

Abstract
Bruton's tyrosine kinase (BTK) plays a key role in B cell receptor signaling and is considered a promising drug target for lymphoma and inflammatory diseases. We have determined the X-ray crystal structures of BTK kinase domain in complex with six inhibitors from distinct chemical classes. Five different BTK protein conformations are stabilized by the bound inhibitors, providing insights into the structural flexibility of the Gly-rich loop, helix C, the DFG sequence, and activation loop. The conformational changes occur independent of activation loop phosphorylation and do not correlate with the structurally unchanged WEI motif in the Src homology 2-kinase domain linker. Two novel activation loop conformations and an atypical DFG conformation are observed representing unique inactive states of BTK. Two regions within the activation loop are shown to structurally transform between 310- and α-helices, one of which collapses into the adenosine-5′-triphosphate binding pocket. The first crystal structure of a Tec kinase family member in the pharmacologically important DFG-out conformation and bound to a type II kinase inhibitor is described. The different protein conformations observed provide insights into the structural flexibility of BTK, the molecular basis of its regulation, and the structure-based design of specific inhibitors.
Keywords: activation loop, Bruton's tyrosine kinase, DFG-out, helix C, phosphorylation, protein crystallography, R406, structure-based drug design
Introduction
Bruton's tyrosine kinase (BTK) plays a key role in B cell antigen receptor signaling. It is also essential for multiple signaling pathways in other hematopoietic cell types. After activation by upstream Src family kinases, BTK phosphorylates phospholipase γC which results in calcium mobilization and consequently activation of NF-κB and mitogen-activated protein kinase signaling pathways. Numerous mutations in BTK have been reported to cause defects in B cell maturation and mature B cell function resulting in X-linked agammaglobulinemia. Inhibition of BTK activity with small molecules has indicated that BTK is a potential therapeutic target for the treatment of inflammatory disorders, autoimmunity, thromboembolism, B-lineage leukemias, and lymphomas.1–4
BTK is a member of the Tec family of nonreceptor tyrosine kinases. It contains an N-terminal pleckstrin homology domain, followed by a Tec homology, a Src homology (SH) 3, an SH2, and a C-terminal protein kinase domain. Activation of BTK is initiated by phosphorylation of Y551 in the activation loop (amino acids 542–559) of the kinase domain, followed by autophosphorylation of Y223 in the SH3 domain.1–4 The first crystal structure of BTK kinase domain has previously been reported for the unphosphorylated apo form (PDB accession number 1K2P).5 It shows an activation loop that resembles the reported active state conformation of the phosphorylated Src kinase family member lymphocyte-specific protein tyrosine kinase [Fig. 1(A)]. In the BTK apo structure, helix C (amino acids 439–452) is in a distal position (“helix C-out”) associated with the inactive state of the enzyme. Later, crystal structures of unphosphorylated wild-type BTK kinase domain in complex with the inhibitor B43 (PDB accession number 3GEN) and the phospho-mimetic Y551E mutant BTK kinase domain in complex with Dasatinib (PDB accession number 3K54) were reported.6 B43 stabilizes a helix-C out protein conformation where the fully ordered activation loop is occluding access of the protein substrate binding site [Fig. 1(B)]. In the Dasatinib-bound structure of the Y551E BTK mutant the activation loop is not resolved. Helix C is in the adenosine-5′-triphosphate (ATP)-proximal position (“helix C-in”) allowing the formation of the conserved K430–E445 salt bridge indicative of the kinase active state. The described BTK–ligand complexes all adopt the so-called “DFG-in” conformations typical for kinases complexed with “type I” kinase inhibitors. No crystal structure of BTK in the “DFG-out” conformation complexed with a “type II” inhibitor has been reported. The DFG-out conformation does not allow ATP binding to the enzyme, and it is often associated with slow binding kinetics.7–9
Figure. 1.

Four distinct activation loop conformations representing inactive states of BTK. Shown are ribbon representations of (A) apo form, PDB accession number 1K2P, 2.1 Å resolution, and complexes with (B) B43, PDB accession number 3GEN, 1.6 Å, (C) compound 1, 1.85 Å, and (D) compound 2, 2.55 Å, and molecular surface representations of (E) compound 1 and (F) compound 2. Ligands bound to the ATP site and selected protein side chains are displayed in ball-and-sticks representation. AH1: activation loop helical region 1, amino acids 542–547. AH2: activation loop helical region 2, amino acids 549–554.
Here we report the crystal structures of unphosphorylated BTK kinase domain in complex with six inhibitors that differ in size, shape, and chemical scaffold (Fig. 2). Two novel activation loop conformations are observed and the first Tec family kinase structure in the DFG-out conformation is reported. We show that independent of the phosphorylation state of the BTK kinase domain, a broad range of conformational states can be stabilized by different ligands bound to the ATP pocket. The obtained insights into the conformational flexibility of BTK further enable the structure-based design of drugs for the treatment of inflammatory diseases and cancer.
Figure. 2.
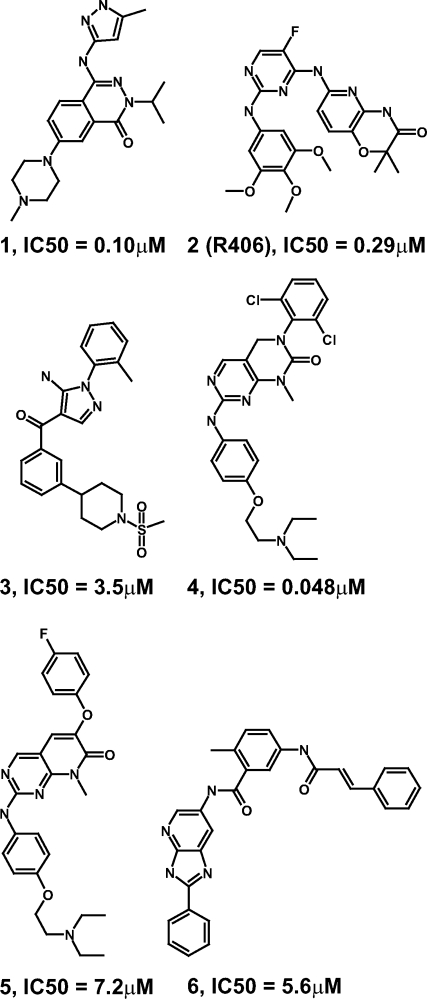
Structural formulas of cocrystallized BTK inhibitors. IC50 values have been determined using a radioactive enzyme assay monitoring BTK product formation.27
Results and Discussion
Compound 1 induces collapse of activation loop into ATP binding pocket
Compound 1, a reported inhibitor of the protein kinases Aurora-A,10 BTK (half maximal inhibitory concentration (IC50) = 100 nM) and SYK,11 forms three hydrogen bonds via its amino-pyrazole moiety to the BTK hinge region [Fig. 3(A)], similar as observed for an analogous compound bound to the Tec kinase family member ITK.12 The phthalazinone core of compound 1 is stacked between V427 in the N-terminal BTK lobe and G480 in the C-terminal lobe. The methyl-piperazine group is solvent exposed.
Figure. 3.

Binding of compounds 1–6 to the ATP pocket of BTK. All protein–ligand and selected protein–protein hydrogen bonds are indicated as black dashes. 2Fo–Fc electron density maps contoured at 1σ around the ligands are displayed in magenta.
Interestingly, a novel conformation of the activation loop is stabilized by compound 1. Amino acids 542–547 form a short α-helix and collapse into the ATP binding pocket [Fig. 1(C)]. In this position, the side chains of the activation loop residues V546 and L547 engage in direct lipophilic interactions with the methyl and isopropyl moieties of compound 1 [Fig. 3(A)]. In the crystal structure of BTK complexed with the inhibitor B43,6 the equivalent activation loop residues form a short 310-helix [Fig. 1(B)]. It has been reported that a 310-helix can gradually transform into an α-helix and vice versa while maintaining a near-helical conformation throughout.13 The same transformation occurs for amino acids 549–554. In the “collapsed” compound 1-bound structure reported here these activation loop residues form a short α-helix [Fig. 1(C)], whereas the same residues from a 310-helix in the B43-bound structure [Fig. 1(B)].
The collapsed BTK protein conformation observed in the compound 1 complex represents an inactive state of the kinase as it lacks major hallmarks of active state conformations: the K430-E445 salt bridge is not formed and the activation loop blocks substrate accessibility [Fig. 1(C)].7–9 The solvent-exposed location of the unphosphorylated Y551 side chain suggests that this activation loop conformation could also be adopted after Y551 phosphorylation. This is in contrast to the conformation of unphosphorylated BTK when complexed with B43, where Y551 phosphorylation would prevent the observed inactive state conformation due to electrostatic repulsion between the phosphate group and the D521 side chain.6
In the discovery of kinase inhibitors for the treatment of chronic diseases like rheumatoid arthritis, unwanted effects due to off-target kinase activity are a major obstacle. Knowledge about protein conformations unique to the kinase(s) to be inhibited provide a valuable starting point for the structure-based design of highly specific kinase inhibitors.7–9 The activation loop conformation observed for BTK when complexed with compound 1 [Fig. 1(C)] has to our knowledge not been reported for any protein kinase. The loop's position not only fundamentally changes the size and shape of the ATP binding pocket, but also creates two novel binding subpockets not present in other kinase crystal structures: A deeply buried subpocket of acidic nature gated by the side chains of the activation loop residues L547 and E550, and a surface-exposed subpocket of basic potential around M596, the catalytic loop residue R525 and the activation loop residue K558 [Fig. 1(E)]. Both subpockets could be explored for the structure-based design of inhibitor specificity for BTK.
Compound 2 (R406) stabilizes atypical DFG conformation
R788/Fostamatinib, a prodrug of compound 2 (R406), is an orally bioavailable SYK inhibitor that reduced disease activity in a phase II clinical trial with rheumatoid arthritis patients.14 Compound 2 inhibits SYK enzyme activity in vitro with an IC50 = 41 nM.15 It has been reported that compound 2 also inhibits about 15 other kinases with less than 10-fold selectivity.16 Here we show that compound 2 inhibits the activity of BTK with an IC50 = 290 nM. It is therefore possible yet unlikely that inhibition of BTK is contributing significantly to the clinical efficacy or adverse events observed for R788/Fostamatinib. Compound 2 forms two hydrogen bonds to the BTK hinge region via its diamino-pyrimidine moiety [Fig. 3(B)], which is sandwiched between the side chains of A428 in the N-terminal kinase lobe and L528 in the C-terminal lobe. The trimethoxy-phenyl moiety of compound 2 is, partially solvent exposed, positioned between the side chains of L408 and G480. The pyrido-oxazinone side chain of compound 2 forms multiple lipophilic interactions with the Gly-rich loop of BTK.
The overall binding mode and small molecule conformation of compound 2 bound to BTK are similar as observed in the crystal structure of compound 2 bound to SYK.17 Major differences between the BTK and SYK complex structures are additional interactions in the BTK bound structure between compound 2 and the fully resolved activation loop which adopts a so far unreported conformation [Fig. 1(D)]. The side chains of the loop residues L542, V546, and L547 are in van der Waals contact with the oxazinone moiety of compound 2, and its NH is forming a hydrogen bond with the backbone carbonyl of L542 [Fig. 3(B)]. For the L547 side chain to be positioned between compound 2 and the C-terminal kinase lobe [Fig. 1(F)], the DFG tripeptide has to adopt an atypical conformation. The D539 side chain is occupying the space filled in the DFG-in conformation by the F540 side chain. In the atypical DFG conformation stabilized by compound 2, the F540 side chain is positioned between the side chains of F517 and the helix C residues E445, V448, and M449.
The activation loop structure stabilized by compound 2 represents yet another inactive state conformation of BTK. Again, the K430–E445 salt bridge is not formed; instead, the K430 side chain engages in a hydrogen bond with the R544 backbone carbonyl, and the helix C residue E445 forms a salt bridge with the R554 side chain [Fig. 3(B)]. The same salt bridge is present in the crystal structures of unphosphorylated BTK kinase domain in its apo5 [Fig. 1(A)] and B43-bound6 [Fig. 1(B)] states, even though the protein main chain arrangements supporting this salt bridge differ significantly between the three structures. Both salt bridge residues differ in position by up to 9 Å while maintaining their interaction. It has been suggested that the E445–R544 salt bridge observed in the BTK apo structure plays a role in the stabilization of the inactive state of BTK.5 The proposed mechanistic model postulates that after phosphorylation of Y551, R544 switches its binding partner from E445 to p-Y551, consequently releasing E445 to engage in the active state salt bridge with K430. This hypothesis is further strengthened by the conserved nature of the E445–R544 salt bridge among structurally distinct inactive state BTK conformations.
Compounds 3 and 4 bind to the active state conformation of BTK
Compound 3 belongs to the amino-pyrazole class of p38α inhibitors.18 It inhibits BTK with modest potency (IC50 = 3.5 μM). Compound 3 forms two hydrogen bonds to the BTK hinge backbone and one with the side chain of the so-called gatekeeper residue T474 [Fig. 3(C)]. The phenyl moiety of compound 3 is positioned in the back pocket between T474 and the K430–E445 salt bridge. The solvent-exposed sulfonyl-piperidine-phenyl front group of compound 3 is sandwiched between the N-terminal strand of the Gly-rich loop and the C-terminal lobe residues G480/C481.
Compound 4 inhibits multiple protein kinases including lymphocyte-specific protein tyrosine kinase and p38α (Goldstein et al., manuscript in preparation). It is the most potent inhibitor of BTK (IC50 = 0.048 μM) described in this article. Compound 4 binds to the hinge of BTK via its amino-pyrimidine moiety [Fig. 3(D)]. The dichloro-phenyl group of compound 4 is occupying the backpocket and the phenoxy group is sandwiched between L408 and G480. The diethylamino moiety of compound 4 is highly solvent exposed and poorly resolved in the electron density indicating mobility.
The conformations of unphosphorylated, inactive BTK kinase domain complexed with compounds 3 and 4 are virtually identical and basically the same as observed in the crystal structure of the phospho-mimetic Y551E mutant BTK in complex with Dasatinib.6 This shows that—at least for the isolated kinase domain—activation loop phosphorylation is not a prerequisite for BTK to adopt the active state conformation characterized by the conserved K430–E445 salt bridge.
Compound 5 induces the helix C-out conformation of BTK
Compound 5 (IC50 = 7.2 μM) belongs to the phenoxy-pyridone class of p38α inhibitors (Goldstein et al., manuscript in preparation). It binds to the BTK hinge region in the same way as the structurally related but ∼100-fold more potent compound 4 [Fig. 3(D,E)]. Compound 5 contains an additional oxygen linker atom between the bicyclic core and the halogen-substituted phenyl side chain (Fig. 2). In p38α, inhibitors with this linker addition induce a rotamer switch of the threonine gatekeeper residue, allowing the phenoxy side chain to enter into an enlarged back pocket (Goldstein et al., manuscript in preparation). This conformational change has been observed in p38α for several inhibitor classes.19 In BTK, however, the phenoxy side chain of compound 5 does not induce a rotamer switch of the gatekeeper residue T474, and consequently it cannot enter into an enlarged back pocket. Instead, the phenoxy side chain of compound 5 is protruding into the DFG region of BTK [Fig. 3(E)]. It causes a S538 rotamer switch and induces the helix C-out conformation. By steric hindrance, the phenoxy moiety of compound 5 prevents the helix C side chain E445 from forming a salt bridge with K430 as observed for the shorter compound 4 [Fig. 3(D)]. Consequently, the helix C-in conformation is not accessible when compound 5 is bound to BTK. Interestingly, addition of a single oxygen atom linker to the bound inhibitor shifts BTK from an active to an inactive state conformation. In p38α, however, the same linker atom has very different structural consequences.
Compound 6 binds to DFG-out conformation of BTK
It has been postulated that type II protein kinase inhibitors that bind to the DFG-out conformation provide a better starting point for the design of highly specific inhibitors compared to type I inhibitors that bind to the DFG-in conformation.7–9 Here we describe the first crystal structure of a Tec kinase family member binding a type II kinase inhibitor and adopting the DFG-out conformation. Compound 6, a type II Src kinase family inhibitor of the azabenzimidazole class20 inhibits BTK with IC50 = 5.6 μM. It binds to the kinase hinge sequence via its azabenzimidazole core [Fig. 3(F)]. The directly attached phenyl ring is sandwiched between L408 and G480. Interestingly, this feature is shared by all structures described in this article, despite the broad range of hinge-binding chemical scaffolds [Fig. 3(A–F)]. The tolyl moiety of compound 6 is positioned between the side chains of the gatekeeper residue T474 and K430 [Fig. 3(F)]. The adjacent primary amide linker forms hydrogen bonds with the BTK side chains E445 and S538. The terminal styrene moiety of compound 6 induces the DFG-out conformation by displacing the F540 side chain.
Compounds 1–6 bind to “active” WEI conformation
The W395A mutation in the conserved WEI sequence (amino acids 395–397) of the SH2-kinase linker region has been shown to dramatically reduce BTK kinase activity.21 Based on comparison with crystal structures of Src family kinase members it has been proposed that the WEI conformation observed in the Dasatinib-bound BTK structure represents an active state while the WEI conformation observed in the B43-bound structure represents an inactive state. This hypothesis was further supported by the fact that B43 was cocrystallized with unphosphorylated BTK kinase domain while phospho-mimetic Y551E mutant BTK was required to cocrystallize Dasatinib.6 Interestingly, in the six crystal structures reported here, the WEI sequences adopt the conformation assigned to the active state of BTK. Considering that these structures were determined with unphosphorylated BTK, and considering the lack of correlation between the observed WEI conformation and the conformations of other regulatory elements like helix C or the activation loop, assignment of BTK activation states due to WEI conformations seems difficult.
Conclusions
The crystal structures of BTK kinase domain in complex with six distinct ATP competitive inhibitors described here demonstrate that bound small molecules can stabilize diverse kinase protein conformations. The stabilization of kinase conformations can occur independently of the phosphorylation state, presence of regulatory domains, or physiological binding partners. We consider this a property of protein kinases in general and not a phenomenon specific to BTK. The diversity of protein conformations observed with different bound inhibitors highlights the challenge of drawing meaningful conclusions from comparisons of X-ray crystal structures of any kinase where in addition to the parameter of interest, for example, phosphorylation, the bound ligand is varied.
The BTK–ligand structures reported here, in combination with previously known structures,5,6 illustrate that BTK can be stabilized in at least four distinct protein conformations all representing catalytically inactive states. Which of these conformations are physiologically relevant for the regulation of BTK activity in vivo remains to be determined. The structural information we and others provide can serve as starting point for further investigations.
In general, the different BTK conformations stabilized by diverse inhibitors demonstrate that cocrystallization with ligands of different shapes, sizes, and chemical scaffolds can be an excellent tool to explore the conformational flexibility of a protein at near-atomic resolution. ATP binding pockets that differ significantly in shape, size, and electrostatic potential have been revealed for BTK, providing a rich set of targets for the structure-based design of small molecule inhibitors. The availability of a diverse ensemble of experimentally determined BTK protein structures significantly increases the chance to successfully dock small molecule BTK inhibitors for which no complex crystal structures have been reported, for example, the leflunomide metabolite analog LFM-A13.22
Materials and Methods
Protein production
Full-length BTK cDNA was used to amplify the region encoding the kinase domain with boundaries of 387–659 by polymerase chain reaction. The product was cloned into a baculovirus transfer vector derived from pVL1392 that was modified to confer a 6x-histidine tag and tobacco etch virus protease cleavage site. The truncated BTK gene was confirmed by DNA sequencing and cotransfected into Sf-9 cells with Baculogold-Bright baculovirus DNA. Baculovirus was expanded by subsequent passages in increasing size of culture. Small-scale expression analysis was performed during virus expansion by small-scale purification of expressed BTK protein by PhyTip immobilized metal ion affinity chromatography.
For large-scale expression, Sf-9 cells were grown to 1.0–1.5 × 106 cells/mL in Sf-900 II SFM medium (Invitrogen) at 27°C and infected with recombinant Baculovirus at 0.3 multiplicity of infection. Following infection, the culture was supplemented with 4% virus feed solution consisting of 1.25% glutamine, 12.5% chemically defined lipid concentrate (Invitrogen) and 12.5× Yeastolate (Invitrogen). Cells were harvested 3 days postinfection at 4000 rpm for 10 min in a Sorvall RC12BP centrifuge. Sf-9 cell pellets were suspended in lysis buffer [50 mM N-(2-Hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) sodium salt (Na-HEPES) (pH 8.0), 300 mM NaCl, 10% glycerol, 1 mM dithiothreitol supplemented with 1× Roche complete protease inhibitors] at a ratio of 100 mL buffer to each pellet from a 1-L culture. The cells were then lysed on ice by passing through a microfuidizer (Microfluidics) at a pressure of 80 psi. Most cell debris was removed by centrifugation at 250,000g.
Two 1 mL metal affinity columns (Nickel HiTrap columns, Life Technologies) were linked in tandem and equilibrated with lysis buffer supplemented with 20 mM imidazole but not protease inhibitors. The lysate from 1 to 2 L of culture fluid was loaded onto the columns at a flow rate of 1 mL/min. Once the lysate was loaded and the column washed with the equilibration buffer until the UV adsorption returned to baseline levels, tobacco etch virus protease (2000 U in 2 mL equilibration buffer) was loaded onto the columns. The mobile phase flow was stopped, and the columns were incubated overnight at room temperature to release the bound BTK from its polyhistidine tag. A third 1 mL Nickel HiTrap column, which had been washed with equilibration buffer, was placed downstream of the columns treated with protease to capture any BTK that remained uncleaved. Protein that was washed from all three columns was collected in 0.5 mL fractions. All fractions containing protein, as judged by UV adsorption, were collected and pooled. A Superdex 200 16/60 size exclusion column (Life Technologies) was equilibrated with buffer [50 mM HEPES (pH 8), 250 mM NaCl, 5% glycerol, 10 mM MgCl2, and 2 mM dithiothreitol]. BTK from the metal affinity columns were loaded onto the size exclusion column at a flow rate of 1 mL /min. After collecting 30 mL buffer in 2 mL fractions, peak fractions were pooled. The pools that contained BTK were concentrated to 10 mg/mL and flash frozen at −80°C.
Crystallization, data collection and processing
Two hours prior to crystallization, BTK protein at 10 mg/mL concentration was incubated on ice with 10 mM tris(2-carboxyethyl)phosphine (pH of 100 mM stock solution adjusted to 7.0 with NaOH), 0.52 mM inhibitor, and 2.6% dimethyl sulfoxide. BTK crystals were obtained at 20°C in hanging drops by mixing 0.5 μL of protein solution with 0.5 μL of 0.1M Na-HEPES (pH 8.0), 33% PEG3350. Crystals were prepared for flash cooling by gradually exchanging the crystallization drop solution to 20% glycerol, 27% PEG3350, 0.08M Na-HEPES (pH 8.0), and 0.1 mM inhibitor.
Synchrotron data collection was provided by Reciprocal Space Consulting, LLC (Oakland, CA) at beam line 9-2 of the Stanford Synchrotron Radiation Laboratory (Palo Alto, CA) and beam line 5.0.2 of the Advanced Light Source (Berkeley, CA) using ADSC Quantum 315 CCD detectors. The diffraction images were processed with DENZO, and the intensities were scaled with SCALEPACK.23
Molecular replacement, structure refinement, and model building
The structure of the first BTK–ligand complex was solved with the molecular replacement program PHASER24 using the crystal structure of murine BTK kinase domain5 (PDB accession number 1K2P) as search model. The solution found was a BTK monomer in space group P21212 with the unit cell dimensions α = 71 Å, β = 105 Å, and γ = 38 Å. The same crystal form has also been obtained for a BTK kinase domain variant containing eight additional amino acids at the N-terminus.6 The model was refined against the experimental data, and electron density maps were calculated using REFMAC5.25 Subsequent BTK–ligand structures were determined by rigid body refinement. The structure models were built with the graphics software MOLOC.26 Statistics on data processing and structure refinement are listed in Table I. Illustrations of the final BTK crystal structures were created using the PyMOL Molecular Graphics System (Schrödinger, LLC).
Table I.
Statistics for X-ray Data Processing and Model Refinement
| Compound | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| PDB accession no. | 3PIX | 3PIY | 3PIZ | 3PJ1 | 3PJ2 | 3PJ3 |
| Data processing | ||||||
| Cell dimensions | ||||||
| a (Å) | 70.0 | 70.9 | 71.3 | 71.7 | 71.4 | 71.6 |
| b (Å) | 104.4 | 105.0 | 105.3 | 104.8 | 104.8 | 105.4 |
| c (Å) | 38.0 | 38.1 | 38.1 | 38.2 | 38.0 | 38.1 |
| Wavelength (Å) | 0.9795 | 0.9795 | 0.9795 | 0.9804 | 0.9795 | 1.0000 |
| Resolution (Å) | 40.00−1.85 | 40.00−2.55 | 40.00−2.20 | 40.00−2.00 | 50.00−1.75 | 40.00−1.85 |
| Total no. reflections | 376,882 | 98,448 | 171,910 | 251,581 | 197,275 | 504,429 |
| No. unique reflectionsa | 22,572 (1492) | 9,339 (805) | 13,820 (909) | 20,034 (1881) | 27,449 (1755) | 24,080 (1560) |
| Multiplicitya | 5.2 (3.6) | 4.9 (3.3) | 4.0 (2.1) | 6.9 (5.0) | 3.9 (2.2) | 5.7 (1.8) |
| Completeness (%)a | 92.6 (63.2) | 95.4 (85.5) | 91.6 (61.8) | 99.6 (96.8) | 92.7 (60.0) | 95.0 (63.1) |
| I/σ(I)a | 13.7 (2.1) | 6.1 (2.1) | 9.0 (2.3) | 13.2 (2.8) | 15.6 (2.4) | 8.4 (1.9) |
| Rsymab (%) | 7.6 (44.8) | 18.1 (50.3) | 13.5 (27.8) | 13.5 (39.3) | 6.7 (23.5) | 15.7 (71.7) |
| Refinement | ||||||
| Resolution (Å) | 37.96−1.85 | 38.04−2.55 | 38.04−2.21 | 59.2−2.00 | 42.22−1.75 | 38.12−1.85 |
| No. reflections | 21,384 | 8860 | 13,089 | 18,971 | 26,007 | 22,792 |
| No. atoms | 2373 | 2227 | 2123 | 2213 | 2220 | 2114 |
| Rcrystc (%) | 22.7 | 22.5 | 22.5 | 22.1 | 22.9 | 23.3 |
| Rfreec (%) | 26.6 | 29.5 | 28.2 | 25.7 | 26.3 | 25.4 |
| Rmsd bond length (Å) | 0.007 | 0.014 | 0.008 | 0.007 | 0.006 | 0.007 |
| Rmsd bond angles (°) | 1.09 | 1.49 | 1.01 | 0.98 | 0.96 | 0.95 |
| Mean B value (Å2) | 30.0 | 28.3 | 38.6 | 24.7 | 31.6 | 29.7 |
| Water molecules | 148 | 38 | 72 | 174 | 142 | 96 |
Number in parenthesis are values for the highest of 10 resolution shells.
Rsym = Σhkl |〈I〉 – I|/Σhkl|I|.
Rcryst = Σhkl|〈Fo〉 – Fc|/Σhkl|Fo|. Rfree is calculated the same way using a random 5% test set of reflections.
Glossary
Abbreviations:
- AH
activation loop helical region
- ATP
adenosine-5′-triphosphate
- BTK
Bruton's tyrosine kinase
- IC50
half maximal inhibitory concentration
- Na-HEPES
N-(2-Hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) sodium salt
- SH
Src homology.
References
- 1.Brunner C, Muller B, Wirth T. Bruton's tyrosine kinase is involved in innate and adaptive immunity. Histol Histopathol. 2005;20:945–955. doi: 10.14670/HH-20.945. [DOI] [PubMed] [Google Scholar]
- 2.Lindvall JM, Blomberg KE, Valiaho J, Vargas L, Heinonen JE, Berglof A, Mohamed AJ, Nore BF, Vihinen M, Smith CI. Bruton's tyrosine kinase: cell biology, sequence conservation, mutation spectrum, siRNA modifications, and expression profiling. Immunol Rev. 2005;203:200–215. doi: 10.1111/j.0105-2896.2005.00225.x. [DOI] [PubMed] [Google Scholar]
- 3.Pan Z. Bruton's tyrosine kinase as a drug discovery target. Drug News Perspect. 2008;21:357–362. doi: 10.1358/dnp.2008.21.7.1255308. [DOI] [PubMed] [Google Scholar]
- 4.Uckun FM, Qazi S. Bruton's tyrosine kinase as a molecular target in treatment of leukemias and lymphomas as well as inflammatory disorders and autoimmunity. Expert Opin Ther Pat. 2010;20:1457–1470. doi: 10.1517/13543776.2010.517750. [DOI] [PubMed] [Google Scholar]
- 5.Mao C, Zhou M, Uckun FM. Crystal structure of Bruton's tyrosine kinase domain suggests a novel pathway for activation and provides insights into the molecular basis of X-linked agammaglobulinemia. J Biol Chem. 2001;276:41435–41443. doi: 10.1074/jbc.M104828200. [DOI] [PubMed] [Google Scholar]
- 6.Marcotte DJ, Liu YT, Arduini RM, Hession CA, Miatkowski K, Wildes CP, Cullen PF, Hong V, Hopkins BT, Mertsching E, Jenkins TJ, Romanowski MJ, Baker DP, Silvian LF. Structures of human Bruton's tyrosine kinase in active and inactive conformations suggest a mechanism of activation for TEC family kinases. Protein Sci. 2010;19:429–439. doi: 10.1002/pro.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cowan-Jacob SW, Mobitz H, Fabbro D. Structural biology contributions to tyrosine kinase drug discovery. Curr Opin Cell Biol. 2009;21:280–287. doi: 10.1016/j.ceb.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 8.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 9.Jacobs MD, Caron PR, Hare BJ. Classifying protein kinase structures guides use of ligand-selectivity profiles to predict inactive conformations: structure of lck/imatinib complex. Proteins. 2008;70:1451–1460. doi: 10.1002/prot.21633. [DOI] [PubMed] [Google Scholar]
- 10.Boyd E, Brookfield F, Georges G, Goller B, Huensch S, Rueger P, Rueth M, Scheiblich S, Schuell C, Von Der Saal W, Warne J, Weigand S. 2006. Preparation of novel phthalazinone derivatives as Aurora-A kinase inhibitors for use against illnesses such as cancer. Patent Application WO2006032518 A1.
- 11.Goldstein DM, Rueth M. 2007. Methods of inhibiting BTK and SYK protein kinases using (pyrazolylamino)phthalazines and their preparation. Patent Application US2007219195 A1.
- 12.Kutach AK, Villasenor AG, Lam D, Belunis C, Janson C, Lok S, Hong LN, Liu CM, Deval J, Novak TJ, Barnett JW, Chu W, Shaw D, Kuglstatter A. Crystal structures of IL-2-inducible T cell kinase complexed with inhibitors: insights into rational drug design and activity regulation. Chem Biol Drug Des. 2010;76:154–163. doi: 10.1111/j.1747-0285.2010.00993.x. [DOI] [PubMed] [Google Scholar]
- 13.Toniolo C, Benedetti E. The polypeptide 310-helix. Trends Biochem Sci. 1991;16:350–353. doi: 10.1016/0968-0004(91)90142-i. [DOI] [PubMed] [Google Scholar]
- 14.Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grossbard EB, Magilavy DB. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med. 2010;363:1303–1312. doi: 10.1056/NEJMoa1000500. [DOI] [PubMed] [Google Scholar]
- 15.Braselmann S, Taylor V, Zhao H, Wang S, Sylvain C, Baluom M, Qu K, Herlaar E, Lau A, Young C, Wong BR, Lovell S, Sun T, Park G, Argade A, Jurcevic S, Pine P, Singh R, Grossbard EB, Payan DG, Masuda ES. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J Pharmacol Exp Ther. 2006;319:998–1008. doi: 10.1124/jpet.106.109058. [DOI] [PubMed] [Google Scholar]
- 16.Norman P. A novel Syk kinase inhibitor suitable for inhalation: R-343(?)–WO-2009031011. Expert Opin Ther Pat. 2009;19:1469–1472. doi: 10.1517/13543770903059281. [DOI] [PubMed] [Google Scholar]
- 17.Villasenor AG, Kondru R, Ho H, Wang S, Papp E, Shaw D, Barnett JW, Browner MF, Kuglstatter A. Structural insights for design of potent spleen tyrosine kinase inhibitors from crystallographic analysis of three inhibitor complexes. Chem Biol Drug Des. 2009;73:466–470. doi: 10.1111/j.1747-0285.2009.00785.x. [DOI] [PubMed] [Google Scholar]
- 18.Goldstein DM, Alfredson T, Bertrand J, Browner MF, Clifford K, Dalrymple SA, Dunn J, Freire-Moar J, Harris S, Labadie SS, La FJ, Lapierre JM, Larrabee S, Li F, Papp E, McWeeney D, Ramesha C, Roberts R, Rotstein D, San PB, Sjogren EB, So OY, Talamas FX, Tao W, Trejo A, Villasenor A, Welch M, Welch T, Weller P, Whiteley PE, Young K, Zipfel S. Discovery of S-[5-amino-1-(4-fluorophenyl)-1H-pyrazol-4-yl]-[3-(2,3-dihydroxypropoxy)ph enyl]methanone (RO3201195), an orally bioavailable and highly selective inhibitor of p38 MAP kinase. J Med Chem. 2006;49:1562–1575. doi: 10.1021/jm050736c. [DOI] [PubMed] [Google Scholar]
- 19.Goldstein DM, Kuglstatter A, Lou Y, Soth MJ. Selective p38alpha inhibitors clinically evaluated for the treatment of chronic inflammatory disorders. J Med Chem. 2010;53:2345–2353. doi: 10.1021/jm9012906. [DOI] [PubMed] [Google Scholar]
- 20.Engh R, Hertenberger H, Honold K, Masjost B, Rueger P, Schaefer W, Scheiblich S, Schwaiger M. 2007. Azabenzimidazole derivatives, their manufacture and use as anti-cancer agents. Patent Application WO2007017143.
- 21.Guo S, Wahl MI, Witte ON. Mutational analysis of the SH2-kinase linker region of Bruton's tyrosine kinase defines alternative modes of regulation for cytoplasmic tyrosine kinase families. Int Immunol. 2006;18:79–87. doi: 10.1093/intimm/dxh351. [DOI] [PubMed] [Google Scholar]
- 22.Mahajan S, Ghosh S, Sudbeck EA, Zheng Y, Downs S, Hupke M, Uckun FM. Rational design and synthesis of a novel anti-leukemic agent targeting Bruton's tyrosine kinase (BTK), LFM-A13 [alpha-cyano-beta-hydroxy-beta-methyl-N-(2,5-dibromophenyl)propenamide] J Biol Chem. 1999;274:9587–9599. doi: 10.1074/jbc.274.14.9587. [DOI] [PubMed] [Google Scholar]
- 23.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CWJ, Sweet RM, editors. Macromolecular crystallography, part A. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 24.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 25.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 26.Gerber PR. Peptide mechanics: a force field for peptides and proteins working with entire residues as small unites. Biopolymers. 1992;32:1003–1017. [Google Scholar]
- 27.Dinh M, Grunberger D, Ho H, Tsing SY, Shaw D, Lee S, Barnett J, Hill RJ, Swinney DC, Bradshaw JM. Activation mechanism and steady state kinetics of Bruton's tyrosine kinase. J Biol Chem. 2007;282:8768–8776. doi: 10.1074/jbc.M609920200. [DOI] [PubMed] [Google Scholar]