

Abstract
YbbR domains are widespread throughout Eubacteria and are expressed as monomeric units, linked in tandem repeats or cotranslated with other domains. Although the precise role of these domains remains undefined, the location of the multiple YbbR domain-encoding ybbR gene in the Bacillus subtilis glmM operon and its previous identification as a substrate for a surfactin-type phosphopantetheinyl transferase suggests a role in cell growth, division, and virulence. To further characterize the YbbR domains, structures of two of the four domains (I and IV) from the YbbR-like protein of Desulfitobacterium hafniense Y51 were solved by solution nuclear magnetic resonance and X-ray crystallography. The structures show the domains to have nearly identical topologies despite a low amino acid identity (23%). The topology is dominated by β-strands, roughly following a “figure 8” pattern with some strands coiling around the domain perimeter and others crossing the center. A similar topology is found in the C-terminal domain of two stress-responsive bacterial ribosomal proteins, TL5 and L25. Based on these models, a structurally guided amino acid alignment identifies features of the YbbR domains that are not evident from naïve amino acid sequence alignments. A structurally conserved cis-proline (cis-Pro) residue was identified in both domains, though the local structure in the immediate vicinities surrounding this residue differed between the two models. The conservation and location of this cis-Pro, plus anchoring Val residues, suggest this motif may be significant to protein function.
Keywords: structural homolog, cis-proline, functional annotation, tandem domain
Introduction
Following closely on the heels of genome sequencing projects, the protein structure initiative has aimed to generate three-dimensional models of proteins with uncharacterized structures in an effort to provide representative structures in hundreds of protein families. It is anticipated that, in many cases, these can provide structural platforms to guide the annotation of genes with unknown function.1,2 One of the 1200+ protein families selected for structural description, YbbR, shows little sequence similarity to other families and was predicted to represent a unique, undescribed portion of protein structural space.
YbbR domains were selected for study by the Northeast Structural Genomics (NESG) Consortium (http://www.nesg.org) based on their occurrence in a large, structurally uncharacterized domain family and the identification genes coding these domains in human gut metagenomics sequencing projects. YbbR domains were first identified as subunits of the larger YbbR protein of Bacillus subtilis. These domains are found in Eubacteria and are particularly common in genera of Gram-positive bacteria from the phylum Firmicutes but can be found in Gram-negative genera as well. These domains appear in soil-borne bacteria and extremophiles as well as human pathogens such as Bacillus anthracis, Clostridium botulinum, Leptospira interrogans, and Staphlyococcus aureus. YbbR and YbbR-like proteins are classified as pfam3 Protein Family PF07949, and occur in seven different arrangements from one lone domain to two, three, or four repeats or one, two, or three sequential domains following a DisA nucleotide-binding checkpoint domain (http://www.pfam.sanger.ac.uk). The ybbR locus of B. subtilis defines the common four domain, sequential repeat arrangement found in many ybbR-containing genomes and is located in the glmM operon immediately upstream of the glucosamine-1-phosphate mutase gene glmM.4
Despite the broad distribution of these domains and the essential role of the glmM operon in peptidoglycan biosynthesis, and thus cell growth and division,5 the function(s) of the YbbR protein and the YbbR domains are unknown. YbbR domains are potentially important to the development of new antibacterial agents for two reasons: first, the proximity of ybbR to the glmM gene suggests that targeting YbbR domains may be part of a strategy to interrupt peptidoglycan synthesis, and second, the suggestion that YbbR domains are in vivo substrates for a surfactin-type phosphopantetheinyl transferase (Sfp-PPTase),6 an important activator of nonribosomal peptidic virulence factor biosynthesis and a currently recognized antibiotic target,7,8 suggests additional importance for targeting these domains in drug discovery efforts.
To structurally define the YbbR domains, structures of two of four predicted YbbR domains from the 437-residue YbbR-like protein Dhaf_0833 from Desulfitobacterium hafniense were determined using solution nuclear magnetic resonance (NMR) spectroscopy and X-ray crystallography. D. hafniense is an anaerobic Gram-positive bacterium (phylum Firmicutes, class Clostridia) best known for its ability to degrade halogenated organic compounds.9,10 Based on these structural studies, the global topology of the YbbR domain was identified, and unique structurally conserved characteristics common to the YbbR domain family were revealed. Structural similarity to proteins outside of the YbbR-like family was noted, possibly indicating more widespread occurrence of this domain in bacterial proteins.
Results
Structure-prediction tools utilizing YbbR domain primary sequences (mGenTHREADER and a BLAST search of the PDB) failed to identify significant homology to proteins deposited in the PDB. Plasmids containing different YbbR domains from different organisms were prepared, expressed in E. coli, and evaluated for structural characterization using solution NMR spectroscopy and X-ray crystallography. Of these, Domains I and IV of the YbbR-like protein of D. hafniense expressed with a high yield were soluble, stable, and either gave positive results in crystal trials or yielded 15N-heteronuclear single quantum coherence (HSQC) spectra consistent with proteins suitable for structural studies. Gel filtration chromatography indicated both Domains I and IV were monomeric (data not shown). 1H, 13C, and 15N resonances of Domains I and IV were assigned and used to generate distance-dependent nuclear Overhauser effect (NOE) restraints. These distance constraints were combined with backbone dihedral angle estimates and residual dipolar coupling (RDC)-derived orientational constraints (Table I) for calculating structural models. In parallel, crystals of Domain I were obtained and found to diffract to 1.9 Å. A complete dataset was collected, and the phases were determined using single wavelength anomalous diffraction of the selenium atoms incorporated as selenomethionine residues.
Table I.
Summary of Conformationally Restricting Experimental Constraints for NMR Structures
| Domain I | Domain IV | |
|---|---|---|
| NOE-based distance constraints | ||
| Total | 775 | 1127 |
| Intraresidue [i = j] | 157 | 308 |
| Sequential [|i − j| = 1] | 223 | 372 |
| Medium range [1 < |i − j| < 5] | 73 | 148 |
| Long-range [|i − j| ≥ 5] | 322 | 299 |
| NOE constraints per constrained residue | 9.0 | 12.8 |
| Dihedral-angle constraints | 65 | 98 |
| Residual dipolar coupling constraints | ||
| Phage | ||
| N: number (Q score) | n.d. | 41 (0.1) |
| N-Co: number (Q score) | n.d. | 49 (0.1) |
| Gel | ||
| H-N: number (Q score) | 64 (0.4) | 43 (0.2) |
| PEG | ||
| H-N: number (Q score) | 61 (0.2) | n.d. |
| Total number of restricting constraints | 965 | 1358 |
| rmsd of backbone atoms | ||
| Ordered regionsa | 0.6 Å | 0.7 Å |
| Entire polypeptide | 2.1 Å | 2.1 Å |
| NOE violations per model >0.5Å | 0.7 | 1.6 |
| Dihedral angle violations per model >10° | 0 | 0 |
| rmsd | ||
| Bond length | 0.014 Å | 0.021 Å |
| Bond angle | 1.4° | 1.5° |
| Ramachandran analysis | ||
| Most favored region (%) | 89.3% | 88.5% |
| Allowed region (%) | 10.7% | 9.4% |
| Generously allowed (%) | 0.0% | 1.9% |
| PSVS11Z-scores | ||
| Verify3D51 | −2.41 | −4.17 |
| Prosall52 | −1.36 | −2.61 |
| Procheck (phi-psi)53 | −1.97 | −2.56 |
| MolProbity54 | −1.61 | −0.47 |
Ordered regions were Domain I (46): 9–17,24–26,31–37,50–55,66–72,75–78,82–91 and Domain IV (76): 9–24, 27–64, 67–68, 71–87, 89–91. n.d., not determined.
Models of the YbbR domains calculated from solution NMR measurements converged to an ensemble of structures with an root mean squared deviation (rmsd) for ordered backbone atoms of 0.6 Å for Domain I and 0.7 Å for Domain IV [Table I, Fig. 1(A,C)]. Protein Structure Validation Software Suite (PSVS) analysis11 verified the high quality of these models with global structure quality Z-scores for the critical indices similar to those observed in high-resolution X-ray crystal structures (Table I). It is important to note that the ensemble rmsd determined by the PSVS package reflects atoms in regions of the protein that are constrained in the structure calculation (ordered residues). Residues that are not constrained due to a lack of measurable NOEs or RDCs appear disordered in the structural ensemble but should not be considered dynamic based on this classification. By this measure, the Domain IV models had a higher proportion of “ordered residues” with 76 residues used for the calculation compared with 46 residues with the models of Domain I (see the footnote in Table I). The X-ray diffraction derived model was likewise of high quality and has good global structure quality Z-scores [Fig. 1(E), Table II]. The NMR and X-ray structures of Domain I overlaid with a backbone rmsd over structured residues to 1.1 Å. Despite the moderate sequence identity between Domains I and IV of 23%, these domains were structurally very similar and the lowest energy models from NMR methods for the two domains overlaid with a global backbone rmsd of 2.2 Å [Fig. 1(F)].
Figure. 1.
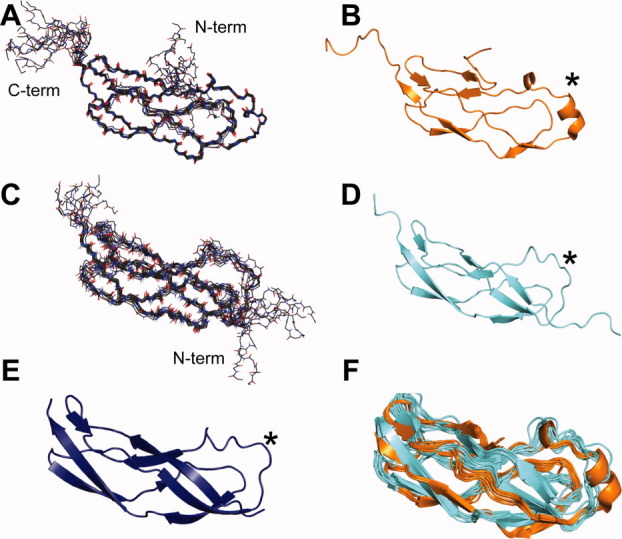
Structures of Domain I and IV from the Ybbr family protein of Desulfitobacterium hafniense. Panels A and C: The ensemble of 10 structures for Domain IV and I, respectively, as calculated from solution NMR spectroscopy-observed constraints. Nitrogen positions are depicted as blue sticks, and oxygen with red sticks. Panels B, D, and E: Ribbon diagrams of the lowest energy solution NMR structures of Domain IV (orange) and I (cyan) and the 1.9-Å X-ray crystal structure of Domain I (deep blue), respectively, show a unique topology that is identical for both of these domains. Panel F: An overlay of the solution structures of Domain I and IV show the similarity of these two molecules. The star marks the homology-modeled site of phosphopantetheine modification is identified by Walsh and coworkers (2005).
Table II.
Summary of Crystallographic Information for Domain I
| Space group | P43212 |
| Molecules per asymmetric unit | 1 |
| VM | 2.11 Å3 Da−1 |
| Unit cell parameters | a = b = 44.8 Å c = 86.0 Å, α = β = γ = 90° |
| Resolutiona | 50–1.9 (1.93–1.90) Å |
| Unique reflections | 15491 |
| Completeness | 100 (100) |
| Redundancy | 17.7 (14.3) |
| Rmerge | 0.058 (0.412) |
| Rcryst | 0.224 |
| Rfree | 0.258 |
| No. of protein atoms | 669 |
| No. of water molecules | 83 |
| Average B factor | |
| Protein Main chain | 23.1 Å2 |
| Protein Side chain | 32.5 Å2 |
| Water molecules | 37.3 Å2 |
| rmsd | |
| Bond lengths | 0.006 Å |
| Bond angles | 1.60° |
| Ramachandran statistics | |
| Most favored region | 91.8% |
| Allowed region | 6.8% |
| Generously allowed | 1.4% |
| PSVS11Z-scores | |
| Verify3D51 | 0.96 |
| Prosall52 | −1.16 |
| Procheck (phi-psi)53 | −1.18 |
| MolProbity54 | −2.40 |
values in parentheses are for the highest resolution shell.
The YbbR domains adopt the structure of elongated bent cylinders, roughly twice as long as their diameter, as shown in Figure 1(B,D,E). These domains are comprised almost entirely of β-pleated sheets and loops, though Domain IV had one short α-helical segment [Fig. 1(B)]. The β-sheets were mostly antiparallel with a section of parallel β-sheet [Fig. 2(A)] as exemplified by residues 10–11 and 49–50 of Domain IV. The N-terminal segment of Domain I is well defined and nestled between two strands [Fig. 1(D)]; however, resonances belonging to the seven N-terminal residues of Domain IV were not observed in the spectra, and therefore, these residues did not converge in the ensemble of models [Fig. 1(A)]. The Domain IV N-terminal sequence was shortened relative to that of Domain I, as a result of construct optimization considerations used to enhance success in protein sample production. In this case, the shortening may have precluded formation of stabilizing interactions and may account for the difference in rigidity.
Figure. 2.
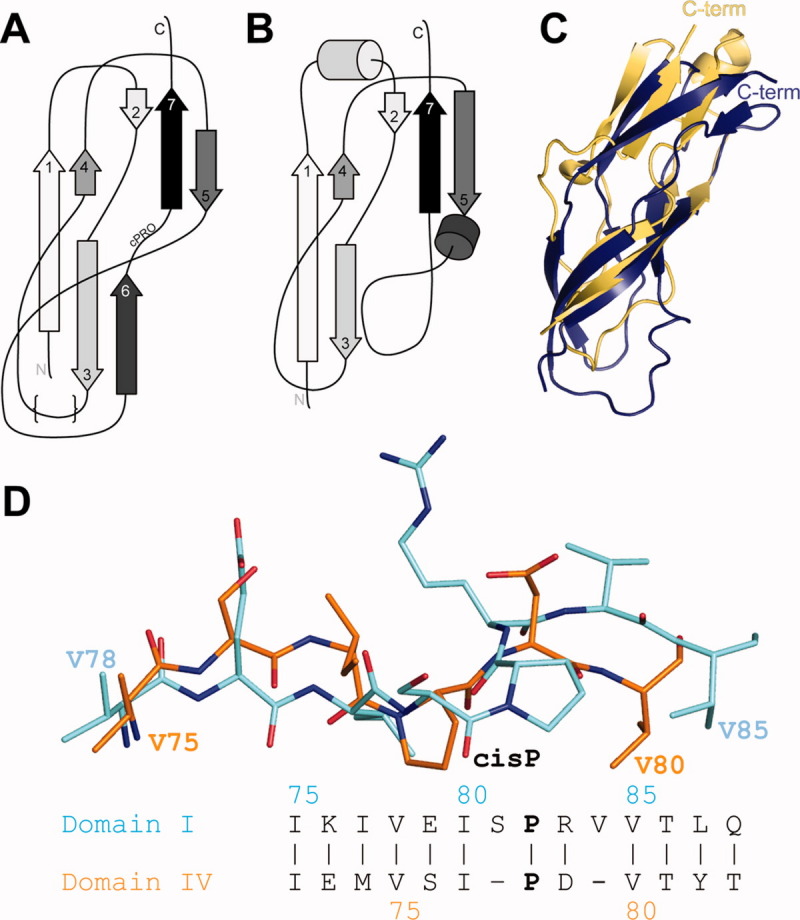
Unique structural features of Domains I and IV are conserved and are similar to the C-terminal domain of the TL5 ribosome-associated protein from Thermus thermophilus. Panel A: The unique topology of these domains forms a “figure 8”-like fold. The structurally variable region is depicted with {}s. Panel B: The topology of the TL5 protein is very similar, though one b-strand is missing and two additional helices are present. Panel C: An overlay of the Domain I X-ray structure (deep blue) and TL5 C-terminal domain (light orange) shows considerable differences. Panel D: The presence of a Pro residue preceded by a cis peptide bond is observed in structures of YbbR Domains I and IV though residues in the immediate vicinity are not conserved. However, the location of hydrophobic amino acid sidechains preceding the cis-Pro is conserved. Domain coloring is the same as in Figure 1.
The topology of the YbbR domain fold can be described as a “figure 8” with some strands coiling around the domain perimeter and others crossing the center as shown in Figure 2(A). A search for structurally similar proteins using the DALI and MarkUs bioinformatics tools12,13 returned many matches to small segments of the YbbR domains. However, only two proteins from this list were topologically similar to the entire YbbR domain: the C-terminal domain of the TL5 and L25 ribosomal proteins from Thermus thermophilus14,15 and Deinococcus radiodurans,16 respectively. The amino acid sequence of L25 is 7.8 and 13.6% identical with Domain I and IV, respectively, and TL5 is 21.3 and 17% identical, respectively. TL5 and the Domain I X-ray structure can be superimposed with a backbone rmsd of 2.2 Å [Fig. 2(C)]. TL5 and Domain IV overlaid with an rmsd for backbone atoms of 3.8 Å. The general strand topology is conserved between the TL5 protein and both YbbR domains including the parallel β-sheets, though the loop regions are substantively different, and the TL5 protein lacks the sixth beta strand observed in the YbbR domains [Fig. 2(A,B)].
An interesting feature of the YbbR domains, not present in the TL5 and L25 proteins, is the presence of a cis-proline (cis-Pro) residue near the C-terminus (Domain I P82, Domain IV P78). Although the location of this cis-Pro conformation is conserved, the local structure in the immediate vicinity surrounding the cis-Pro differs in Domains I and IV, as shown in Figure 2(D). Domain IV has a shorter sequence surrounding the cis-Pro, and the extra residues in Domain I introduced bulges in the strand relative to Domain IV. Despite these differences, the locations of preceding and following Val residues (V78 and V85 for Domain I and V75 and V80 for Domain IV) were identical.
Three regions of Domain IV gave rise to weak or absent NMR signals, unlike Domain I, which had nearly uniform signal intensities. Weak NMR signals may reflect macromolecular dynamics and are often suggestive of functional involvement.17,18 Measurements of residue-specific rotational correlation times from cross-correlation experiments for Domain IV were relatively uniform as shown in Figure 3(A), though the C-terminus had markedly reduced values, suggestive of motion on the ps-ns timescale. In addition, the average value of 4.7 ns for the ordered residues of Domain IV (7–87) was indicative of a monomeric form and consistent with the gel filtration chromatography result. Measurements of spin relaxation rates (R1s and R2s) are likewise informative of macromolecular dynamics but, unlike the correlation time measurements from cross-correlation experiments, R2s can be sensitive to slow, μs-ms timescale motions. Decay rates for transverse magnetization (R2) of Domain IV amide nitrogen signals revealed greater values for residues in three regions of the protein (residues of the N-terminus, S28 and Y65) that also had reduced or absent signals in some of the backbone assignment experiments [Fig. 3(B)]. This behavior is not observed for Domain I [Fig. 3(C)] and may reflect instability in Domain IV due to the shorter N-terminal relative to Domain I. These measurements are consistent with slow-timescale dynamics within Domain IV; however, the structural basis for these motions is not clear.
Figure. 3.
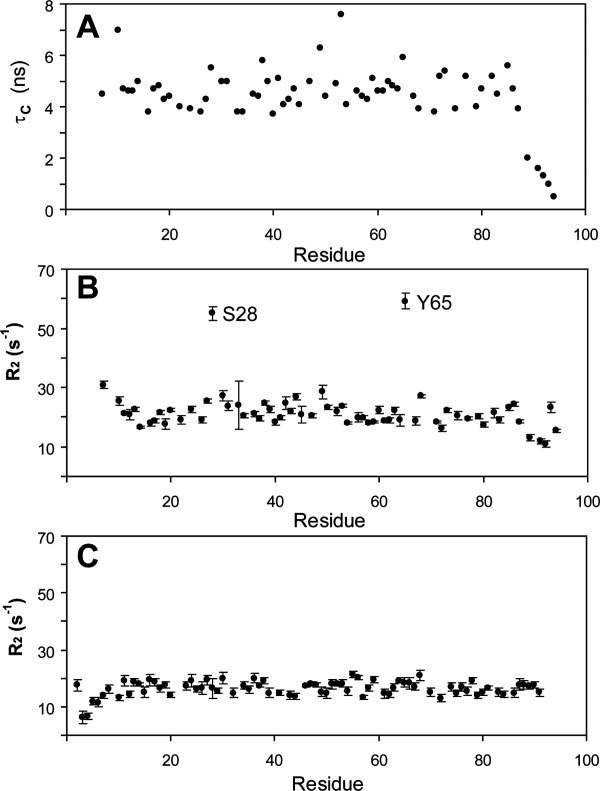
NMR spin relaxation measurements identify dynamic regions of Domain IV that are not present in Domain I. Panel A: A measurement of the rotational correlation time of each NH moiety of Domain IV showed a relatively homogenous distribution with an average for ordered residues of 4.7 ns, suggesting ps-ns timescale motions of the backbone are uniform excepting the highly mobile C-terminal residues. Panel B: Measurements of the R2 spin relaxation rate identified three regions of Domain IV with significantly larger values, suggesting the presence slow μs-ms timescale motions. These residues likewise have greatly reduced or absent intensity in three-dimensional heteronuclear backbone assignment experiments when compared to other residues in the protein. Panel C: Similar transverse relaxation measurements of Domain I (shown) and analysis of triple-resonance experiments identified no regions with enhanced relaxation rates.
Discussion
The models of Domains I and IV provide structural templates for homology modeling of other YbbR family proteins that may help to identify unusual conserved structural features, which reflect domain function. A refined amino acid alignment for the four YbbR domains of the D. hafniense protein based on the domain structures presented here shows conserved features of the domains as shown in Figure 4. Primarily because of the low sequence identity across all four of the domains (2.5 vs. 4.3% in the structure-based alignment) as judged using a nonstructure based alignment, these features are not evident from a simple amino acid sequence alignment and highlight the importance of structural information in identifying features potentially relevant to function. The presence of a cis-Pro residue and the position of the Val residues identified above suggest that these features are conserved to preserve a local surface characteristic, because they alone are unlikely to dictate the shape of the entire domain.
Figure. 4.
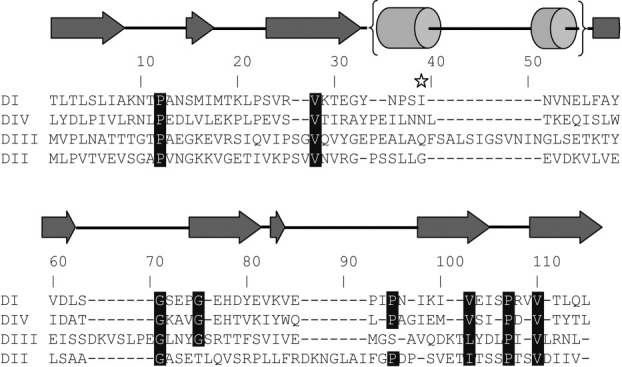
A structure-based sequence alignment of the four YbbR domains in the D. hafniense YbbR protein highlights regions of conservation not observed in a naïve amino acid alignment. Structurally conserved features are highlighted. Secondary structure features are shown above the alignment and are based on the domain structures presented herein. The homology modeled site of Sfp PPTase modification is shown with a star.
The similarity of the YbbR domains to the TL5 and L25 ribosomal proteins may offer insight into the biochemical function of the YbbR domains. TL5 and L25 are believed to bind to the 5S ribosome through their N-terminal domains, but both have a functionally uncharacterized C-terminal domain with a topology similar to YbbR.14 In a crystal structure of the D. radiodurans ribosome (PDB: 2zjr), for example, the L25 protein is engaged with the 5S ribosome, and the C-terminal domain does not appear to interact with any component of the ribosome; it instead lies on the periphery of the holomolecule.16 D. radiodurans mutants lacking L25 are still viable, though growth is reduced.19 L25 and TL5, also known as CTC proteins in different systems, are stress response factors,20,21 though again, the role of the C-terminal domain in this response remains undefined.
The modular often repeated arrangement of YbbR-containing genes likewise is suggestive of function. Similar arrangements of repeated SH2/3 or PDZ protein–protein interaction domains are frequently observed in eukaryotic systems.22 Multidomain scaffolds often serve to organize larger protein complexes and coordinate specific events, much like the Shank family of proteins in the nervous system of higher organisms.23 Similar scaffold proteins have been described in bacterial proteomes including FtsZ, a scaffold for cytokinesis machinery,24 and CheW, a scaffold for signal transduction in flagellar motor regulation.25 Alternatively, YbbR repeats may not bind proteins but another biomolecule such as a carbohydrate or other cell wall/membrane component, working in parallel to obtain a high avidity interaction. Monomeric lectins, for example, often interact weakly with cognate ligands but commonly oligomerize to enhance affinity in manner that could likewise be achieved by sequential repetition.26
As mentioned in the introduction, the third domain of the YbbR protein from B. subtilis has been identified as a substrate for a Sfp-PPTase.6 Walsh and coworkers identified the site of modification as the Ser residue in a D-S-E-L-F motif. The structure and location of this modification may now be studied with a homology model of the B. subtilis YbbR domain (not shown) built using the structures of Domains I and IV presented here. Based on this model, the Ser acceptor residue from the B. subtilis YbbR domain would reside at the solvent-exposed C-terminus of a helix found between β-strands 3 and 4 of the Domain IV structure presented here. The location of this residue, mapped onto the Domain IV structure and aligned sequences, is shown in Figures 1 and 4. This location is similar in terms of local secondary structure and position on the protein/solvent boundary to the phosphopantatheine acceptor site on acyl carrier protein.27 Interestingly, both Domain I and III of the D. hafniense protein contain a Ser residues in this vicinity that may be substrates for this type of modification (Fig. 4). Although in vivo modification of YbbR domain has not been demonstrated, these data are consistent with a conserved structural site for Sfp-PPTase modification. The potential of some, but not all YbbR domains, to accept this modification suggests these domains may assume a variety of roles in situ.
Other proteins displaying structures similar to the YbbR domains have appeared in the Protein Data Bank (PDB) since Domains I and IV were deposited and contain the unique elements observed in the YbbR structures. One protein from Streptococcus thermophilus (PDB: 2kxy; Liu and Prestegard, unpublished) is 18% identical at the amino acid level to Domain IV but shares the topology and cis-Pro architecture. Another protein from Bacillus halodurans is only 15% identical but shares a nearly identical topology (PDB: 2kq1; Wu, Szyperski et al., unpublished). In addition, chemical shifts from the B. halodurans protein (Biological Magnetic Resonance Data Bank 16576) provide evidence for a cis-Pro residue similar to that described herein, based on the Cβ and Cγ chemical shifts.28
Future genetic and biochemical studies will be required to unambiguously define the role of YbbR domains in a host organism. The structural and sequence conservation in a wide assortment of bacteria combined with the location in the genome, the relationship to a general stress response protein, and role of some domains as an acceptor of phosphopantetheinylation combine to suggest an important role for these proteins in the function and survival of bacteria that express YbbR domains.
Materials and Methods
NESG targets DhR29a (Domain IV) and DhR29b (Domain I) were cloned, expressed, and purified based on the standard procedures of NESG to produce a uniformly 15N/13C-enriched protein samples for NMR spectroscopy and selenomethionine-labeled samples for X-ray crystallography.29,30 DNA encoding residues 32–118 (Domain I; NESG ID DhR29B) or 321–409 (Domain IV; NESG ID DhR29A) from the YbbR family protein Dhaf_0833 of D. hafniense Y51 was cloned into a pET21b vector with a noncleavable C-terminal hexa-His tag. These pET expression vectors for Domain I (NESG DhR29B-31-118-21.1) and Domain IV (NESG DhR29A-321-409-21.2) are freely available from the PSI Materials Repository (http://www.psimr.asu.edu/). Transformed cells were cultured at 37°C in MJ9 minimal medium30 containing (15NH4)2SO4 and U-13C-glucose as the sole nitrogen and carbon sources. Proteins were purified using an AKTAexpress FPLC apparatus with a two-step protocol consisting of HisTrap HP affinity and HiLoad 26/60 Superdex 75 gel filtration chromatography. The purity of purified proteins (>98%) was verified with sodium dodecyl sulfate polyacrylamide gel electrophoresis and matrix-assisted laser-desorption ionization time of flight mass spectrometry. Extensive cloning, expression, purification, and biophysical characterization data are on-line in the NESG Spine database (http://www.spine.nesg.org/target.cgi?id=DhR29).31
Solution NMR spectroscopy and refinement
The backbone assignment and NOE experiments for Domain IV and all RDC experiments were collected at the University of Georgia on a Varian Inova 600-MHz spectrometer equipped with a cryogenically cooled probe. The backbone assignment and NOE pulse sequences were supplied by Varian as part of the BioPack distribution. The backbone and NOE experiments for Domain I were collected at Pacific Northwest National Laboratory on a Varian Inova 600- and 750-MHz spectrometers with 5-mm room temperature probes. The NMR spectra of Domain IV were collected using protein concentrations of 0.9–1.1 mM in a buffer containing 20 mM MES, 200 mM sodium chloride, 5 mM calcium chloride, 0.02% sodium azide, 10 mM dithiothreitol, 1x protease inhibitors, 10% D2O, and 50 μM DSS, pH 6.5 at 25 °C. NMR spectra for Domain I were collected using similar protein concentrations in a buffer containing 20 mM ammonium acetate, 200 mM sodium chloride, 10 mM calcium chloride, 0.02% sodium azide, and 5% D2O, pH 4.5 at 25°C. Sequence-specific backbone resonance assignments were determined using HNCO, HNCA, HN(CO)CA, HNCACB, and CBCA(CO)NH experiments. CCONH, HBHA(CO)NH, and HCCH-TOCSY experiments were used for sidechain assignment. The NOE distance constraints for structure calculations were derived from 15N-edited NOESY-HSQC and 13C-edited NOESY-HSQC (for the aliphatic region) taken with mixing times of 100 ms. Stereospecific valine and leucine methyl proton assignments were obtained using the method of Neri et al.32 NMR data were processed using NMRPipe and analyzed using NMRViewJ (Domain IV) or SPARKY (Domain I).33,34 Backbone resonance assignment completeness for Domains I and IV, respectively, was: 100 and 90% for NH, 91 and 84% for Co, 100 and 94% for Ca, and 100 and 95% for Cb. These values do not include the N-terminal Met or the His6 Tag residues.
For RDC measurements Domain I was partially aligned in a positively charged (50% 3-acrylamidopropyl-trimethylammonium chloride + 50% acrylamide) compressed gel medium.35 The positively charged gel was cast in a 3.2-mm diameter plastic tube. The polymerized gel was first washed extensively in deionized water followed by washing with protein buffer to equilibrate pH. Finally, the gel was washed with deionized water to remove buffer. The swelled gel (∼7-mm diameter) was trimmed to a length of 35 mm and dried in open air for 2 days. The gel pellet was swollen in a 5-mm Shigemi NMR tube using the protein solution. The plunger of the Shigemi tube was fixed at a height of 14 mm from the bottom of the tube.
Domain I was also partially aligned in 4.2% (v/v) C12E5 polyethylene glycol (PEG) bicelle using previously published protocols.36 Specifically, 16% (v/v) C12E5 PEG stock was prepared by mixing 50 μL of PEG (Sigma Aldrich), 50 μL of D2O, 200 μL of protein buffer (0.02% NaN3, 10 mM DTT, 5 mM CaCl2, 200 mM NaCl, 1x Protease Inhibitors, 20 mM MES pH 6.5, 10% D2O, 50 μM DSS), and 16 μL of hexanol. The alignment medium was then prepared by mixing 55 μL of 16% PEG stock with 145 μL of protein and 20 μL of D2O to reach 4.2%.
Domain IV was partially aligned in a negatively charged (50% 2-acrylamido-2-methyl-1-propanesulfonic acid + 50% acrylamide) compressed gel medium.35 Preparation of the negatively charged gel was identical to the positively charged gel. Domain IV was also aligned in 12.5 mg/mL Pf1 phage (ASLA Biotech) using previous published protocols.37 The alignment medium was prepared by mixing 55 μL of Pf1 phage PEG stock with 145 μL of protein and 20 μL of D2O to reach 12.5 mg/mL concentration. In all cases, RDCs of 1H-15N amide pairs were measured using a J-modulated experiment.38 N—CO RDCs were measured according to Liu and Prestegard.39
Backbone dihedral angles for Domains I and IV were predicted using TALOS based on the assigned chemical shifts of HA, CA, CB, CO, and N.40 Residue-specific rotational correlation times were estimated using the dipole-dipole/CSA cross-correlated relaxation of backbone amides by the method of Liu and Prestegard.41 Antiphase HzNx R2 rates were measured in an isotropic medium and fitted simultaneously with the isotropic 1J values. The structure calculations were initially done using CYANA.42 NOEs and RDCs from regions of Domain IV with motional influences, as characterized by the R2 values, were not included in structure refinement. During the optimization stage for the NOE distance and dihedral angle constraints, 50 structures were calculated by CYANA and 20 structures with the lowest target energies were selected for analysis. Starting Da and R values for each partially aligning medium were calculated from principle order parameters determined in PALES.43 The structural refinement was performed using NOE distance, dihedral angle, and orientation constraints using XPLOR-NIH; the top 10 structures with the lowest NOE violations of the 50 calculated structures were selected for final structure deposition.44 The results of these refinements are summarized in Table I. The quality of the structural models was verified using the Protein Structure Validation Suite (PSVS) software (http://www.psvs.nesg.org).11 Structural ensembles were deposited in the PDB as 2l5n (Domain I) and 2l3u (Domain IV). Chemical shift, NOESY peak list, and NOESY fid data have been deposited in the Bio MagResDB (Domain I-16570 and Domain IV-16568). RDC data are available at http://www.spine.nesg.org/rdc.cgi. The rmsd determinations were made using CHIMERA45 or PyMOL (Schrödinger, LLC). Alignments of the NMR and X-ray models of Domain I included 74 Cα atoms. Alignments of TL5 with Domains I and IV included the best 34 pairs of Cα atoms.
Crystallization, data collection, and structure determination
The selenomethionine derivative of Domain I (DhR29B) was crystallized by the hanging drop vapor diffusion method at room temperature. The protein sample (∼ 15 mg/mL) was mixed with an equal volume (2 + 2 μL) of a precipitant solution containing 0.1M KBr, 0.1M Bis-tris (pH 7.0), and 30% (w/v) PEG 8000. Crystals grew to an approximate size of 0.4 mm × 0.8 mm × 0.8 mm in 2 weeks. A single-wavelength anomalous diffraction dataset was collected at beamline X4A of the Brookhaven National Laboratory from a flash-cooled crystal after soaking the crystal in a cryoprotectant solution containing 20% (v/v) glycerol with the mother liquor. Data were processed, scaled, and merged using the HKL2000 program package.46 The structure was determined by SHELX.47 ARP/wARP48 and COOT49 were used for model building. Several cycles of simulated annealing, minimization, and B-factor refinement were carried out using the CNS program package.50 The R-factor, R-free, and geometry were checked during refinements. The R-free was calculated based on 10% of randomly selected data excluded from the refinement. The solvent molecules located in the difference electron-density maps (Fo–Fc) were included in the final refinement. The crystallographic statistics for data collection and refinement are summarized in Table II. The resulting model was deposited in the PDB as 3lyw.
Acknowledgments
The authors thank Yizhou Liu for assistance with the J-modulated RDC experiments and R. Belote, C. Ciccosanti, and K. Hamilton, for assistance in cloning and purifying the proteins. NMR data for Domain I were collected at the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the U.S. Department of Energy's Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory.
Glossary
Abbreviations:
- glmM
phosphoglucosamine mutase
- HSQC
heteronuclear single quantum coherence
- NESG
Northeast Structural Genomics Consortium
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- PDB
Protein Data Bank
- PSVS
Protein Structure Validation Suite software
- RDC
residual dipolar coupling
- rmsd
root mean squared deviation
- Sfp-PPTase
surfactin-type phosphopantetheinyl transferase.
References
- 1.Burley SK, Joachimiak A, Montelione GT, Wilson IA. Contributions to the NIH-NIGMS protein structure initiative from the PSI production centers. Structure. 2008;16:5–11. doi: 10.1016/j.str.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaroszewski L, Li ZW, Krishna SS, Bakolitsa C, Wooley J, Deacon AM, Wilson IA, Godzik A. Exploration of uncharted regions of the protein universe. PLoS Biol. 2009:e1000205. doi: 10.1371/journal.pbio.1000205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glanzmann P, Gustafson J, Komatsuzawa H, Ohta K, Berger-Bachi B. glmM operon and methicillin-resistant glmM suppressor mutants in Staphylococcus aureus. Antimicrob Agents Chemother. 1999;43:240–245. doi: 10.1128/aac.43.2.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mengin-Lecreulx D, vanHeijenoort J. Characterization of the essential gene glmM encoding phosphoglucosamine mutase in Escherichia coli. J Biol Chem. 1996;271:32–39. doi: 10.1074/jbc.271.1.32. [DOI] [PubMed] [Google Scholar]
- 6.Yin J, Straight PD, McLoughlin SM, Zhou Z, Lin AJ, Golan DE, Kelleher NL, Kolter R, Walsh CT. Genetically encoded short peptide tag for versatile protein labeling by Sfp phosphopantetheinyl transferase. Proc Natl Acad Sci USA. 2005;102:15815–15820. doi: 10.1073/pnas.0507705102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chalut C, Botella L, de Sousa-D'Auria C, Houssin C, Guilhot C. The nonredundant roles of two 4′-phosphopantetheinyl transferases in vital processes of Mycobacteria. Proc Natl Acad Sci USA. 2006;103:8511–8516. doi: 10.1073/pnas.0511129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yasgar A, Foley TL, Jadhav A, Inglese J, Burkart MD, Simeonov A. A strategy to discover inhibitors of Bacillus subtilis surfactin-type phosphopantetheinyl transferase. Mol Biosyst. 2010;6:365–375. doi: 10.1039/b913291k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christiansen N, Ahring BK. Desulfitobacterium hafniense sp nov, an anaerobic, reductively dechlorinating bacterium. Intl J System Bacteriol. 1996;46:442–448. [Google Scholar]
- 10.Nonaka H, Keresztes G, Shinoda Y, Ikenaga Y, Abe M, Naito K, Inatomi K, Furukawa K, Inui M, Yukawa H. Complete genome sequence of the dehalorespiring bacterium Desulfitobacterium hafniense Y51 and comparison with Dehalococcoides ethenogenes 195. J Bacteriol. 2006;188:2262–2274. doi: 10.1128/JB.188.6.2262-2274.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007;66:778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- 12.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petrey D, Fischer M, Honig B. Structural relationships among proteins with different global topologies and their implications for function annotation strategies. Proc Natl Acad Sci USA. 2009;106:17377–17382. doi: 10.1073/pnas.0907971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gryaznova OI, Davydova NL, Gongadze GM, Jonsson BH, Garber MB, Liljas A. A ribosomal proteins for Thermus thermophilus is homologous to a general shock protein. Biochimie. 1996;78:915–919. doi: 10.1016/s0300-9084(97)86713-2. [DOI] [PubMed] [Google Scholar]
- 15.Fedorov R, Meshcheryakov V, Gongadze G, Fomenkova N, Nevskaya N, Selmer M, Laurberg M, Kristensen O, Al-Karadaghi S, Liljas A, Garber M, Nikonov S. Structure of ribosomal protein TL5 complexed with RNA provides new insights into the CTC family of stress proteins. Acta Cryst D. 2001;57:968–976. doi: 10.1107/s0907444901006291. [DOI] [PubMed] [Google Scholar]
- 16.Harms JM, Wilson DN, Schluenzen F, Connell SR, Stachelhaus T, Zaborowska Z, Spahn CMT, Fucini P. Translational regulation via L11: molecular switches on the ribosome turned on and off by thiostrepton and micrococcin. Mol Cell. 2008;30:26–38. doi: 10.1016/j.molcel.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 17.Mittermaier A, Kay LE. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312:224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 18.Palmer AG, III, Kroenke CD, Loria JP. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 2001;339:204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 19.Korepanov AP, Gongadze GM, Garber MB, Court DL, Bubunenko MG. Importance of the 5 S rRNA-binding ribosomal proteins for cell viability and translation in Escherichia coli. J Mol Biol. 2007;366:1199–1208. doi: 10.1016/j.jmb.2006.11.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gardan R, Duche O, Leroy-Setrin S, Labadie J, Consortiu ELG. Role of ctc from Listetia monocytogenes in osmotolerance. Appl Environ Microbiol. 2003;69:154–161. doi: 10.1128/AEM.69.1.154-161.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmalisch M, Langbein I, Stulke J. The general stress protein Ctc of Bacillus subtilis is a ribosomal protein. J Mol Microbiol Biotech. 2002;4:495–501. [PubMed] [Google Scholar]
- 22.Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 23.Sheng M, Kim E. The Shank family of scaffold proteins. J Cell Sci. 2000;113:1851–1856. doi: 10.1242/jcs.113.11.1851. [DOI] [PubMed] [Google Scholar]
- 24.Adams DW, Errington J. Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat Rev Microbiol. 2009;7:642–653. doi: 10.1038/nrmicro2198. [DOI] [PubMed] [Google Scholar]
- 25.McNally DF, Matsumura P. Bacterial chemotaxis signaling complexes: formation of a CheA/CheW complex enhances autophosphorylation and affinity for CheY. Proc Natl Acad Sci USA. 1991;88:6269–6273. doi: 10.1073/pnas.88.14.6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varki A. Essentials of glycobiology. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2009. p. xxix. 784. [PubMed] [Google Scholar]
- 27.Kim Y, Prestegard JH. Refinement of the NMR structures for acyl carrier protein with scalar coupling data. Proteins. 1990;8:377–385. doi: 10.1002/prot.340080411. [DOI] [PubMed] [Google Scholar]
- 28.Schubert M, Labudde D, Oschkinat H, Schmieder P. A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on C-13 chemical shift statistics. J Biomol NMR. 2002;24:149–154. doi: 10.1023/a:1020997118364. [DOI] [PubMed] [Google Scholar]
- 29.Acton TB, Gunsalus KC, Xiao R, Ma LC, Aramini J, Baran MC, Chiang YW, Climent T, Cooper B, Denissova NG, Douglas SM, Everett JK, Ho CK, Macapagal D, Rajan PK, Shastry R, Shih LY, Swapna GV, Wilson M, Wu M, Gerstein M, Inouye M, Hunt JF, Montelione GT. Robotic cloning and protein production platform of the Northeast Structural Genomics Consortium. Methods Enzymol. 2005;394:210–243. doi: 10.1016/S0076-6879(05)94008-1. [DOI] [PubMed] [Google Scholar]
- 30.Jansson M, Li YC, Jendeberg L, Anderson S, Montelione BT, Nilsson B. High-level production of uniformly 15N- and 13C-enriched fusion proteins in Escherichia coli. J Biomol NMR. 1996;7:131–141. doi: 10.1007/BF00203823. [DOI] [PubMed] [Google Scholar]
- 31.Goh CS, Lan N, Echols N, Douglas SM, Milburn D, Bertone P, Xiao R, Ma LC, Zheng D, Wunderlich Z, Acton T, Montelione GT, Gerstein M. SPINE 2: a system for collaborative structural proteomics within a federated database framework. Nucleic Acids Res. 2003;31:2833–2838. doi: 10.1093/nar/gkg397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neri D, Szyperski T, Otting G, Senn H, Wuthrich K. Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional 13C labeling. Biochemistry. 1989;28:7510–7516. doi: 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- 33.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 34.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 35.Cierpicki T, Bushweller JH. Charged gels as orienting media for measurement of residual dipolar couplings in soluble and integral membrane proteins. J Am Chem Soc. 2004;126:16259–16266. doi: 10.1021/ja046054g. [DOI] [PubMed] [Google Scholar]
- 36.Ruckert M, Otting G. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J Am Chem Soc. 2000;122:7793–7797. [Google Scholar]
- 37.Hansen MR, Mueller L, Pardi A. Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nature Struct Biol. 1998;5:1065–1074. doi: 10.1038/4176. [DOI] [PubMed] [Google Scholar]
- 38.Tjandra N, Grzesiek S, Bax A. Magnetic field dependence of nitrogen-proton J splittings in N-15-enriched human ubiquitin resulting from relaxation interference and residual dipolar coupling. J Am Chem Soc. 1996;118:6264–6272. [Google Scholar]
- 39.Liu YZ, Prestegard JH. Measurement of one and two bond N-C couplings in large proteins by TROSY-based J-modulation experiments. J Magn Res. 2009;200:109–118. doi: 10.1016/j.jmr.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 41.Liu YZ, Prestegard JH. Direct measurement of dipole-dipole/CSA cross-correlated relaxation by a constant-time experiment. J Magn Res. 2008;193:23–31. doi: 10.1016/j.jmr.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 43.Zweckstetter M, Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]
- 44.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Res. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 45.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 46.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 47.Sheldrick GM. A short history of SHELX. Acta Cryst. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 48.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 49.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 50.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Cryst D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 51.Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Macromol Cryst B. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 52.Sippl MJ. Recognition of errors in 3-dimensional structures of proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 53.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck—a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 54.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]