

Abstract
Staphylococci use cell wall-anchored proteins as adhesins to attach to host tissues. Staphylococcus saprophyticus, a uropathogenic species, has a unique cell wall-anchored protein, uro-adherence factor A (UafA), which shows erythrocyte binding activity. To investigate the mechanism of adhesion by UafA, we determined the crystal structure of the functional region of UafA at 1.5 Å resolution. The structure was composed of three domains, designated as the N2, N3, and B domains, arranged in a triangular relative configuration. Hemagglutination inhibition assay with domain-truncated mutants indicated that both N and B domains were necessary for erythrocyte binding. Based on these results, a novel manner of ligand binding in which the B domain acts as a functional domain was proposed as the adhesion mechanism of S. saprophyticus.
Keywords: UafA, Staphylococcus saprophyticus, MSCRAMMs, hemagglutination, crystal structure
Introduction
Staphylococcus saprophyticus is a leading gram-positive uropathogen of uncomplicated urinary tract infection in young and middle-aged women.1–5 S. saprophyticus is a notable uropathogen infection with which is not associated with indwelling catheters, although other staphylococci are often clinically isolated with the presence of indwelling catheters, suggesting that S. saprophyticus has the ability to adhere to the urinary tract.6 Adhesion to the surface of host cells is the crucial initial step in microbial infections, and this is mediated by bacterial cell-surface proteins named MSCRAMMs (Microbial Surface Components Recognizing Adhesive Matrix Molecules).7,8 Most MSCRAMMs on gram-positive pathogens are covalently anchored to the cell wall peptidoglycans via an LPXTG motif. Recent genome analysis of the S. saprophyticus type strain ATCC 15305 revealed that this bacterium possesses only one cell wall-anchored protein, uro-adherence factor A (UafA), while other staphylococci have more than 10 cell wall-anchored proteins, suggesting that colonization by S. saprophyticus involves a mechanism distinct from other staphylococci.9,10 In addition, S. saprophyticus has a redundant uro-adaptive transport system and significantly high urease activity.10 It has been suggested that S. saprophyticus can survive in the human urinary tract due to a combination of these characteristic features.10
In general, MSCRAMMs contain an N-terminal signal peptide, a functional region called the A region, a repeat region of about 20 kDa called the B region, and a region of low sequence complexity called the R region. The C-terminus of MSCRAMMs includes a cell wall–membrane-spanning region (WM region) containing the LPXTG motif, which is necessary for recognition and covalent linkage to cell wall peptidoglycan by sortase.8,11 The crystal structures of the A region of clumping factor A (ClfA),12,13 fibrinogen binding protein (SdrG),14,15 and collagen binding protein (CNA)16 have been determined, and shown to commonly adopt a DE-variant-type immunoglobulin (Ig)-fold. Based on the structures in complex with their specific ligands, two ligand binding mechanisms, the “dock, lock, and latch model”13,14 and the “collagen hug model,”16 have been proposed. In both binding models, the ligand molecule is captured between the N1 and N2 subdomains in the A region, and the binding is completed by a “latch” in which the C-terminal extension of the N2 subdomain is inserted into a trench on the N1 subdomain and forms a β-sheet. These two mechanisms differ in their ligand-free form, i.e., the trench on the N1 subdomain is not filled with a latch in the apo form of the “dock lock and latch model,” whereas the latch is located in the trench and forms a β-sheet with the N1 subdomain in the apo-form of the “collagen hug model.”
In addition, the crystal structure of two tandemly connected B domains of CNA has also been determined. The B domain is composed of two similar IgG-like subdomains.17 These two subdomains share sequence identity of 41% and can be superposed with a root mean squared deviation (RMSD) of 1.28 Å. In addition, two B domains tandemly connected have very similar structures. As the B region neither binds collagen nor influences the collagen binding activity of the A region, the B repeats are thought to act as a stalk that projects the A region at the surface of bacterial cells.18 Although structures of individual domains were revealed as described above, no structure containing both N and B domains has yet been reported.
Similar to other MSCRAMMs, UafA from S. saprophyticus contains an N-terminal signal peptide, a 72-kDa A region followed by a 13-kDa B domain, a 148-kDa Ser-Glu-rich R region, and a WM region containing the LPXTG motif (Fig. 1).10 The A region of UafA consists of three subdomains: N1, N2, and N3. The N2 and N3 domains show 32% sequence similarity with each other. As S. saprophyticus possesses UafA as a unique MSCRAMM, it is likely that UafA contributes significantly to adhesion to the urinary tract, which would be a severe environment for bacterial colonization due to intensive fluid flux. UafA has been shown to play a significant role in the hemagglutination activity of S. saprophyticus.10 However, the precise adhesion mechanism and candidate ligands of UafA remain to be elucidated.
Figure. 1.
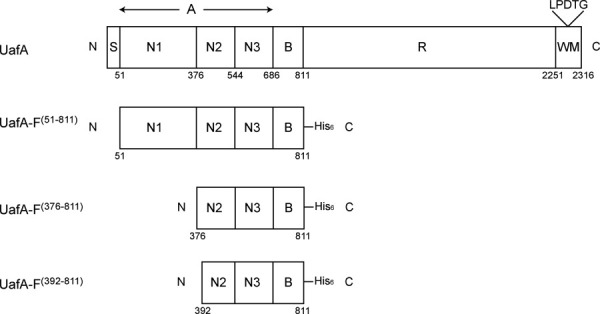
Domain organization of UafA and partial proteins used for structure determination. S, signal peptide; A, A region; N1, N2, N3, domains in the A region; B, B region of about 20 kDa; R, region of low sequence complexity. Numbers indicate the residue number of each position.
In this study, we investigated the adhesion mechanism of UafA by crystal structure analysis and hemagglutination inhibition assay of domain-truncated UafAs. Based on the results, we propose a novel binding mechanism using the B domain for adhesion of S. saprophyticus.
Results
Self-truncation of N1 domain in the functional domain of UafA
We first cloned and expressed the region from Ala51 to Thr811, which contains A and B domains, as a functional region of UafA, UafA-F(51-811), with a His6 tag at the C-terminus (Fig. 1). Although the molecular weight of UafA-F(51-811) predicted from the sequence was about 86 kDa, that of the purified protein was estimated as about 50 kDa by SDS-PAGE. This partial protein, named UafA-F50 kDa, could bind with Ni-affinity resin and was detected on Western blotting using anti-His tag antibody, indicating that the N-terminal region was truncated. The N-terminal sequences of UafA-F50 kDa determined by the Edman degradation method showed that the cleavage site is between Lys375 and Phe376, indicating that the whole N1 domain is truncated. This result agreed well with those of time-of-flight mass spectrometry analysis indicating that the molecular mass of the purified protein was 49.24 kDa. These results indicated that the N1 domain of UafA was specifically and spontaneously digested, although it is unclear whether the N1 domain is digested by proteases from Escherichia coli or by its self-splicing activity. Similar fragmental truncation of the N1 domain was also observed in clumping factors A and B, which are the MSCRAMMs from S. aureus.19
It has been reported that UafA is involved in hemagglutination.10 To determine whether UafA-F50 kDa has erythrocyte binding activity, the hemagglutination inhibitory activity of UafA-F50 kDa was examined (Fig. 2). Erythrocytes formed large aggregates mediated by the hemagglutination activity of S. saprophyticus (2nd well from the left in Fig. 2). However, when UafA-F50 kDa was added, hemagglutination was significantly inhibited and erythrocytes precipitated to the bottom of the well (3rd well from the left in Fig. 2). The inhibition of hemagglutination was dependent on the concentration of UafA-F50 kDa added (Fig. 2), suggesting that UafA-F50 kDa can bind to erythrocytes by itself, and that the N1 domain is not necessary for hemagglutination.
Figure. 2.
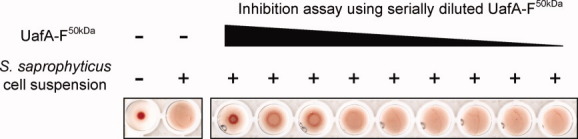
Hemagglutination inhibition activity of UafA-F50 kDa. Erythrocytes were incubated with S. saprophyticus cells and 2-fold serially diluted UafA-F50 kDa. As controls, erythrocytes without S. saprophyticus cells and UafA-F50 kDa are also shown.
Crystal structure of functional region of UafA
As UafA-F50 kDa, which showed erythrocyte binding activity as described above, is a truncated fragment of UafA-F(51-811) in E. coli cells, and the DISOPRED2 disorder prediction server (http://bioinf.cs.ucl.ac.uk/disopred/) suggested that the N1 domain is unstructured. The region from Phe376 to Thr811, designated UafA-F(376-811), was cloned and prepared to determine the crystal structure of the functional domain (Fig. 1). The structure was determined by the single-wavelength anomalous diffraction (SAD) method using selenium as the anomalous scatterer and refined at a resolution of 2.45 Å. In this structure, 18 residues at the N-terminus, the C-terminal 10 residues containing the His6 tag, and a region between Lys706 and Ser715, Gln769 and Ser779, and Asp796 and Asp798 were disordered. The final model consisted of 396 residues, 1 glycerol, and 113 water molecules. As the disorder of the N-terminus was serious, a mutant derivative UafA-F(392-811), in which the N-terminal 17 residues of UafA(376-811) were deleted, was prepared to improve the resolution (Fig. 1). The crystal structure was determined at 1.50 Å resolution and all residues except the N-terminal 3 and C-terminal 7 residues were modeled. The final model consisted of 418 residues, two glycerol molecules, five potassium ions, and 526 water molecules. The revealed structure consisted of three domains, corresponding to N2 (Ala392–Phe541), N3 (Thr547–Val688), and B (Phe695–Asp829) domains [Fig. 3(A)]. All three domains were composed mainly of β-strands and had an Ig-like fold. In particular, N2 and N3 had a similar β-sandwich structure composed of two β-sheets and were superposed with an RMSD of 3.09 Å for 101 Cα atoms. The topology of these two domains is identical to the DEv-IgG fold reported for the ligand binding domains of other MSCRAMMs, ClfA,12,13 CNA (16), and SdrG.14,15 The two β-sheets in the N2 domain are composed of β-strands A, B, and E, and C, D1, D2, F, and G, respectively, whereas those in the N3 domain are composed of β-strands A, B, D, and E, and C, D1, D2, F, and G, respectively [Fig. 3(B); the names of the β-strands correspond to those in the structure of SdrG]. The region corresponding to β-strand D in the N2 domain of SdrG did not participate in β-sheet formation and formed a loop (loop D). Although loop D was located alongside β-strand E, the distance between them was too great for formation of the hydrogen bonds required for a β-sheet. This absence of β-strand D from β-sheet formation was also observed in the ligand-free structure of ClfA (Supporting Information Fig. 1).12 In addition, the relative configuration of N2 and N3 was similar to those of other MSCRAMMs (Supporting Information Fig. 1).13,14,16 The B domain was connected to the N3 domain by a loop of Arg689–Gly694 (hereafter referred to as loop N3-B), and located beside the N2 domain. The B domain consists of two β-sheets, which are composed of β-strands A, D, I, and F, and β-strands B, C, E, G, and H, respectively [Fig. 3(B)]. The structure of the B domain was generally similar to that of subdomains in the B region of CNA except for β-strands G and H, which were absent in CNA, and β-strand F, which was directly connected to β-strand I by a short loop in CNA [Fig. 5(A)]. Other significant differences were observed in the loop structures. A long loop connecting β-strands F and G (loop F-G (B)) was not present in CNA, and the location of a long loop connecting β-strands A and B (loop A-B (B)) was significantly different [Fig. 5(A)].17 These structural differences were mainly located on one side of the B domain opposite the N2 domain [Fig. 5(B)]. Interestingly, these loops were stabilized by bound potassium ions K1, K2, and K3 [Fig. 5(B), see below].
Figure. 3.
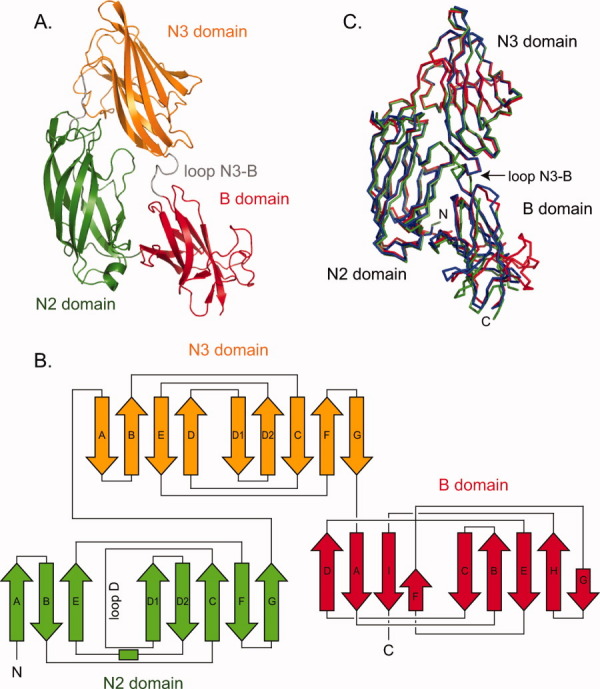
Crystal structure of functional region of UafA. (A) Overall structure of UafA-F(392-811). N2, N3, and B domains are shown in green, orange, and red, respectively. The N3-B loop is shown as a gray loop. (B) Topology diagrams of UafA(392-811). Arrows and boxes represent β-strands and α-helixes, respectively. The colors correspond to those in Figure 3(A). (C) Superposition of three structures. UafA-F(376-811), P21 form UafA-F(392-811) (1.5 Å resolution), and P212121 form UafA-F(392-811) (1.7 Å resolution) are shown in blue, red, and green, respectively. Cα atoms in N3 domains are superposed.
Figure. 5.
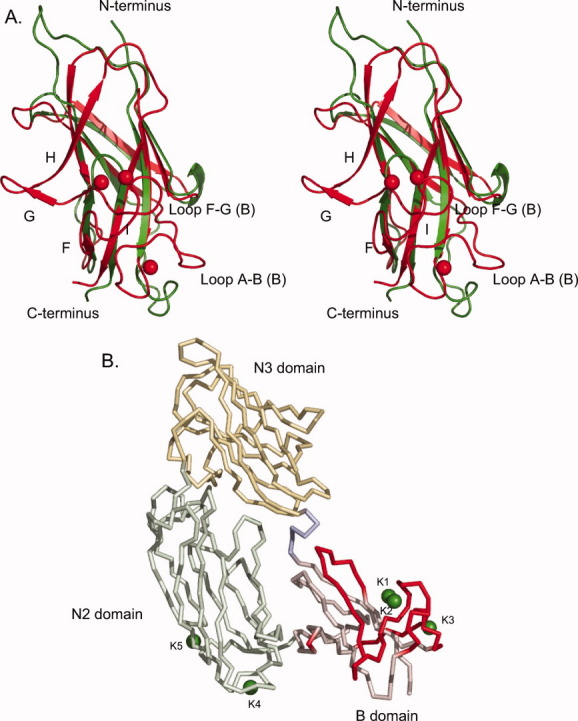
Structure comparison of the B domain of UafA with that of CNA. (A) Stereo diagram of the B domain of UafA superposed onto that of CNA. Gly694–Asp809 of UafA (red) and Ser538–Thr623 of CNA (green) are superposed. Potassium ions bound in the B domain of UafA are also shown as red balls. (B) Residues absent in CNA or of which Cα are more than 4 Å apart from the position of the corresponding residue in CNA are shown in red. The bound potassium atoms are also shown as green balls.
At the interface between N2 and N3 domains, the interactions of 17 residues (Thr423, Ser459, Arg460, Lys461, Lys514, Glu515, Thr518, Asp520 of the N2 domain, and Asn545, Asp553, Ile554, Asn555, Leu557, Asp608, Ser609, Ser611, and Ile685 of the N3 domain) were observed. N2 and B domains also interacted with 15 residues (Ala392, Glu442, Arg460, Arg464, Arg474, Thr475, Thr493, and Asn494 in the N2 domain, and Ala693, Asp728, Thr730, Arg733, Thr736, Asn749, and Thr751 in the B domain). In contrast, there was no significant interaction between N3 and B domains.
Superposition of the N3 domains or B domains from the three structures determined in this study—the C2 structure of UafA-F(376-811) at 2.45 Å resolution, the P21 structure of UafA-F(392-811) at 1.5 Å resolution, and the P212121 structure of UafA-F(392-811) at 1.7 Å resolution—indicated flexibility in loop N3-B [Figs. 3(C) and 4]. In the structure of P21 crystals that grew in 30% PEG MME 2000 and 70 mM potassium thiocyanate, five potassium ions (K1, 2, 3, 4, and 5) were bound to UafA-F(392-811). K1 and K2 were positioned in close proximity, and bound with Asp775, Asp780, and Asp778 in loop F-G (B) of the B domain (Figs. 4, 5, and Supporting Information Fig. S3). K3 bound with Asp709 and Asp714 in loop A-B (B) of the B domain (Figs. 4, 5, and Supporting Information Fig. S3). These loops were disordered in the structures of crystals grown without potassium ions. In addition, the directions of β-strand G and the following loop were significantly different. These results suggested that loops in the B domain are flexible and potassium ions stabilize the B domain. Other potassium ions bound at the surface of the N2 domain.
Figure. 4.
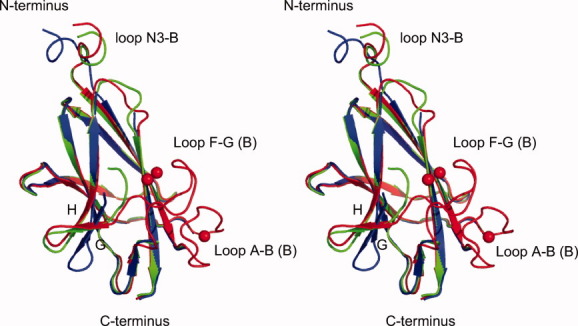
Stereo diagram of the superposed B domains in UafA-F(376-811), P21 form UafA-F(392-811), and P212121 form UafA-F(392-811). The colors correspond to those in Figure 3(C). The bound potassium ions are also shown as red balls.
Functional analysis of the B domain for inhibition of hemagglutination by S. saprophyticus
UafA-F50 kDa inhibited hemagglutination by S. saprophyticus (Fig. 2). To examine the contribution of the B domain to inhibition of hemagglutination, four domain-truncated mutants of UafA-F(376-811) were prepared based on the structural information [Fig. 6(A)], and their hemagglutination inhibitory activities were examined [Fig. 6(B)]. As loop N3-B contributes significantly to ligand binding in the fibrinogen binding adhesion protein SdrG, two partial proteins containing N2, N3, and N3-B loop were also prepared (UafA-#2 and #3). As shown in Figure 6(B), only UafA-F(376-811) containing N2, N3, and B domains showed significant activity, and the other four truncated proteins showed no activities, indicating that in addition to the N2 and N3 domains, the B domain is also necessary for inhibition of hemagglutination by S. saprophyticus.
Figure. 6.
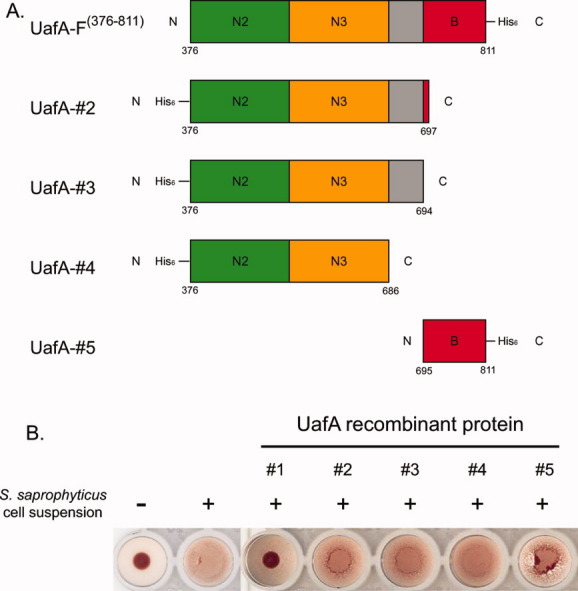
Hemagglutination inhibition activity of the mutants. (A) Domain organization of each mutant prepared for functional analysis. The N3-B loop is shown as a gray box. (B) Hemagglutination inhibition activity of each mutant. Erythrocytes were incubated with S. saprophyticus cells and 100 μg of the truncated protein. Positive control, erythrocytes with S. saprophyticus cells; Negative control, erythrocytes without S. saprophyticus cells or UafA.
Discussion
Function of the B domain in hemagglutination
In this study, we determined the crystal structures of the functional region of UafA, and revealed the arrangement of N2, N3, and B domains. This is the first study to reveal the structure of the functional region including both N and B domains. Hemagglutination inhibition assay using truncated UafAs revealed that both N and B domains are necessary for the function, which is distinct from other MSCRAMMs reported previously.18 In the cases of the CNA from S. aureus, fibrinogen binding protein ClfA from S. aureus, and fibrinogen binding protein SdrG from Staphylococcus epidermidis, the affinity of the N domain region for their ligands is independent of the B domain.13,14,16 The B domains in these proteins are thought to act as a stalk to present the ligand binding the A domain away from the bacterial cell surface [Fig. 7(B)]. In contrast, the results of the present study indicated that the B domain of UafA plays a role in ligand binding, although the individual structures of these domains in the functional region of UafA are similar to those of other MSCRAMMs. Interestingly, UafA has only one B domain downstream of the N domain repeats, whereas several B domains are repeated in CNA and SdrG. Instead, UafA possesses a Ser-Glu-rich region of low complexity (R region) with a molecular mass of 148 kDa after the B domain. Neither CNA nor SdrG has such a long region of low complexity, although there is a Ser-Asp repeat region of about 19 kDa downstream of the B repeats in SdrG. Taken together, these results strongly suggest that the Ser-Glu-rich region acts as a stalk in UafA in place of the B repeats in other MSCRAMMs. The Ser-Glu-rich region is predicted to be an intrinsically disordered domain by the DISOPRED2 disorder prediction server. Therefore, the Ser-Glu-rich region can efficiently present the functional domain away from the cell surface. S. saprophyticus has several capsular polysaccharide synthesis-related genes in the staphylococcal chromosomal cassette region.10 The long Ser-Glu-rich region in UafA may be more suitable for presenting the functional region outside the thick capsular polysaccharide than using the B repeats as a stalk.
Figure. 7.
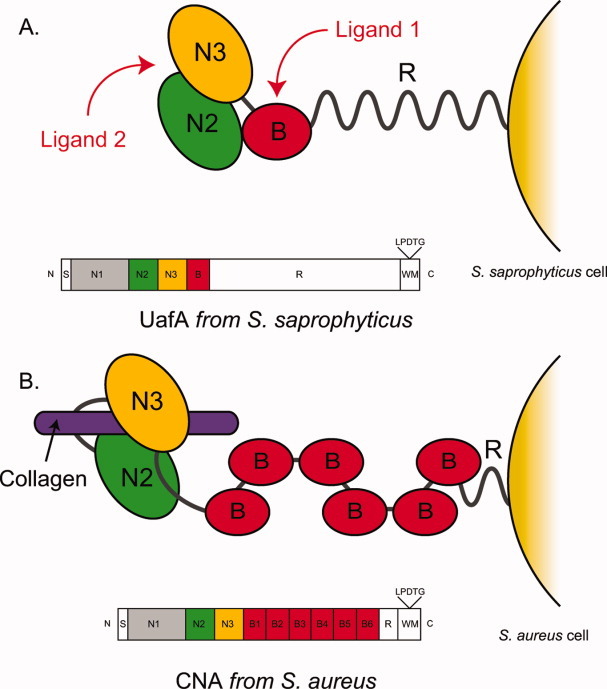
Hypothetical ligand binding model of UafA. The ligand binding mechanism of UafA (A, present study) and that of CNA (B16) are shown. N2, N3, and B domains are shown in green, yellow, and red, respectively. Collagen, which is a ligand of CNA, is also shown as a purple bar. The domain organization of each protein is also shown.
Deduced ligand binding mechanism
Two distinct ligand binding mechanisms have been proposed for MSCRAMMs, i.e., the “collagen hug model” of CNA16 and the “dock, lock, and latch model” of SdrG15 and ClfA.13 In both mechanisms, the ligand molecule is captured between two tandemly connected N domains, and the connecting loop N3-B between the N and B domains acts as a latch to enclose the captured ligand by intruding into the space between β-strands E and D and completing β-sheet formation in the first N domain. This β-sheet completion by the loop N3-B is coupled with ligand binding in the “dock, lock, and latch” mechanism, whereas the β-sheet is constructed even in the absence of the ligand in the “collagen hug” model. In the structure of UafA, the relative allocations of N2 and N3 domains were similar to both CNA and SdrG, but the loop N3-B did not participate in β-sheet formation (Supporting Information Fig. 1), suggesting that the structural features of the N2 and N3 domains in UafA are rather similar to those of SdrG, which adopts the “dock, lock, and latch” mechanism. The absence of the long linker between N2 and N3 also confirms the similarity in domain arrangement of UafA and those of proteins that adopt “dock, lock, and latch” mechanism (Supporting Information Fig. S1). A TYFTDYVD-like motif, which was proposed as an important sequence in the N2 domain to form a latch pocket in this mechanism,14 was conserved at the identical position of UafA as a sequence of 489KYTFTNYVD497. Significant structural flexibility was observed in the N3-B loop, which would act as the latch [Figs. 3(C) and 4]. These structural features strongly indicate the latch formation of UafA on ligand binding.
In comparison of the B domain with that of CNA (Fig. 5), a large structural difference was observed in β-strands G and H, loop F-G, and loop A-B. Briefly, β-strands G and H and loop F-G were not present in the B domain of CNA, and the location of loop A-B was significantly different between UafA and CNA. As only UafA requires the B domain for binding, it is possible that these structurally different regions conferred a ligand recognition property on the B domain of UafA. Structural superposition of B domain in the presence and absence of potassium ions showed that loops in this region are flexible and were stabilized by potassium ions (Fig. 4). Potassium ions may contribute to ligand recognition as well as integrins, which use metal ions for ligand binding.20 Curiously, the structurally different regions are located on the opposite side of the interface between the B and N2 domains [Fig. 5(B)]. Assuming that the B domain captures a specific ligand with this side far from N2, other ligands would be necessary for N2 and N3 domains to bind with the ligand specifically by a mechanism similar to those reported for other MSCRAMMs. UafA may recognize more than two types of ligand with the N and B domains, respectively. A previous report and the present study indicated that UafA is involved in hemagglutination.10 However, the ligand molecule recognized by UafA has not yet been elucidated. We examined hemagglutination of proteinase K-treated erythrocytes to confirm whether the ligand of UafA is a protein located at the surface of erythrocytes. The proteinase K-treated erythrocytes showed hemagglutination to the same extent as untreated erythrocytes, suggesting that the ligand molecule of UafA is not a protein (Supporting Information Fig. S2). An unidentified specific ligand(s) located at the erythrocyte surface—possibly low molecular weight compounds, such as saccharides or lipids—would be recognized by UafA using the N and B domains. It was reported that Streptococcus suis uses an adhesin that recognizes the disaccharide galactosyl-α 1-4-galactose to bind to host cells.21,22 In addition, botulinum neurotoxin was reported to bind to its target presynaptic neuronal cells in the host tissue by recognizing both sugar parts of gangliosides with low affinity and membrane protein with high affinity.23 To discuss the ligand recognition mechanism in which the B domain participates, further studies are required to identify the ligands and determine the structure of UafA in complex with these ligands.
Experimental Procedures
Construction of expression vector for the functional domains of UafA and truncated proteins
Gene fragments encoding the desired regions of the deduced functional domain of UafA (Figs. 1 and 6) were amplified using KOD-Plus DNA polymerase (Toyobo, Osaka, Japan) with S. saprophyticus genomic DNA as the template and the synthesized primer sets shown in Supporting Information Table. The PCR products were inserted into the NcoI and XhoI sites of the pET28b or pET26b vector (Merck, Whitehouse Station, NJ). A His6 tag was fused at the C- or N-termini of all fragments for purification. The correctness of the DNA sequences was confirmed with an ABI 310 Genetic Analyzer (Applied Biosystems, Tokyo, Japan).
Expression and purification of functional domains of UafA
All functional domains used in this study were prepared by the same method as described below. E. coli strain B834 (DE3), harboring the expression vector and pT-RIL (Stratagene, Madison, WI), was grown at 37°C in LB medium supplemented with 25 μg mL−1 kanamycin and 34 μg mL−1 chloramphenicol until the early stationary phase. To induce expression of the desired protein, isopropyl β-d-thiogalactopyranoside was added to a final concentration of 0.5 mM, and the culture was further incubated for 18 h at 25°C with shaking at 150 rpm. The selenomethionine derivative of the functional domain of UafA was obtained by the same method as described above except using M9 medium supplemented with 1 mM selenomethionine instead of LB medium.
Cells were harvested by centrifugation at 5000 × g for 10 min at 4°C, and then disrupted using a sonicator (Branson, Danbury, CT) in 30–40 mL of 20 mM Tris-HCl (pH 8.0), 300 mM NaCl. The cell debris was removed by centrifugation at 40000 × g, 30 min, 4°C, and the supernatant was loaded onto a 1-mL HisTrap HP column (GE Healthcare Biosciences AB, Uppsala, Sweden) preequilibrated with 50 mM Tris-HCl (pH 8.0), 300 mM NaCl. The column was washed with 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, and then the adsorbed protein was eluted with a 0 – 0.5 M stepwise gradient of imidazole in 50 mM Tris-HCl (pH 8.0), 300 mM NaCl. Fractions containing the desired protein were dialyzed against 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, 1 mM EDTA, and then further purified on a HiLoad 26/60 Superdex 200-pg column (GE Healthcare Biosciences AB). Fractions containing the desired protein were collected and used for further experiments.
Crystallization
Purified UafA-F(376-811) was dialyzed against 50 mM Tris-HCl (pH 8.0), 50 mM NaCl, and then concentrated to 12 mg mL−1. Initial crystallization conditions were screened by the sparse matrix method at 20°C using Crystal Screen kits, Index I & II, PEG/Ion Screen, Grid Screen PEG6000, and Grid Screen PEG/LiCl (Hampton Research, Laguna Hills, CA). Crystals of Se-Met-substituted UafA-F(376-811) suitable for further structural analyses were grown by the hanging-drop vapor diffusion method from 300 mM trilithium citrate, 20% PEG3350. Crystals of native UafA-F(376-811) were grown from 200 mM triammonium citrate and 15% PEG3350.
Purified UafA-F(392-811) was dialyzed against 50 mM Tris-HCl (pH 8.0) and 50 mM NaCl, and then concentrated to 10 mg mL−1. Two types of UafA-F(392-811) crystal with different crystallization conditions and space groups were obtained by the sitting-drop vapor diffusion method. The crystallization conditions were as follows: 1) 30% PEG MME 2000, and 70 mM potassium thiocyanate, and 2) 130 mM triammonium citrate, and 22.5% PEG3350.
X-ray diffraction
All X-ray diffraction experiments were performed at SPring-8 (Harima, Japan). X-ray diffraction data of Se-Met UafA-F(376-811) were collected on the beamline BL38B2 at SPring-8. For SAD phasing, a wavelength of 0.9791 Å was chosen on the basis of the fluorescence spectrum of the Se K absorption edge. Diffraction data sets were collected up to a resolution of 3.00 Å. The crystals of Se-Met-substituted UafA-F(376-811) belonged to the space group P21, with three molecules of UafA-F(376-811) in an asymmetric unit.
X-ray diffraction data of native UafA-F(376-811) up to a resolution of 2.45 Å were collected on the beamline BL38B2 at SPring-8. The crystals of native UafA-F(376-811) belonged to the space group C2, and the asymmetric unit contained one molecule of UafA-F(376-811).
X-ray diffraction data of UafA-F(392-811) were collected on the beamline BL41XU at SPring-8. The diffraction data at 1.50 Å resolution were obtained from crystals grown in 30% PEG MME 2000 and 70 mM potassium thiocyanate. The crystal belonged to the space group P21, and the asymmetric unit contained one molecule of UafA-F(392-811). The diffraction data of crystals grown in 130 mM triammonium citrate and 22.5% PEG3350 were collected up to a resolution of 1.70 Å. The crystal belonged to the space group P212121, and the asymmetric unit contained one molecule of UafA-F(392-811).
All diffraction data sets were indexed, integrated, scaled, and merged using the HKL2000 program package.24 The data collection and processing statistics are summarized in Table I.
Table I.
X-ray Data Collection and Refinement Statistics
| UafA-F(376-811) | UafA-F(392-811) | UafA-F(392-811) | ||
|---|---|---|---|---|
| Se-Met Peak | Native | in P21 form | in P212121 form | |
| Data collection | ||||
| Space group | P21 | C2 | P21 | P212121 |
| Cell dimensions | a = 82.6 | a = 76.53 | a = 37.9 | a = 64.8 |
| b = 112.6 | b = 181.98 | b = 126.9 | b = 73.6 | |
| c = 99.7 | c = 81.80 | c = 45.8 | c = 110.9 | |
| β = 108.4 | β = 115.41 | β = 96,7 | ||
| Beamline | SPring-8 BL38B2 | Spring-8 BL38B2 | SPring-8 BL41XU | Spring-8 N;41XU |
| Resolution (Å)a | 5-3.00 (3.11-3.00) | 50-2.45 (2.54-2.45) | 50-1.50 (1.55-1.50) | 50-1,70 (1.76-1.70) |
| Wavelength (Å) | 0.9791 | 1.00000 | 1.00000 | 1.00000 |
| Rsym(%)ab | 12.8 (34.6) | 6.1 (23.1) | 4.8 (14.9) | 4.8 (40.8) |
| Completeness (%)a | 98.9 (90.9) | 98.7 (90.4) | 99.9 (100) | 99.3 (94.9) |
| Observed reflections | 224566 | 87224 | 259912 | 427053 |
| Unique reflections | 34538 | 18642 | 68192 | 58776 |
| Multiplicitya | 6.5 (4.5) | 4.7 (4.0) | 3.8 (3.7) | 7.3 (6.5) |
| Refinement and model quality | ||||
| Resolution range (Å) | 20-2.45 | 20-1.50 | 20-1.70 | |
| No. of reflections | 17271 | 61383 | 52901 | |
| R-factorc | 0.206 | 0.166 | 0.191 | |
| Rfree-factor | 0.246 | 0.187 | 0.216 | |
| Total protein atoms | 3082 | 3321 | 3121 | |
| Total ligand atoms | 6 | 17 | 6 | |
| Total water atoms | 113 | 526 | 320 | |
| Average B-factor (Å2) | 30.7 | 11.69 | 25.82 | |
| Rms deviation from ideality | ||||
| Bond lengths (Å) | 0.006 | 0.009 | 0.012 | |
| Bond angles (°) | 0.87 | 1.2 | 1.4 | |
| Ramachandran plot | ||||
| Residues in most favored regions(%) | 90.8 | 88.8 | 91.0 | |
| Residues in additional allowed regions (%) | 8.9 | 10.9 | 8.7 | |
| Residues in generously allowed regions (%) | 0.3 | 0.3 | 0.3 | |
The values in parentheses refer to data in the highest resolution shell.
Rsym = Σh Σi|Ih,I − <Ih>|/ΣhΣi|Ih,i|, where <Ih> is the mean intensity of a set of equivalent reflections.
R-factor = Σ|Fobs − Fcalc|/Σ Fobs, where Fobs and Fcalc are observed and calculated structure factor amplitudes, respectively.
Structure solution and refinement
The SAD method performed using the programs SHELX25 and SOLVE26 identified eight of 12 selenium sites at 3.8 Å resolution. Using these selenium sites, the initial phases were calculated with the program SOLVE at a resolution of 4.2 Å, and then extended up to 3.0 Å resolution by solvent flattening and noncrystallographic symmetry averaging with the program RESOLVE.27–30 After phase improvement, the initial models including 86% of the N2–N3 region and 50% of the B-domain were built manually. The phases of native UafA-F(376-811) crystals up to a resolution of 2.45 Å were determined using the program DM-MULTI31 by multi-crystal averaging between Se-Met and native crystals. Finally, 396 of 446 residues (exceptions were the N-terminal 18 residues, C-terminal 10 residues, and residues 706–716 and 769–779) were built by RESOLVE, LAFIRE,32 and manual building with the program O.33 The final model was refined with the program REFMAC5.34 A random 7% of all observed reflections were set aside for cross-validation analysis and used for monitoring throughout the refinement by calculating the free R value (Rfree). The crystallographic R and Rfree values and averaged B-factor converged to 20.6%, 24.6%, and 30.7 Å2, respectively.
Crystal structures of UafA-F(392-811) were determined by the molecular replacement method with the program MOLREP35 using the crystal structure of UafA-F(376-811) as a search model. For refinement, a random 10% of all observed reflections were set aside for cross-validation analysis and used for calculating Rfree. The final models were refined with the program REFMAC5. The crystallographic R and Rfree values and averaged B-factor for P21 and P212121 forms converged to 16.6%, 18.7%, and 11.69 Å2, and 19.1%, 21.6%, and 25.82 Å2, respectively. The refinement statistics are summarized in Table I.
Preparation of cell suspension
S. saprophyticus C1 strains were grown in 2 mL of MH-broth at 37°C with shaking for 18 h. The harvested cells were washed with PBS, and resuspended with 2 mL of PBS.
Hemagglutination inhibition activity assay
Aliquots of 100 μg of each domain of UafA were mixed with 20 μL of 5% sheep erythrocyte suspension on 96-well U-bottomed titer plates. The mixtures were kept under static conditions at room temperature for 5 min, and then 50 μL of cell suspension was added. After 2-h incubation, the inhibition of hemagglutination by the presence of these domains was confirmed.
Acknowledgments
We thank Ms. N. Hirano of Hokkaido University for her technical assistance. We also thank Dr. Y. Kitago of Hokkaido University for help with structure analysis, and beam line scientists of Photon Factory and SPring8 for their help with the X-ray diffraction experiments.
References
- 1.Torres Pereira A. Coagulase-negative strains of staphylococcus possessing antigen 51 as agents of urinary infection. J Clin Pathol. 1962;15:252–253. doi: 10.1136/jcp.15.3.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meers PD, Whyte W, Sandys G. Coagulase-negative staphylococci and micrococci in urinary tract infections. J Clin Pathol. 1975;28:270–273. doi: 10.1136/jcp.28.4.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallmark G, Arremark I, Telander B. Staphylococcus saprophyticus: a frequent cause of acute urinary tract infection among female outpatients. J Infect Dis. 1978;138:791–797. doi: 10.1093/infdis/138.6.791. [DOI] [PubMed] [Google Scholar]
- 4.Kahlmeter G. An international survey of the antimicrobial susceptibility of pathogens from uncomplicated urinary tract infections: the ECO.SENS Project. J Antimicrob Chemother. 2003;51:69–76. doi: 10.1093/jac/dkg028. [DOI] [PubMed] [Google Scholar]
- 5.Raz R, Colodner R, Kunin CM. Who are you—Staphylococcus saprophyticus? Clin Infect Dis. 2005;40:896–898. doi: 10.1086/428353. [DOI] [PubMed] [Google Scholar]
- 6.von Eiff C, Peters G, Heilmann C. Pathogenesis of infections due to coagulase-negative staphylococci. Lancet Infect Dis. 2002;2:677–685. doi: 10.1016/s1473-3099(02)00438-3. [DOI] [PubMed] [Google Scholar]
- 7.Patti JM, Hook M. Microbial adhesins recognizing extracellular matrix macromolecules. Curr Opin Cell Biol. 1994;6:752–758. doi: 10.1016/0955-0674(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 8.Foster TJ, Hook M. Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 1998;6:484–488. doi: 10.1016/s0966-842x(98)01400-0. [DOI] [PubMed] [Google Scholar]
- 9.Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K, Nagai Y, Lian J, Ito T, Kanamori M, Matsumaru H, Maruyama A, Murakami H, Hosoyama A, Mizutani-Ui Y, Takahashi NK, Sawano T, Inoue R, Kaito C, Sekimizu K, Hirakawa H, Kuhara S, Goto S, Yabuzaki J, Kanehisa M, Yamashita A, Oshima K, Furuya K, Yoshino C, Shiba T, Hattori M, Ogasawara N, Hayashi H, Hiramatsu K. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet. 2001;357:1225–1240. doi: 10.1016/s0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- 10.Kuroda M, Yamashita A, Hirakawa H, Kumano M, Morikawa K, Higashide M, Maruyama A, Inose Y, Matoba K, Toh H, Kuhara S, Hattori M, Ohta T. Whole genome sequence of Staphylococcus saprophyticus reveals the pathogenesis of uncomplicated urinary tract infection. Proc Natl Acad Sci USA. 2005;102:13272–13277. doi: 10.1073/pnas.0502950102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazmanian SK, Ton-That H, Schneewind O. Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol Microbiol. 2001;40:1049–1057. doi: 10.1046/j.1365-2958.2001.02411.x. [DOI] [PubMed] [Google Scholar]
- 12.Deivanayagam CC, Wann ER, Chen W, Carson M, Rajashankar KR, Hook M, Narayana SV. A novel variant of the immunoglobulin fold in surface adhesins of Staphylococcus aureus: crystal structure of the fibrinogen-binding MSCRAMM, clumping factor A. EMBO J. 2002;21:6660–6672. doi: 10.1093/emboj/cdf619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganesh VK, Rivera JJ, Smeds E, Ko YP, Bowden MG, Wann ER, Gurusiddappa S, Fitzgerald JR, Hook M. A structural model of the Staphylococcus aureus ClfA-fibrinogen interaction opens new avenues for the design of anti-staphylococcal therapeutics. PLoS Pathog. 2008;4:e1000226. doi: 10.1371/journal.ppat.1000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ponnuraj K, Bowden MG, Davis S, Gurusiddappa S, Moore D, Choe D, Xu Y, Hook M, Narayana SV. A “dock, lock, and latch” structural model for a staphylococcal adhesin binding to fibrinogen. Cell. 2003;115:217–228. doi: 10.1016/s0092-8674(03)00809-2. [DOI] [PubMed] [Google Scholar]
- 15.Bowden MG, Heuck AP, Ponnuraj K, Kolosova E, Choe D, Gurusiddappa S, Narayana SV, Johnson AE, Hook M. Evidence for the “dock, lock, and latch” ligand binding mechanism of the staphylococcal microbial surface component recognizing adhesive matrix molecules (MSCRAMM) SdrG. J Biol Chem. 2008;283:638–647. doi: 10.1074/jbc.M706252200. [DOI] [PubMed] [Google Scholar]
- 16.Zong Y, Xu Y, Liang X, Keene DR, Hook A, Gurusiddappa S, Hook M, Narayana SV. A ‘Collagen Hug’ model for Staphylococcus aureus CNA binding to collagen. EMBO J. 2005;24:4224–4236. doi: 10.1038/sj.emboj.7600888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deivanayagam CC, Rich RL, Carson M, Owens RT, Danthuluri S, Bice T, Hook M, Narayana SV. Novel fold and assembly of the repetitive B region of the Staphylococcus aureus collagen-binding surface protein. Structure. 2000;8:67–78. doi: 10.1016/s0969-2126(00)00081-2. [DOI] [PubMed] [Google Scholar]
- 18.Rich RL, Demeler B, Ashby K, Deivanayagam CC, Petrich JW, Patti JM, Narayana SV, Hook M. Domain structure of the Staphylococcus aureus collagen adhesin. Biochemistry. 1998;37:15423–15433. doi: 10.1021/bi981773r. [DOI] [PubMed] [Google Scholar]
- 19.Deivanayagam CC, Perkins S, Danthuluri S, Owens RT, Bice T, Nanavathy T, Foster TJ, Hook M, Narayana SV. Crystallization of ClfA and ClfB fragments: the fibrinogen-binding surface proteins of Staphylococcus aureus. Acta Cryst. 1999;D55:554–556. doi: 10.1107/s0907444998012426. [DOI] [PubMed] [Google Scholar]
- 20.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tikkanen K, Haataja S, Francois-Gerard C, Finne J. Purification of a galactosyl-alpha 1–4-galactose-binding adhesin from the gram-positive meningitis-associated bacterium Streptococcus suis. J Biol Chem. 1995;270:28874–28878. doi: 10.1074/jbc.270.48.28874. [DOI] [PubMed] [Google Scholar]
- 22.Tikkanen K, Haataja S, Finne J. The galactosyl-(alpha 1–4)-galactose-binding adhesin of Streptococcus suis: occurrence in strains of different hemagglutination activities and induction of opsonic antibodies. Infect Immun. 1996;64:3659–3665. doi: 10.1128/iai.64.9.3659-3665.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin R, Rummel A, Binz T, Brunger AT. Botulinum neurotoxin B recognizes its protein receptor with high affinity and specificity. Nature. 2006;444:1092–1095. doi: 10.1038/nature05387. [DOI] [PubMed] [Google Scholar]
- 24.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 25.Sheldrick GM, Hauptman HA, Weeks CM, Miller R. Uson I Ab initio phasing. In: Arnold E, editor. F. Dordrecht: Kluwer Academic Publishers; 2001. pp. 333–351. International tables for crystallography. [Google Scholar]
- 26.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Cryst. 1999;D55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terwilliger TC, Berendzen J. Evaluation of macromolecular electron-density map quality using the correlation of local r.m.s. density. Acta Cryst. 1999;D55:1872–1877. doi: 10.1107/S090744499901029X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Terwilliger TC. Maximum-likelihood density modification. Acta Cryst. 2000;D56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Cryst. 2003;D59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terwilliger TC. Automated side-chain model building and sequence assignment by template matching. Acta Cryst. 2003;D59:45–49. doi: 10.1107/S0907444902018048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cowtan K. Dm: an automated procedure for phase improvement by density modification. Joint CCP4 and ESFEACBM Newsletter on Protein Crystallography. 1994;31:34–38. [Google Scholar]
- 32.Yao M, Zhou Y, Tanaka I. LAFIRE: software for automating the refinement process of protein-structure analysis. Acta Cryst. 2006;D62:189–196. doi: 10.1107/S0907444905038965. [DOI] [PubMed] [Google Scholar]
- 33.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 34.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 35.Vagin A, Teplyako A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]