇆ 2U) with a ΔG for unfolding of 16.4 kcal/mol, which is about 2 kcal/mol lower than that obtained for P61A using urea (Table I).
Figure. 4.
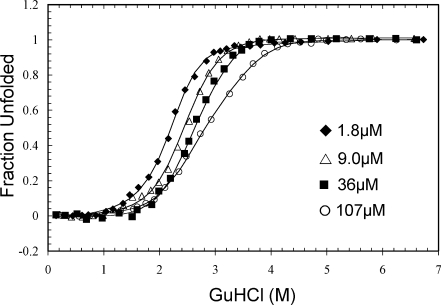
Representative GuHCl-induced denaturation data of P61A FIS monitored by far-UV CD. The solid line curve fits were generated by global fitting the data using Excel and are based on the N2 ⇆ I2 ⇆ 2U denaturation mechanism (Table I). The data are displayed as fraction unfolded, which is defined by Eq. (2). These denaturation experiments were all performed in 10 mM PB at pH 7.4 with 0.1M NaCl at 20°C.
The CD-monitored unfolding curves for P61A FIS exhibit a smaller degree of concentration-independence in the lower half of the unfolding transition, which is indicative of an intermediate step in the unfolding process (N2 ⇆ I2). The transitions are clearly concentration-dependent at higher GuHCl concentrations, consistent with a subsequent dimer dissociation step (I2 ⇆ 2U). As only one of the two overlapping steps is dependent on protein concentration, the apparent m-value derived from two-state fits decreases with increasing protein concentration.30 At the highest protein concentration used (107 μM), the denaturation curve has a slight biphasic profile that indicates a significant population of dimeric intermediate. This minor biphasic profile was not seen in the urea-induced denaturation curves for P61A FIS.30 Perhaps the high ionic strength of GuHCl destabilizes the electrostatic network involving the C-terminus of FIS, thereby decoupling the N2 ⇆ I2 and I2 ⇆ 2U transitions. A similar effect with GuHCl was previously observed for engineered cysteine mutants of FIS.31 This would also be consistent with the 1.6 and 2.2 kcal/mol higher stability of WT and P61A, respectively, when using urea as the denaturant (Table I).
The denaturation cooperativity of P61A/Y95W FIS is strongly dependent on protein concentration
It was expected that having a Trp residue at the C-terminus would allow clear detection of the P61A FIS dimeric intermediate involving disrupted C- and D-helices, because residue 95 is part of Helix D. At concentrations of P61A/Y95W FIS ranging from 1.8–107 μM, the fluorescence denaturation transitions appear to be two-state with modest concentration-dependence [Fig. 5(A)]. In contrast, when the denaturation of the protein was monitored by far-UV CD, the beginning of the transition showed little concentration-dependence, whereas the latter part of the transition was highly concentration-dependent [Fig. 5(B)]. The CD data is consistent with a denaturation mechanism involving a dimeric intermediate in which at least one-third of the secondary structure is lost, followed by dimer dissociation and unfolding of the rest of the protein. When comparing the fluorescence and CD data, it can be seen that at 1.8 μM, the transitions are not significantly different [Fig. 5(A,B)]. However, at 107 μM, the fluorescence-detected transition largely matches the first-half of the CD transition, indicating that this first denaturation step involves the disruption of the C-terminus [Fig. 5(C)]. As the second CD-monitored transition is concentration-dependent, the Cm moves to the right with increasing protein concentration, thereby decoupling it from the concentration-independent transition.
Figure. 5.
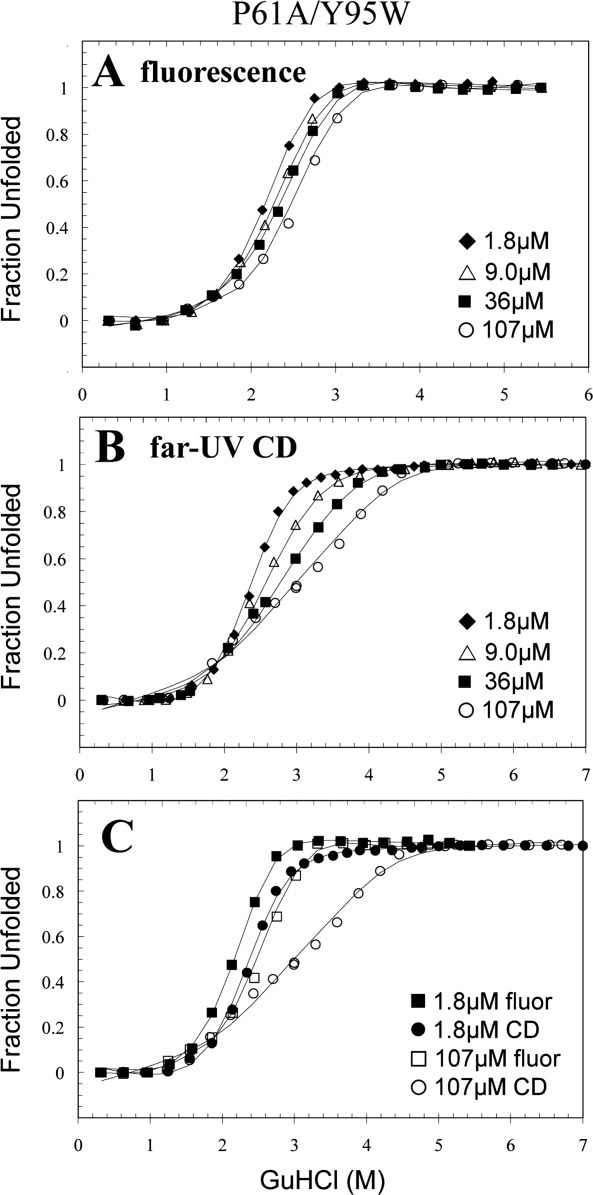
Representative GuHCl-induced denaturation data of P61A/Y95W FIS monitored by fluorescence (A) and far-UV CD (B). An overlay of the highest and lowest concentration used in both methods are shown in (C). The solid line curve fits were generated by global fitting the data using Excel and are based on the N2 ⇆ I2 ⇆ 2U denaturation mechanism (Table I). The data are displayed as fraction unfolded, which is defined by Eq. (2). These denaturation experiments were all performed in 10 mM PB at pH 7.4 with 0.1M NaCl at 20°C.
Global fit of the P61A/Y95W fluorescence and CD data to a three-state model (N2 ⇆ I2 ⇆ 2U) yielded a total ΔG of 15.1 kcal/mol, which is slightly lower than the 16.4 kcal/mol obtained for P61A FIS (Table I). Most of the destabilizing effect is related to the first transition (i.e., C-terminus), which might explain why the CD data of P61A/Y95W is more biphasic than that of P61A (Fig. 4). This destabilizing effect is consistent with prior observations involving Y95F13 and Y95W (unpublished results) mutations in WT FIS and appears to be caused by the loss of the tyrosine side chain hydroxyl group, which is optimally positioned to participate in the electrostatic network present in the C-terminus.30
The Trp69 probe is in a strategic location to monitor both denaturation events in P61A/Y69W FIS
In contrast to P61A/Y95W, the fluorescence-detected denaturation of P61A/Y69W exhibited a clear concentration-independent transition, followed by a concentration-dependent transition, indicating that Trp69 is strategically positioned to separately monitor the disruption of the native state and the subsequent unfolding of the dimeric intermediate [Fig. 6(A)]. The P61A/Y95W CD data show that the conformational change occurring at the C-terminus is accompanied by the loss of secondary structure, yet P61A/Y69W FIS more effectively decoupled the CD-monitored transitions at high protein concentration [Fig. 6(B)]. This may be caused by a higher stability of the dimeric intermediate of P61A/Y69W FIS (Table I). That is, although the ΔG of the first transition of P61A/Y69W is between that of P61A and P61A/Y95W, the dimeric intermediate structure is about 2.0 kcal/mol more stable than in P61A and P61A/Y95W. This results in a higher Cm for the second transition of P61A/Y69W, and therefore, a better decoupling of the N2 ⇆ I2 and I2 ⇆ 2U transitions compared with P61A/Y95W and P61A. This implies that the intermolecular ring stacking interaction of Trp69 with Phe39 is more favorable compared with Tyr69 in WT FIS.
Figure. 6.
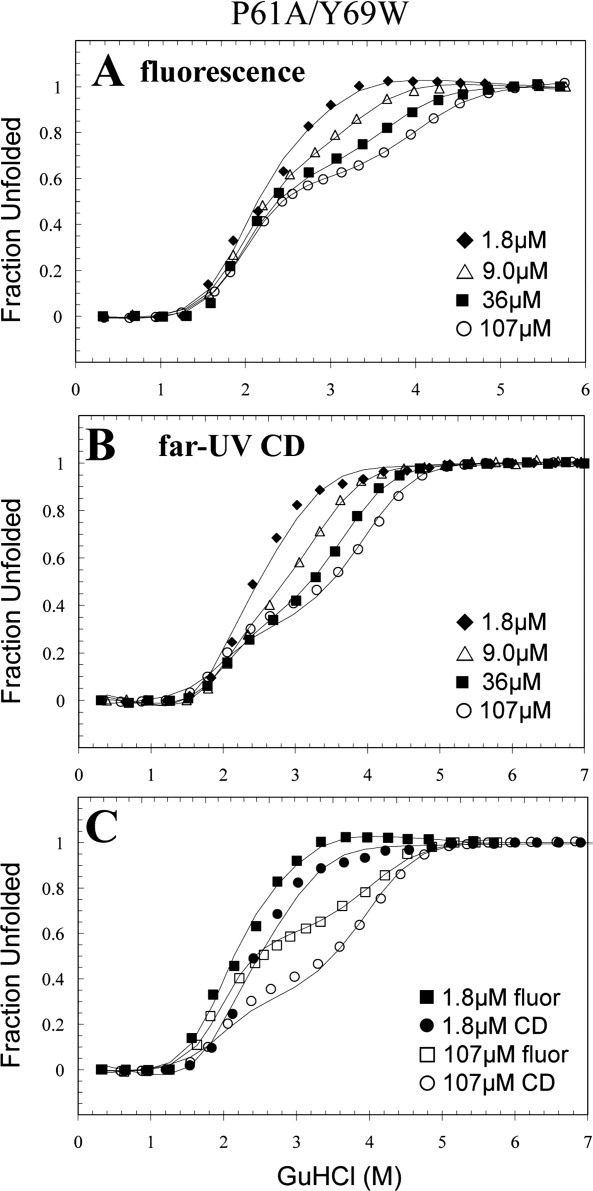
Representative GuHCl-induced denaturation data of P61A/Y69W FIS monitored by fluorescence (A) and far-UV CD (B). An overlay of the highest and lowest concentration used for both methods are shown in (C). The solid line curve fits were generated by global fitting the data using Excel and are based on the N2 ⇆ I2 ⇆ 2U denaturation mechanism (Table I). The data are displayed as fraction unfolded, which is defined by Eq. (2). These denaturation experiments were all performed in 10 mM PB at pH 7.4 with 0.1M NaCl at 20°C.
The shape of the fluorescence and CD data for P61A/Y69W are similar, suggesting that both methods are able to monitor each denaturing event [Fig. 6(C)]. At 107 μM FIS, both denaturation curves are biphasic with a plateau at ∼3.0M GuHCl. However, the curves do not fully superimpose because the loss of fluorescence (∼50%) associated with the formation of the dimeric intermediate (I2) is much higher than the corresponding loss of CD signal (∼30%). Thus, despite its position on the surface of the protein, Trp69 turns out to be an excellent probe for both transitions even at low FIS concentrations, providing data that is consistent with and complementary to the Trp95 data.
Trp38 is an excellent probe for the formation of the dimeric intermediate, but is insensitive to dimer dissociation
The position of the Tyr38 side chain in a hydrophobic pocket within the dimer and without direct interactions with the C-terminus supported the hypothesis that the fluorescence of Trp38 would be exclusively sensitive to the dissociation of the dimeric intermediate. Surprisingly, the fluorescence-monitored denaturation transition of P61A/Y38W exhibited a two-state cooperative transition that was independent of protein concentration [Fig. 7(A)]. The lack of protein concentration dependence of the fluorescence transition, and its occurrence in the 2–3M GuHCl range is consistent with the formation of the dimeric intermediate observed for P61A/Y95W and P61A/Y69W FIS. Remarkably, the cooperative and two-state nature of this transition suggests that it is exclusively monitoring the formation of the dimeric intermediate and not its dissociation.
Figure. 7.
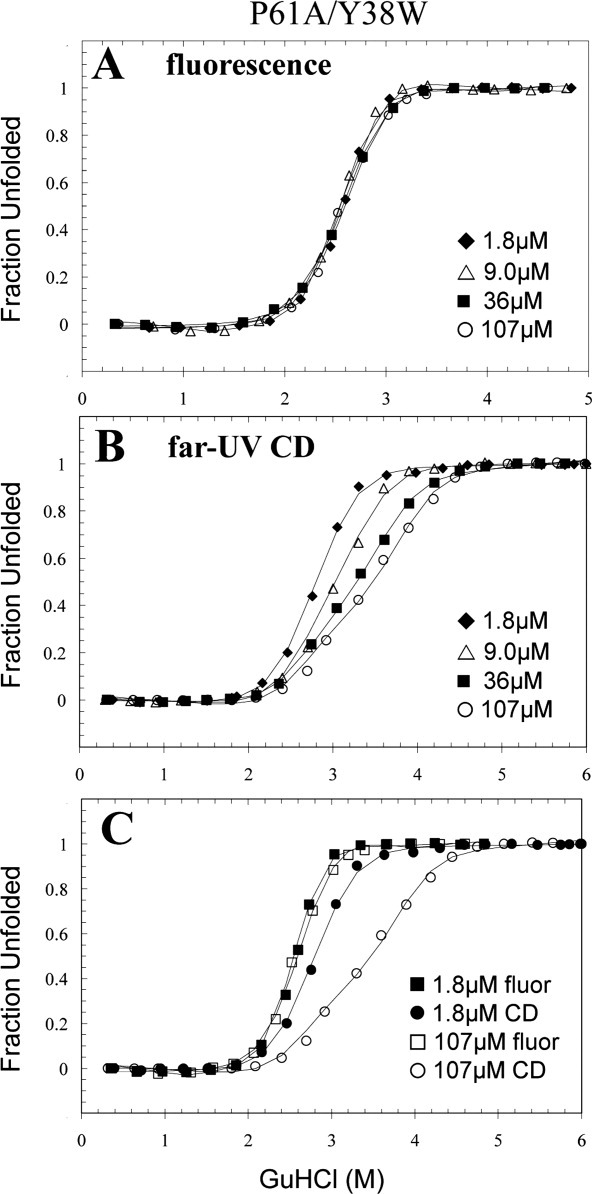
Representative GuHCl-induced denaturation data of P61A/Y38W FIS monitored by fluorescence (A) and far-UV CD (B). An overlay of the highest and lowest concentration used for both methods are shown in (C). The solid line curve fits were generated by global fitting the data using Excel and are based on the N2 ⇆ I2 ⇆ 2U denaturation mechanism (Table I). The data are displayed as fraction unfolded, which is defined by Eq. (2). These denaturation experiments were all performed in 10 mM PB at pH 7.4 with 0.1M NaCl at 20°C.
The CD data of P61A/Y38W FIS is similar to that of P61A/Y95W and P61A/Y69W FIS in that the first part of the transition is less concentration-dependent than the second part, but the P61A/Y38W curves appear to be more two-state-like [Fig. 7(B)]. This difference is likely caused by a stabilizing effect of the Y38W mutation. Global analysis of the P61A/Y38W data shows that this variant is ∼9, 7, 6, and 4.5 kcal/mol more stable than WT, P61A/Y95W, P61A, and P61A/Y69W FIS, respectively (Table I). Trp38 not only lacks a hydroxyl group but also is able to reach deeper than Tyr into the hydrophobic pocket. These added interactions combined with the stabilizing effect of the P61A mutation (∼3.6 kcal/mol) further stabilize the A- and B-helices that make up most of the hydrophobic core of the FIS protein. However, the stabilizing effect of the Y38W mutation propagates throughout the protein affecting both the N and I2 states (Table I). Consistent with this observation, the fluorescence and CD transitions of P61A/Y38W FIS begin above 2M GuHCl, whereas those of P61A/Y95W and P61A/Y69W FIS begin at a lower GuHCl concentration. This might explain the more cooperative N2 ⇆ I2 transition and higher m-value for P61A/Y38W (Table I).
Interestingly, an overlay of the fluorescence and CD data shows an almost complete decoupling of the first (N2 ⇆ I2) and second (I2 ⇆ 2U) transitions at 107 μM FIS [Fig. 7(C)], facilitated by the enhanced stability of the dimeric intermediate in P61A/Y38W FIS (Table I). This information, together with the lack of protein concentration dependence of the fluorescence-monitored titrations of P61A/Y38W, show that the formation of the dimeric intermediate not only involves a major disruption of the C-terminus but is also accompanied by a conformational change in the environment of Trp38 that results in the quenching of its fluorescence. As the dimeric intermediate of P61A/Y38W is more stable than the native state of WT FIS (Table I), it is likely that the conformational change is small, but significant enough to expose Trp38 to the solvent or polar quenching groups within the protein. One possibility is that breakage of the K94-E52 salt-bridge disrupts the N-terminus of Helix-B (res 50–70), propagating the effect to the preceding A-B loop (res 42–49) and the C-terminus of Helix A (res 26–41). The tremendous importance of this salt-bridge interaction to the stability and conformation of FIS was demonstrated by a K94A FIS mutant, which exhibited a 16° decrease in Tm27 and a high tendency to aggregate during purification (unpublished results). Another scenario is that the conformational changes near Tyr69, as demonstrated by the P61A/Y69W mutant, might propagate to its ring-stacking partner Phe39, thereby also affecting the environment of Trp38. Furthermore, it is conceivable that the larger Trp residue might also make direct contact with residues of the C-helix, such as Met80, which is less than 6 Å apart from Tyr38.
General insight into using engineered Trp residues to probe protein denaturation
Trp residues are often engineered into proteins by replacing Tyr or Phe residues because Trp yields higher fluorescence efficiency.3,5,32–34 This type of mutation preserves the aromatic ring and usually has minor effects on the overall structure of the protein. As Trp fluorescence is highly sensitive to the environment, the location of Trp residues within the protein needs to be carefully considered before engineering them into a protein. The polarity of the Trp environment is mostly responsible for its fluorescent intensity. If the Trp residue is partially or mostly solvent exposed in the native state, there will be little difference in fluorescence intensity and λmax [e.g. P61A/Y69W, Fig. 2(B)] between the folded and unfolded states due to solvent quenching in the native state. For this reason, buried Trp residues shielded from the solvent are usually efficient probes for conformational changes in protein [e.g. P61A/Y38W, Fig. 2(A)]. Another factor that affects Trp fluorescence is the presence of nearby quencher residues. It has been shown that certain residues such as lysine, histidine, cysteine, glutamic acid, and tyrosine can act as quenchers with varying degrees of efficiency through electron transfer over a distance of 10 Å.1 Hence, the close proximity of quencher residues might decrease the fluorescence signal of the native state, resulting in an increase in fluorescence upon unfolding [e.g. P61A/Y95W, Fig. 2(C)].
Besides affecting the fluorescence, the environment of a Trp residue can also affect the local or global protein stability. For instance, hydrogen bonds have been shown to contribute to the overall stability of a protein.35–37 Moreover, their stabilizing effects may be synergistic in the context of complex networks of hydrogen bonds and salt bridges. If the hydroxyl group of a Tyr residue is involved in hydrogen bonding, its replacement for Trp will result in the loss of this interaction, which may lead to a destabilizing effect. This appears to be the case for P61A/Y95W, because Y95 interacts intramolecularly with K94 and E59, and a Y95F mutation was shown to decrease the stability of FIS by ∼1.8 kcal/mol.13 The engineering of Trp residues may also be destabilizing if the space is not large enough to accommodate the bulkier side chain. When this happens, the stability and denaturation pathway may be altered, perhaps causing an intermediate to become populated.38 In an extreme case, the destabilizing effect may cause aggregation and not allow the protein to fold. This scenario was encountered for the P61A/Y51W FIS mutant, which we were not able to purify due to its aggregation.
It is interesting that in this set of mutants, the following trend was observed for the efficiency of the Trp residues at detecting the dimeric intermediate: P61A/Y38W > P61A/Y69W > P61A/Y95W. This trend is the opposite of what was expected when simply considering the location of each mutant and the proposed role of the C-terminus. Although our study has shown that it is often difficult to predict the optimum location to engineer a Trp residue to serve as a conformational probe, it has shown some important factors to consider. The degree of Trp solvent exposure and the relative position of nearby fluorescence-quenching moieties are the main determining factors in the change of fluorescence signal upon unfolding. The position of the Trp with respect to the domains or cooperative units determines the degree of sensitivity to local or global unfolding events. Our results also show that the effect of the introduced Trp on protein stability should be carefully considered, as the mutation could preferentially affect a region, thereby altering the denaturation cooperativity compared with the wild type protein. Therefore, selecting multiple sterically allowable locations, including the interior, the solvent accessible surface, or a highly charged region might be a better strategy than attempting to predict a single best location. Thus, even for small proteins, the engineering of at least three fluorescence probes in different chemical environments and monitoring various regions of the native state is likely to provide a more comprehensive picture of the presence and structure of denaturation intermediates.
Materials and Methods
Protein expression, purification, and sample preparation
The P61A/Y#W FIS mutants were made by a two-step megaprimer PCR method as previously described.30 All these mutants were overexpressed in Escherichia coli and purified by passing them twice through a SP-Sepharose cation-exchange column. Two NaCl gradients of 0.3 to 1M and 0.3 to 0.7M were used for the first and second separation steps, respectively. The resulting FIS protein was found to be 95% pure based on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis. The concentration of the FIS mutants were determined using ultraviolet (UV) spectroscopy by measuring the absorbance at 276 nm in 8M GuHCl and using extinction coefficient of 9800 M−1 cm−1 for all P61A/Y#W FIS mutants.39 All the chemical denaturation experiments were performed in 10 mM phosphate buffer (PB) at pH 7.4 with 0.1M NaCl at 20°C.
CD experiments
CD experiments were performed with an OLIS CD instrument (Bogart, GA) equipped with a dual-beam optical system. The equilibrium GuHCl denaturation experiments in the far-UV region were monitored at 222 nm to probe the α-helical content; the signal at 255 nm was used as a baseline correction. To ensure that enough signal was provided by the protein sample, different size cuvettes were used according to the protein concentration: 1-cm cell for 1.8 μM, 0.2-cm cell for 9 μM, 0.1-cm cell for 36 and 107 μM. A codilution titration method was used in which a volume of native FIS is incubated in the cuvette, and a small volume was periodically withdrawn and replaced with the same volume from a stock solution of unfolded (8M GuHCl) FIS at identical protein concentration. This resulted in a gradual increase of GuHCl in the cell and eventual denaturation of FIS. After each withdrawal and addition, the sample was allowed to reach equilibrium (less than 1 min) before the CD data was collected. A sample (60 μL) was removed from the cuvette after each CD measurement and placed on an Abbe refractometer to measure the refractive index, which was used to calculate the exact concentration of GuHCl at each point.
Fluorescence experiments
The equilibrium denaturation studies were done on a Hitachi F-4500 spectrophotometer (Tokyo, Japan). An excitation (EX) wavelength of 295 nm was used to selectively excite the tryptophan probe. Full wavelength scans from 300 to 400 nm were recorded for each mutant to determine the wavelength of maximum emission for each mutant. The denaturation of the P61A/Y#W FIS mutants was monitored at the wavelength that showed the maximum difference in signal between the native and unfolded states.
Analysis of denaturation data
The equilibrium denaturation data of the P61A/Y#W FIS mutants demonstrated a clear decoupling between two separate transitions; therefore, the data was fit to a three-state (N2 ⇆ I2 ⇆ 2U) denaturation mechanism. The instrument signal (Y) is a function of this equation
| (1) |
where FN2, FI2, and F2u are the fraction of the native, intermediate, and unfolded states, respectively. YX represents the instrumental signal of the native (YN2), intermediate (YI2), and unfolded (Y2U) states and is a linear function of denaturant concentration, where the slope and ordinate intercept are allowed to vary for the N and U states, and the slope for the intermediate is fixed to zero. When FN2 is defined as 1 − F2U − FI2, the F2U for this three-state mechanism can be defined as
| (2) |
where K1 and K2 are equilibrium constants for N2 ⇆ I2 and I2 ⇆ 2U, respectively, and Pt is the total protein concentration. These equilibrium constants can be defined in terms of the linear free energy model for protein unfolding, ΔG = ΔGH2O − m[denaturant], and the Gibbs free energy equation, ΔG = −RTLn(Keq). ΔGH2O represents the stability of the protein in the absence of denaturant. This yields the following expression for the equilibrium constant:
| (3) |
This expression defines K1 and K2 in terms of their own ΔGH2O and m-values. Each equilibrium constant has a distinct m-value, which is proportional to the amount of surface area exposed upon protein unfolding.
Global fitting of the denaturation data was performed using Microsoft Excel Solver, which uses the generalized reduced gradient method to solve nonlinear optimization problems.40,41 For all protein concentrations of a given mutant, the sum of squared differences between the data and model was minimized by adjusting a single set of global (m-value and ΔG) and local parameters, which define the slope and intercepts of the denaturant dependent signal for each state. The data from both CD and fluorescence detection methods were fit simultaneously to the same set of global parameters. The fitting routine was restarted multiple times from a variety of initial parameters to find the global minimum and test the robustness of the solution. The curves in Figures 4–7 represent the modeled experimental signal calculated from the parameters that yield the best global fit as expressed in Table I. The open source SOLVER-AID Excel macro was used to calculate standard deviations on the fitting parameters by numerically determining partial differentials within the global model using the central difference method.40,41
Acknowledgments
The authors thank Dr. Dmitri Zagorevski for help with mass spectrometry analyses and Dr. Joachim Jaeger for his assistance with PyMol.
Glossary
Abbreviations:
- CD
circular dichroism
- far-UV
far ultraviolet
- FIS
factor for inversion stimulation
- GuHCl
guanidine hydrochloride
- PB
phosphate buffer
- PCR
polymerase chain reaction
- P61A
proline to alanine mutation at position 61
- P61A/Y#W
collective double mutations with a proline to alanine mutation at position 61 and a tyrosine to tryptophan at different positions
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- WT
wild-type.
References
- 1.Chen Y, Barkley MD. Toward understanding tryptophan fluorescence in proteins. Biochemistry. 1998;37:9976–9982. doi: 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- 2.O'Neill JW, Kim DE, Baker D, Zhang KY. Structures of the B1 domain of protein L from Peptostreptococcus magnus with a tyrosine to tryptophan substitution. Acta Cryst D. 2001;57:480–487. doi: 10.1107/s0907444901000373. [DOI] [PubMed] [Google Scholar]
- 3.Dusa A, Kaylor J, Edridge S, Bodner N, Hong DP, Fink AL. Characterization of oligomers during alpha-synuclein aggregation using intrinsic tryptophan fluorescence. Biochemistry. 2006;45:2752–2760. doi: 10.1021/bi051426z. [DOI] [PubMed] [Google Scholar]
- 4.Lakshminarasimhan A, Bhat PJ. Replacement of a conserved tyrosine by tryptophan in Gal3p of Saccharomyces cerevisiae reduces constitutive activity: implications for signal transduction in the GAL regulon. Mol Genet Genomics. 2005;274:384–393. doi: 10.1007/s00438-005-0031-6. [DOI] [PubMed] [Google Scholar]
- 5.Maki K, Cheng H, Dolgikh DA, Roder H. Folding kinetics of staphylococcal nuclease studied by tryptophan engineering and rapid mixing methods. J Mol Biol. 2007;368:244–255. doi: 10.1016/j.jmb.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orlik F, Andersen C, Benz R. Site-directed mutagenesis of tyrosine 118 within the central constriction site of the LamB (maltoporin) channel of Escherichia coli. II. Effect on maltose and maltooligosaccharide binding kinetics. Biophys J. 2002;83:309–321. doi: 10.1016/S0006-3495(02)75171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reynolds RA, Watt W, Watenpaugh KD. Structures and comparison of the Y98H (2.0 A) and Y98W (1.5 A) mutants of flavodoxin (Desulfovibrio vulgaris) Acta Cryst D. 2001;57:527–535. doi: 10.1107/s0907444901002554. [DOI] [PubMed] [Google Scholar]
- 8.Holm MM, Naur P, Vestergaard B, Geballe MT, Gajhede M, Kastrup JS, Traynelis SF, Egebjerg J. A binding site tyrosine shapes desensitization kinetics and agonist potency at GluR2. A mutagenic, kinetic, and crystallographic study. J Biol Chem. 2005;280:35469–35476. doi: 10.1074/jbc.M507800200. [DOI] [PubMed] [Google Scholar]
- 9.Mogensen JE, Ipsen H, Holm J, Otzen DE. Elimination of a misfolded folding intermediate by a single point mutation. Biochemistry. 2004;43:3357–3367. doi: 10.1021/bi0358622. [DOI] [PubMed] [Google Scholar]
- 10.Kostrewa D, Granzin J, Stock D, Choe HW, Labahn J, Saenger W. Crystal structure of the factor for inversion stimulation FIS at 2.0 A resolution. J Mol Biol. 1992;226:209–226. doi: 10.1016/0022-2836(92)90134-6. [DOI] [PubMed] [Google Scholar]
- 11.Yuan HS, Finkel SE, Feng JA, Kaczor-Grzeskowiak M, Johnson RC, Dickerson RE. The molecular structure of wild-type and a mutant Fis proterelationship between mutational changes and recombinational enhancer function or DNA binding. Proc Natl Acad Sci USA. 1991;88:9558–9562. doi: 10.1073/pnas.88.21.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mallik P, Pratt TS, Beach MB, Bradley MD, Undamatla J, Osuna R. Growth phase-dependent regulation and stringent control of fis are conserved processes in enteric bacteria and involve a single promoter (fis P) in Escherichia coli. J Bacteriol. 2004;186:122–135. doi: 10.1128/JB.186.1.122-135.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boswell S, Mathew J, Beach M, Osuna R, Colon W. Variable contributions of tyrosine residues to the structural and spectroscopic properties of the factor for inversion stimulation. Biochemistry. 2004;43:2964–2977. doi: 10.1021/bi035441k. [DOI] [PubMed] [Google Scholar]
- 14.Haffter P, Bickle TA. Purification and DNA-binding properties of FIS and Cin, two proteins required for the bacteriophage P1 site-specific recombination system, cin. J Mol Biol. 1987;198:579–587. doi: 10.1016/0022-2836(87)90201-4. [DOI] [PubMed] [Google Scholar]
- 15.Kahmann R, Rudt F, Koch C, Mertens G. G inversion in bacteriophage Mu DNA is stimulated by a site within the invertase gene and a host factor. Cell. 1985;41:771–780. doi: 10.1016/s0092-8674(85)80058-1. [DOI] [PubMed] [Google Scholar]
- 16.Johnson RC, Bruist MF, Simon MI. Host protein requirements for in vitro site-specific DNA inversion. Cell. 1986;46:531–539. doi: 10.1016/0092-8674(86)90878-0. [DOI] [PubMed] [Google Scholar]
- 17.Ball CA, Johnson RC. Efficient excision of phage lambda from the Escherichia coli chromosome requires the Fis protein. J Bacteriol. 1991;173:4027–4031. doi: 10.1128/jb.173.13.4027-4031.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson JF, Moitoso de, Vargas L, Koch C, Kahmann R, Landy A. Cellular factors couple recombination with growth phase: characterization of a new component in the lambda site-specific recombination pathway. Cell. 1987;50:901–908. doi: 10.1016/0092-8674(87)90516-2. [DOI] [PubMed] [Google Scholar]
- 19.Weinreich MD, Reznikoff WS. Fis plays a role in Tn5 and IS50 transposition. J Bacteriol. 1992;174:4530–4537. doi: 10.1128/jb.174.14.4530-4537.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Travers A, Schneider R, Muskhelishvili G. DNA supercoiling and transcription in Escherichia coli: the FIS connection. Biochimie. 2001;83:213–217. doi: 10.1016/s0300-9084(00)01217-7. [DOI] [PubMed] [Google Scholar]
- 21.Skoko D, Yan J, Johnson RC, Marko JF. Low-force DNA condensation and discontinuous high-force decondensation reveal a loop-stabilizing function of the protein Fis. Phys Rev Lett. 2005;95:208101-1–208101yphen;4. doi: 10.1103/PhysRevLett.95.208101. [DOI] [PubMed] [Google Scholar]
- 22.Gonzalez-Gil G, Bringmann P, Kahmann R. FIS is a regulator of metabolism in Escherichia coli. Mol Microbiol. 1996;22:21–29. doi: 10.1111/j.1365-2958.1996.tb02652.x. [DOI] [PubMed] [Google Scholar]
- 23.Xu J, Johnson RC. Identification of genes negatively regulated by Fis: Fis and RpoS comodulate growth-phase-dependent gene expression in Escherichia coli. J Bacteriol. 1995;177:938–947. doi: 10.1128/jb.177.4.938-947.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelly A, Goldberg MD, Carroll RK, Danino V, Hinton JC, Dorman CJ. A global role for Fis in the transcriptional control of metabolism and type III secretion in Salmonella enterica serovar typhimurium. Microbiology. 2004;150:2037–2053. doi: 10.1099/mic.0.27209-0. [DOI] [PubMed] [Google Scholar]
- 25.Bradley MD, Beach MB, de Koning AP, Pratt TS, Osuna R. Effects of Fis on Escherichia coli gene expression during different growth stages. Microbiology. 2007;153:2922–2940. doi: 10.1099/mic.0.2007/008565-0. [DOI] [PubMed] [Google Scholar]
- 26.Osuna R, Finkel SE, Johnson RC. Identification of two functional regions in Fis: the N-terminus is required to promote Hin-mediated DNA inversion but not lambda excision. EMBO J. 1991;10:1593–1603. doi: 10.1002/j.1460-2075.1991.tb07680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feldman-Cohen LS, Shao Y, Meinhold D, Miller C, Colon W, Osuna R. Common and variable contributions of Fis residues to high-affinity binding at different DNA sequences. J Bacteriol. 2006;188:2081–2095. doi: 10.1128/JB.188.6.2081-2095.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hobart SA, Ilin S, Moriarty DF, Osuna R, Colon W. Equilibrium denaturation studies of the Escherichia coli factor for inversion stimulation: implications for in vivo function. Protein Sci. 2002;11:1671–1680. doi: 10.1110/ps.5050102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan HS, Wang SS, Yang WZ, Finkel SE, Johnson RC. The structure of Fis mutant Pro61Ala illustrates that the kink within the long alpha-helix is not due to the presence of the proline residue. J Biol Chem. 1994;269:28947–28954. doi: 10.2210/pdb1fip/pdb. [DOI] [PubMed] [Google Scholar]
- 30.Hobart SA, Meinhold DW, Osuna R, Colon W. From two-state to three-state: the effect of the P61A mutation on the dynamics and stability of the factor for inversion stimulation results in an altered equilibrium denaturation mechanism. Biochemistry. 2002;41:13744–13754. doi: 10.1021/bi0265224. [DOI] [PubMed] [Google Scholar]
- 31.Meinhold D, Beach M, Shao Y, Osuna R, Colon W. The location of an engineered inter-subunit disulfide bond in factor for inversion stimulation (FIS) affects the denaturation pathway and cooperativity. Biochemistry. 2006;45:9767–9777. doi: 10.1021/bi060672n. [DOI] [PubMed] [Google Scholar]
- 32.Azuaga AI, Canet D, Smeenk G, Berends R, Titgemeijer F, Duurkens R, Mateo PL, Scheek RM, Robillard GT, Dobson CM, van Nuland NA. Characterization of single-tryptophan mutants of histidine-containing phosphocarrier proteevidence for local rearrangements during folding from high concentrations of denaturant. Biochemistry. 2003;42:4883–4895. doi: 10.1021/bi027182p. [DOI] [PubMed] [Google Scholar]
- 33.Diaz-Espinoza R, Garces AP, Arbildua JJ, Montecinos F, Brunet JE, Lagos R, Monasterio O. Domain folding and flexibility of Escherichia coli FtsZ determined by tryptophan site-directed mutagenesis. Protein Sci. 2007;16:1543–1556. doi: 10.1110/ps.072807607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sendak RA, Rothwarf DM, Wedemeyer WJ, Houry WA, Scheraga HA. Kinetic and thermodynamic studies of the folding/unfolding of a tryptophan-containing mutant of ribonuclease A. Biochemistry. 1996;35:12978–12992. doi: 10.1021/bi961280r. [DOI] [PubMed] [Google Scholar]
- 35.Greene RM, Betz SF, Hilgen-Willis S, Auld DS, Fencl JB, Pielak GJ. Changes in global stability and local structure of cytochrome c upon substituting phenylalanine-82 with tyrosine. J Inorg Biochem. 1993;51:663–676. doi: 10.1016/0162-0134(93)85038-a. [DOI] [PubMed] [Google Scholar]
- 36.Ramilo CA, Leveque V, Guan Y, Lepock JR, Tainer JA, Nick HS, Silverman DN. Interrupting the hydrogen bond network at the active site of human manganese superoxide dismutase. J Biol Chem. 1999;274:27711–27716. doi: 10.1074/jbc.274.39.27711. [DOI] [PubMed] [Google Scholar]
- 37.Stanley AM, Fleming KG. The role of a hydrogen bonding network in the transmembrane beta-barrel OMPLA. J Mol Biol. 2007;370:912–924. doi: 10.1016/j.jmb.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 38.Jones S, Reader JS, Healy M, Capaldi AP, Ashcroft AE, Kalverda AP, Smith DA, Radford SE. Partially unfolded species populated during equilibrium denaturation of the beta-sheet protein Y74W apo-pseudoazurin. Biochemistry. 2000;39:5672–5682. doi: 10.1021/bi9923959. [DOI] [PubMed] [Google Scholar]
- 39.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levie RD. Advanced Excel for Scientific Data Analysis. New York, NY: Oxford University Press; 2004. [Google Scholar]
- 41.Meinhold DW. 2006. p. 135. Thesis: Equilibrium Folding Intermediates of the Dimeric Protein FIS Give Insights into Protein Stability and Cooperativity: Troy, NY.