

Abstract
Staphylococcal α-hemolysin is expressed as a water-soluble monomeric protein and assembles on membranes to form a heptameric pore structure. The heptameric pore structure of α-hemolysin can be prepared from monomer in vitro only in the presence of deoxycholate detergent micelles, artificially constructed phospholipid bilayers, or erythrocytes. Here, we succeeded in preparing crystals of the heptameric form of α-hemolysin without any detergent but with 2-methyl-2,4-pentanediol (MPD), and determined its structure. The structure of the heptameric pore was similar to that reported previously. In the structure, two molecules of MPD were bound around Trp179, around which phospholipid head groups were bound in the heptameric pore structure reported previously. Size exclusion chromatography showed that α-hemolysin did not assemble spontaneously even when stored for 1 year. SDS-PAGE analysis revealed that, among the compounds in the crystallizing buffer, MPD could induce heptamer formation. The concentration of MPD that most efficiently induced oligomerization was between 10 and 30%. Based on these observations, we propose MPD as a reagent that can facilitate heptameric pore formation of α-hemolysin without membrane binding.
Keywords: staphylococcal α-hemolysin; 2-methyl-2,4-pentanediol (MPD); heptamer formation; crystal structure
Introduction
Staphylococcal α-hemolysin is a β-barrel pore-forming toxin, which is toxic for a wide range of mammalian cell types, particularly erythrocytes. α-Hemolysin is secreted as a water-soluble monomeric protein with a molecular mass of ∼33 kDa, which binds to target membranes and forms membrane-inserted heptameric pores.1 Based on a wealth of biochemical data and structural information, a consensus model was proposed for α-hemolysin assembly as follows. First, the α-hemolysin binds to the membrane of a target cell as a monomeric protein. Upon binding, the membrane-bound monomer assembles into a nonlytic heptameric prepore, and the membrane-spanning domain, called the stem domain, penetrates the lipid bilayer, eventually forming a membrane-embedded heptamer.1–6
The crystal structure of the heptameric α-hemolysin has been determined, in which each protomer assembles along a noncrystallographic sevenfold axis.7–9 The overall heptameric structure is divided into three domains, that is, the cap, rim, and stem domains. The cap domain is composed of seven β-sandwiches from each protomer. The rim domain consists of four β-strands with a loop-rich structure, and lies beneath the cap domain. The stem domain forms the transmembrane portion of the channel by a 14-strand antiparallel β-barrel. A crevice between the stem and rim domain is thought to comprise the lipid binding domain as lipid head groups were shown to bind at this region.9 The crystal structure of the monomeric α-hemolysin is still to be elucidated, although it can be predicted from the monomeric structures of leukocidin components10–12 and/or Vibrio cholerae cytolysin, which show sequence identity to α-hemolysin, and it was also analyzed by small angle X-ray scattering.13
The oligomeric pore structure can be prepared from monomeric α-hemolysin in vitro. Deoxycholate detergent micelles can induce heptamerization, and are now widely used to prepare heptameric α-hemolysin.14 The crystal structure of heptameric α-hemolysin described earlier was assembled by deoxycholate.8,9 In addition, artificially constructed phospholipid bilayers facilitate oligomerization.15–17 However, atomic force microscopy (AFM) experiments showed that hexameric pores were also formed in phospholipid bilayers.18 These fully assembled pores are insensitive to sodium dodecyl sulfate (SDS) at room temperature, and therefore they were confirmed to be oligomers by SDS-polyacrylamide gel electrophoresis (SDS-PAGE).14,18
In this study, we prepared crystals from monomeric α-hemolysin and determined the structure to reveal the monomeric structure. The revealed structure was, however, heptameric α-hemolysin. Beginning with this unexpected result, we showed that 2-methyl-2,4-pentanediol (MPD) induced heptamerization of α-hemolysin by analogy with deoxycholate.
Results
Crystal structure of α-hemolysin in MPD
We purified monomeric α-hemolysin, prepared crystals using the monomeric protein, and determined the crystal structure. We expected the monomeric protein to be crystallized as neither membranes (natural or artificial) nor deoxycholate, which induce the formation of heptameric pores,14–16 were added. The monomeric state of the protein in the crystallization setup was confirmed by size exclusion chromatography. In contrast to our expectations, however, a heptameric pore structure was determined, which was similar to that of heptamers constructed by deoxycholate (Fig. 1). There were four molecules in the heptameric structure, that is, 28 protomers, of α-hemolysin in an asymmetric unit. By analogy to the heptameric structure of α-hemolysin reported previously by Song et al.,8 our model shows three domains: the cap domain, the rim domain, and the stem domain. The structure of Song et al.8 (PDB ID: 7AHL) and our structure superpose well with a root mean square deviation (r.m.s.d.) of 0.42 Å for 879 Cα atoms in the heptamer. Figure 1(C) shows the average r.m.s.d. of all atoms for one heptamer of our model and 7AHL. The results showed that Trp179 was specifically perturbed. This residue was located at the base of the rim domain [Fig. 2(A)], and faced the stem regions of two adjacent protomers. Interestingly, two molecules of MPD (referenced as MPD1 and MPD2, respectively), which was added as a precipitant in the crystallization buffer, were bound to Trp179 [Fig. 2(B)]. MPD1 was bound in a crevice composed of Met197, Lys198, Thr199, Arg200, and Trp179. In the four heptamers of the asymmetric unit, a total of 734 and 474 interactions shorter than 4 Å were observed for MPD1 and MPD2, respectively (Table I). Table II shows interactions commonly observed in more than 10 protomers. The residue with the largest number of interactions with MPD1 was Arg200 with 168 interactions in 28 protomers, followed by Trp179 with 144 interactions. MPD2 was located in a space among three protomers, which is surrounded by the rim domain in a protomer and stem domains of two adjacent protomers. MPD2 made interactions with Asn178, Trp179, and Gly180 of a protomer, Ser114 in the stem domain of the adjacent protomer, and His144 and Leu146 in the stem domain of the next to adjacent protomer (Table II). Of a total of 474 observed interactions, 58% were formed with Trp179-Gly180 (Table I). The numbers of interactions by Trp179 and Arg200 with MPDs were significantly larger than those of the others, suggesting the importance of these residues in MPD recognition.
Figure. 1.
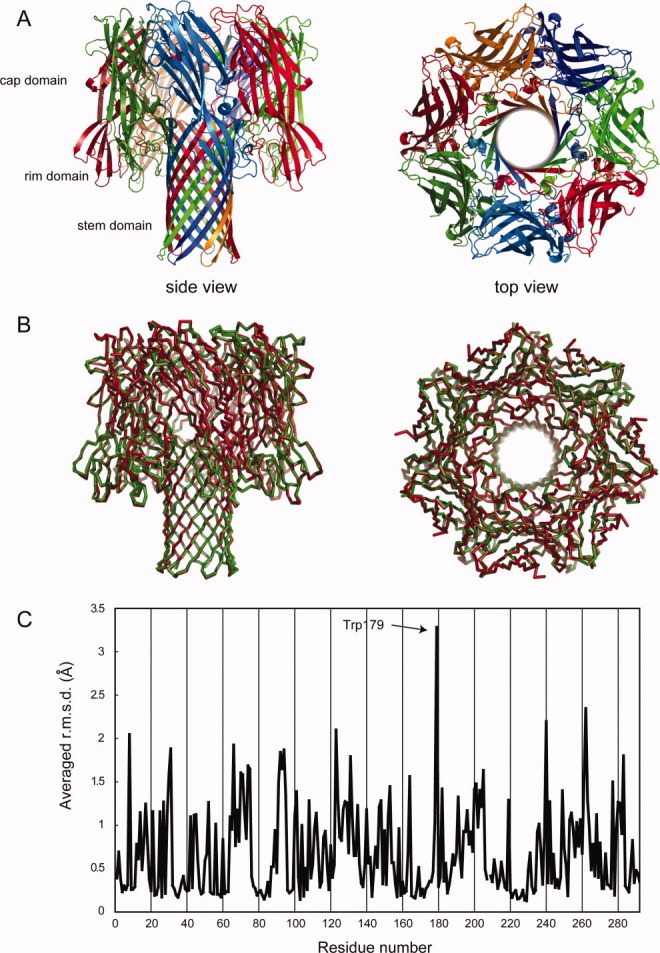
The revealed heptameric structure of α-hemolysin. (A) Overall structure of heptameric structure of α-hemolysin from side (left) and top (right). Protomers are colored individually. (B) Superposition of the heptamer determined in this study (red) and that prepared by deoxycholate (green). Cα traces are shown. (C) The averaged r.m.s.d. of all atoms in each residue between heptamers shown in Fig. 1B. A heptamer was superposed onto the heptamer reported by Song et al. (PDB ID: 7AHL). The r.m.s.d. values are displayed as per-residue averages.
Figure. 2.

MPD binding site. (A) MPD1, MPD2, (orange and red CPK, respectively) and Trp179 (green CPK) in the heptamer. (B) Close-up view of the MPD binding site. Two MPD molecules, Trp179, and Arg200 are shown as sticks. Fo-Fc omit map (contoured at 2.0σ) around the bound MPDs are also shown. Different protomers are distinguished in the ribbon diagrams by different colors. (C) Close-up view of GPC binding site. The bound GPC, Trp179, and Arg200 are shown as sticks. The orientation is aligned to Fig. 2B. The bound DiC3PC was similar to the structure with GPC and is therefore not shown.
Table I.
The Number of Observed Interactions with MPD Within 4Å in Individual Residues
| Residue | Number of interactions with MPD1a | Number of interactions with MPD2a | Total |
|---|---|---|---|
| Tyr112 | 0 | 2 | 2 |
| Ser114 | 0 | 52 | 52 |
| Thr115 | 0 | 6 | 6 |
| Leu116 | 0 | 19 | 19 |
| Ile142 | 0 | 4 | 4 |
| His144 | 0 | 27 | 27 |
| Leu146 | 0 | 39 | 39 |
| Asn176 | 103 | 0 | 103 |
| Gln177 | 56 | 0 | 56 |
| Asn178 | 0 | 49 | 49 |
| Trp179 | 144 | 132 | 276 |
| Gly180 | 0 | 144 | 144 |
| Tyr182 | 9 | 0 | 9 |
| Gln194 | 54 | 0 | 54 |
| Met197 | 85 | 0 | 85 |
| Lys198 | 80 | 0 | 80 |
| Thr199 | 35 | 0 | 35 |
| Arg200 | 168 | 0 | 168 |
| 734 | 474 |
Interactions in all 28 protomers are summed.
Table II.
Interactions of MPD with the Protein
| MPD | Atom | Residue | Atom | Averaged distancea | Number of protomers in which the interaction was observed |
|---|---|---|---|---|---|
| MPD1 | CM | Asn176(A)b | Cγ | 4.12 | 18 |
| Asn176(A) | O | 4.09 | 20 | ||
| Asn176(A) | Nδ2 | 4.10 | 16 | ||
| Asn176(A) | Oδ1 | 4.10 | 16 | ||
| Gln177(A) | Nɛ2 | 4.42 | 10 | ||
| Trp179(A) | Cβ | 4.28 | 12 | ||
| Trp179(A) | Cδ1 | 4.26 | 14 | ||
| Trp179(A) | Cγ | 4.23 | 13 | ||
| Met197(A) | O | 4.29 | 10 | ||
| C1 | Trp179(A) | Cγ | 4.41 | 10 | |
| Gln194(A) | Oɛ1 | 3.83 | 19 | ||
| Arg200(A) | Cδ | 4.52 | 11 | ||
| Arg200(A) | Cγ | 4.33 | 11 | ||
| Arg200(A) | Cζ | 3.93 | 18 | ||
| Arg200(A) | Nɛ | 3.91 | 15 | ||
| Arg200(A) | Nη | 3.76 | 20 | ||
| O2 | Asn176(A) | Nδ2 | 4.06 | 15 | |
| Gln194(A) | Oɛ1 | 4.22 | 13 | ||
| Met197(A) | O | 3.45 | 25 | ||
| C2 | Met197(A) | O | 4.11 | 10 | |
| C3 | Gln177(A) | Nɛ2 | 4.40 | 10 | |
| Lys198(A) | O | 4.11 | 14 | ||
| O4 | Trp179(A) | Cδ1 | 4.27 | 11 | |
| Trp179(A) | Nɛ1 | 4.04 | 17 | ||
| C5 | Gln177(A) | Nɛ2 | 4.44 | 15 | |
| Lys198(A) | O | 3.80 | 17 | ||
| Thr199(A) | Cα | 4.51 | 10 | ||
| MPD2 | CM | Ser114(F) | O | 4.05 | 16 |
| His144(G) | O | 4.01 | 20 | ||
| C1 | Leu146(G) | Cβ | 4.36 | 11 | |
| Leu146(G) | Cδ2 | 4.25 | 13 | ||
| Asn178(A) | O | 4.29 | 18 | ||
| Gly180(A) | C | 4.12 | 18 | ||
| Gly180(A) | N | 4.12 | 18 | ||
| Gly180(A) | O | 3.14 | 24 | ||
| C2 | Gly180(A) | O | 3.75 | 25 | |
| O2 | Gly180(A) | O | 3.49 | 23 | |
| C3 | Ser114(F) | Cβ | 4.29 | 14 | |
| O4 | Asn178(A) | O | 4.14 | 17 | |
| C5 | Trp179(A) | Cα | 4.26 | 14 | |
| Trp179(A) | Cδ2 | 4.09 | 16 | ||
| Trp179(A) | Cɛ3 | 3.70 | 24 | ||
| Trp179(A) | Cζ3 | 4.17 | 15 | ||
| Gly180(A) | N | 4.38 | 12 |
Distances in all 28 protomers were averaged.
Letters in parentheses indicate the chain of the protomer when focusing on MPD mainly interacting with chain A.
Interactions within 4Å that were observed in more than 10 protomers are listed. The number of protomer in which the interaction was observed, and averaged distance in all 28 protomers are also shown. Interactions shown as bold are hydrogen bonds.
Identification of a factor that induced heptamerization
There are two possible explanations for the production of heptamer crystals from a solution of monomeric α-hemolysin. The first is that α-hemolysin can spontaneously assemble into heptamer in solution when stored at high concentration, and the generated heptamers were crystallized. The other is that heptamers were formed during crystallization, facilitated by some component(s) of the crystallization buffer. To evaluate oligomer formation during storage in aqueous solution, the time dependence of changes in molecular weight was examined by size exclusion chromatography (Fig. 3). Comparison of the elution volumes of molecular markers indicated that the purified protein was a monomer, as the deduced molecular mass of monomeric α-hemolysin is ∼33 kDa. Even after incubation for 1 year, no heptamer formation was seen, indicating that α-hemolysin does not form the heptamer spontaneously even on long-term storage at a high concentration. Therefore, it is plausible that the crystallization buffer used in this study induced heptameric pore formation.
Figure. 3.

Time dependence of the change in molecular weight of purified monomeric α-hemolysin. The chromatograms of size exclusion chromatography for samples stored for 1 day, 2 weeks, 4 weeks, 10 weeks, and 1 year are shown. For clarity, the offset in absorption units (AU) is chosen arbitrarily. The elution volumes of molecular weight standards are also shown.
The crystallizing conditions contained 0.2 M ammonium acetate and 35% MPD. The effects of these components on heptamer formation were examined by SDS-PAGE (Fig. 4). As the heptameric pore structure of α-hemolysin is resistant to SDS,4,14 a new band should appear around 200 kDa on SDS-PAGE if heptameric pores are formed. Figure 4(A) shows that SDS-resistant heptamer was formed when 20% MPD was added, and this effect was independent of ammonium acetate [Fig. 4(A) lanes 3 and 4, respectively]. These results indicated that the heptameric pore structure in the crystal was induced by MPD added as a precipitant to the crystallization buffer.
Figure. 4.
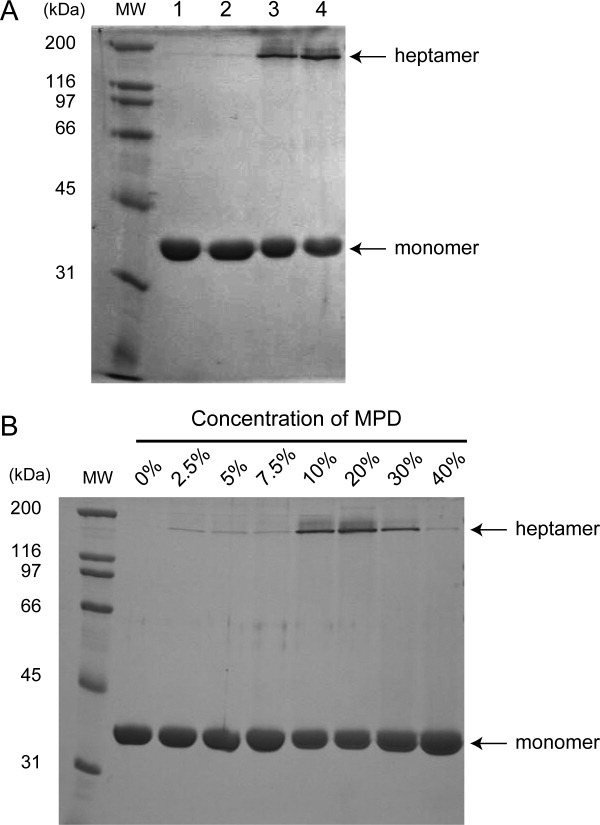
Contribution of MPD on heptamerization. (A) Effects of each component in the crystallization buffer on heptamerization. lane 1; purified monomeric α-hemolysin only, lane 2; 0.2 M ammonium acetate, lane 3; 20% MPD, lane 4; 0.2 M ammonium acetate and 20% MPD, MW; molecular weight marker. (B) Concentration dependence of MPD on heptamerization. The final concentrations of MPD are shown at the top of the gel.
Next, the influence of MPD concentration on oligomer formation was evaluated [Fig. 4(B)]. Although only small amounts of oligomer were formed at concentrations lower than 7.5%, significant amounts of oligomer appeared between 10 and 30% MPD, indicating the dependence of oligomer formation on MPD concentration. However, adding MPD at concentrations in excess of 40% caused a decrease in oligomer formation. Taken together, these observations indicated that a MPD concentration of around 20% showed the most efficient SDS-resistant oligomer formation.
Discussion
The crystal structure revealed in this study was a heptameric pore form despite the use of monomeric α-hemolysin for crystallization (Fig. 1). MPD in the crystallization buffer was shown to induce heptamer formation, whereas monomeric α-hemolysin itself did not form oligomers spontaneously (Figs. 3 and 4). In the crystal structure, two MPD molecules were bound around Trp179 in each protomer (Fig. 2). It should be noted that lipid head groups, GPC and DiC3PC, were shown to bind at the same site as MPD1 [Fig. 2(C)].9 The binding characteristics of GPC and DiC3PC were similar in the reported structure.9 In the structure of the LukF monomer, DiC3PC was bound at Trp177, which corresponds to Trp179 of α-hemolysin.10 Based on the crystal structure of α-hemolysin in complex with GPC and DiC3PC, Galdiero and Gouaux proposed that α-hemolysin penetrates the target membrane by specifically recognizing lipid head groups with Trp179 and Arg200, and perturbs the structure of the lipid bilayer.9 Taken together, we concluded that in the crystal structure presented in this study, MPD1 facilitates heptamerization of α-hemolysin by a mechanism similar to that of lipids in a membrane, like GPC and DiC3PC, that is, it binds specifically with Trp179 and Arg200 and induces heptamerization. In addition, Galdiero and Gouaux. proposed the importance of lipids in mediating interactions between protomers in the heptamer.9 MPD2 was located at a pocket surrounded by three protomers and interacted with residues in the extended stem domain of two protomers (Table II). As the stem domain is folded beside the rim domain before β-barrel formation, we concluded that binding of MPD2 occurred during and/or after the oligomerization process. Then, MPD2 would facilitate interprotomer interactions and stabilize the heptameric pore [Fig. 2(C)]. However, in the crystal structure of the α-hemolysin heptamer in complex with lipid head groups, GPC was not bound at the same site as MPD2.9 α-Hemolysin has been reported to form heptameric pores in artificially constructed phospholipid bilayers, that is, liposomes composed of phosphatidylcholine or sphingomyelin and cholesterol.15–17 Although cholesterol was not essential for the initial binding of α-hemolysin to the lipid bilayer, inclusion of cholesterol in the liposomes enhanced the toxin activity.17 Valeva et al. proposed that cholesterol forms a microdomain with sphingomyelin, the binding and assembly site for α-hemolysin, then facilitates the receptor-less targeting of the toxin.19 In addition, cholesterol has been reported to increase membrane fluidity, which promotes the assembly process of α-hemolysin.20 Inclusion of cholesterol into the liposome causes a conformational change of membrane-bound α-hemolysin molecules. Under these conditions, almost all α-hemolysin lost the N-terminal 8 amino acids by trypsin treatment.20 The N-terminal region (amino-latch) becomes sensitive for proteolysis in the prepore state, and its location changes during pore formation.10,21 Taken together, these observations suggest that cholesterol may facilitate the conformational change of α-hemolysin bound on the lipid bilayer during pore formation. Hence, we postulate an additional novel role of cholesterol in the membrane as a stabilizer or facilitator of α-hemolysin heptamer pore structure. Deoxycholate, which has a steroid structure, is widely used to induce oligomerization of α-hemolysin without a lipid bilayer.14 The heptameric pore structures of α-hemolysin in complex with lipid head groups were prepared using deoxycholate.8,9 Therefore, we suppose that deoxycholate acts instead of cholesterol, and C3-OH and A ring of steroid structure may interact with the rim domain of α-hemolysin for pore formation in the presence of lipid head groups. In the crystal structure reported previously, however, deoxycholate was not bound at the lipid head group binding site.8,9 Deoxycholate would provide an environment similar to the lipid bilayer containing cholesterol, such as a hydrophobic environment surrounded by a hydrophilic surface as micelles, and α-hemolysin may form the stable heptameric pore structure spontaneously in the micelles. However, MPD is unable to form micelles as it has neither a long aliphatic tail nor a hydrophilic head group. In addition, MPD1 was bound specifically at the lipid head group binding site, suggesting that the mechanisms of action by deoxycholate and MPD1 in inducing oligomerization are not identical. On the other hand, as MPD2 would induce oligomer formation by binding with Trp179 at a site different from the lipid head group binding site, MPD2 would facilitate oligomer formation by supplying an environment imitating a biological membrane. In this regard, the hydrophobicity of MPD derived from the methyl or methylene group may help to stabilize hydrophobic stem domains, which act as a transmembrane region. In summary, it is reasonable to conclude that heptamers were formed by MPD1 acting as a lipid head group and that of MPD2 providing a membrane-like environment due to its hydrophobicity like deoxycholate, indicating the importance of lipid recognition for oligomerization. Structure comparison showed significant flexibility of Trp179, which is a phospholipid binding residue [Fig. 1(C)]. Meesters et al. also reported the flexibility at the same site based on molecular dynamics simulation.13 The flexibility around the lipid recognition site may facilitate lipid recognition. To determine the detailed mechanism of action of MPD in pore formation by α-hemolysin, it will be necessary to examine dynamic structural changes in solution using spectrometric methods.
The concentration of MPD that most efficiently induced oligomerization was from 10% to 30% [Fig. 4(B)]. Fortunately, crystals of heptameric α-hemolysin grew from a buffer containing 35% MPD, suggesting that two transitions, that is, heptamer formation and heptamer crystallization, occurred simultaneously during the crystallization procedure. Growth of well-diffracting crystals required more than 6 months. Although crystals grew from a buffer containing a high concentration of MPD several weeks later, they did not diffract well. These results suggest that a high concentration of MPD facilitates crystallization, but adversely affects stable heptamer formation. Both requirements would be fulfilled at 35% MPD.
In the structure of LukF monomer, DiC3PC was bound at the corresponding site to MPD1 of α-hemolysin, as described earlier.10 As MPD1 in this study was bound at the same position as DiC3PC and GPC in the previously reported structure, it is likely that MPD1 also binds at the lipid-head binding pocket in other two-component toxins and facilitates oligomer formation.
Materials and Methods
Construction of the expression vector for α-hemolysin—A DNA fragment encoding α-hemolysin without the signal sequence was amplified using KOD-Plus DNA polymerase (Toyobo, Osaka, Japan), with S. aureus strain Mu50 (ATCC 700699) genomic DNA as the template and the following primers: α-hemolysin-S (5′-CATGCCATGGCAGATTCTGATATTAATATTAAAACCGG-3′), α-hemolysin-AS (5′-CCGCTCGAGATTTGTCATTTCTTCTTTTTCCCAATCG-3′), (recognition sites for restriction enzyme are underlined). The PCR products were inserted into the NcoI and XhoI sites of the pET28-b vector (Merck, Whitehouse Station, NJ). A His6-tag was fused at the C-terminus for the purpose of purification. The construct was verified by sequencing using an ABI 310 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Expression and purification of monomeric α-hemolysin—Transformed Escherichia coli strain B834 (DE3) harboring an α-hemolysin expression vector and pRAREII (Merck) was grown at 37°C in LB medium supplemented with 50 μg mL−1 kanamycin and 34 μg mL−1 chloramphenicol with shaking at 150 rpm until the early stationary phase. To induce expression of the desired protein, isopropyl-β-d-thiogalactopyranoside was added to a final concentration of 0.5 mM, and the culture was continued for 18 h at 25°C.
Cells were harvested by centrifugation at 5000g for 10 min at 4°C, and then disrupted using a sonicator (Branson, Danbury, CT) in 50 mM Tris-HCl (pH 8.0), 300 mM NaCl. During sonication, the sample was kept at 4°C using a NESLAB RTE cooling bath circulator (Thermo Fisher Scientific, Waltham, MA). The cell debris was removed by centrifugation at 40,000g for 30 min at 4°C, and the supernatant was loaded onto a HisTrap column (GE Healthcare Biosciences AB, Uppsala, Sweden) pre-equilibrated with 20 mM Tris-HCl (pH 8.0), 300 mM NaCl. The column was washed with the same buffer and the adsorbed protein was eluted with 50 mL of a 0–0.5 M gradient of imidazole in 20 mM Tris-HCl (pH 8.0), 300 mM NaCl. Fractions containing α-hemolysin were dialyzed against 20 mM Tris-HCl (pH 8.0), 200 mM NaCl, and then further purified on a HiLoad 26/60 Superdex 200-pg column (GE Healthcare Biosciences AB) equilibrated with 20 mM Tris-HCl (pH 8.0), 200 mM NaCl. Fractions of the elution volume from 200 to 220 mL corresponding to the molecular weight of monomeric α-hemolysin were collected and used for further experiments. Purified monomeric α-hemolysin was concentrated to 5 mg mL−1 and stored at 4°C until use.
Crystallization and X-ray diffraction data collection—The initial crystallization conditions were screened at 20°C using Wizard I, Wizard II (Emerald Biostructures, Bainbridge Island, WA), Index (Hampton Research, Laguna Hills, CA), Nextal Classics Suites, and Nextal Cryos Suites (QIAGEN GmbH, Hilden, Germany). Initial crystals were grown under two sets of conditions with the Index kit (No. 51; 0.1 M Bis-Tris pH 6.5, 45% MPD, and 0.2 M ammonium acetate, and No. 52; 0.1 M HEPES pH 7.5, 45% MPD, and 0.2 M ammonium acetate). These crystallizing conditions were optimized by altering the concentrations of MPD and ammonium acetate and the pH. Crystals suitable for further experiments were grown by the sitting-drop vapor diffusion method from a solution containing 0.1 M HEPES (pH 7.5), 0.2 M ammonium acetate, and 35% (v/v) MPD more than 6 month after initial setup. Although crystals were grown several weeks after initial setup, these crystals did not diffract well. In addition, crystals grown from a solution using Bis-Tris instead of HEPES could not be used due to their high mosaicity. X-ray diffraction experiments were performed on the beamline BL5A at Photon Factory (Tsukuba, Japan) under cryogenic conditions (100 K). A diffraction data set was collected up to a resolution of 2.3 Å with a Quantum 315r detector (ADSC, Poway, CA). The α-hemolysin crystal belonged to space group P21 with unit cell parameters a = 102.8 Å, b = 293.9 Å, c = 170.5 Å, β = 92.4°. Diffraction data were indexed, integrated, scaled, and merged using the HKL2000 program package.22
Structure determination and refinement—the structure of α-hemolysin was determined by the molecular replacement method using the program MOLREP23 by using the structure of the protomer in heptameric α-hemolysin (PDB ID: 7AHL) as a search probe. Although monomeric α-hemolysin was used for crystallization, the revealed structure was heptameric α-hemolysin. Four α-hemolysin heptamers, that is, 28 protomers of α-hemolysin, were present in an asymmetric unit. To monitor the refinement, a random 7% subset from all reflections was set aside for calculation of the Rfree factor. The positional and individual B factor refinements were carried out automatically with the program LAFIRE.24 After automatic refinement and model fitting by LAFIRE, several cycles of manual model fitting with Coot25 and refinement with phenix.refine26 were carried out; phenix.refine included anisotropic scaling, bulk solvent correction, NCS restrained coordinate refinement, and TLS refinement. NCS restraints were applied to the nonflexible residues of all chains with the following ranges: 1–12, 19–64, 76–89, 97–124, 136–189, 210–254, and 266–293. Three TLS groups per chain (residues 1–98, 99–167, and 168–295) were determined by TLSMD web server.27 Finally, water molecules were picked automatically by the program phenix.refine, and then ligand molecules were placed manually. The crystallographic Rwork and Rfree values converged to 20.6 and 23.3%,, respectively. The stereochemical qualities of the final refined models were analyzed using the program RAMPAGE.28 The refinement statistics are summarized in Table III.
Table III.
X-ray Data Collection and Refinement Statistics
| Data collection | |
| Space group | P21 |
| Cell dimensions (Å, °) | a = 102.8 |
| b = 293.9 | |
| c = 170.5 | |
| β = 92.4 | |
| Beamline | Photon factory BL5A |
| Resolution (Å)a | 50 – 2.30 (2.34 – 2.30) |
| Wavelength (Å) | 1.00000 |
| Rsym (%)ab | 8.5 (39.9) |
| <I>/<σI>a | 28.3 (3.6) |
| Completeness (%)a | 99.1 (99.0) |
| Observed reflections | 1,503,616 |
| Unique reflections | 439,066 |
| Multiplicitya | 3.4 (3.1) |
| Refinement and model quality | |
| Resolution range (Å) | 20 – 2.3 |
| No. of reflections | 433,754 |
| Rwork (%)c | 19.31 |
| Rfree (%)d | 22.62 |
| Total protein atoms | 65,983 |
| Total ligand atoms | 560 |
| Total water atoms | 2,064 |
| r.m.s.d. from ideal | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 0.911 |
| Dihedral angles (°) | 13.111 |
| Ramachandran plot | |
| Residues in favored regions (%) | 96.0 |
| Residues in allowed regions (%) | 4.0 |
| Residues in outlier regions (%) | 0 |
The values in parentheses refer to data in the highest resolution shell.
Rsym=ΣhΣi|Ih,I–<Ih>|/ΣhΣi|Ih,i|, where <Ih> is the mean intensity of a set of equivalent reflections.
R =Σ|Fobs–Fcalc|/ΣFobs, where Fobs and Fcalc are observed and calculated structure factor amplitudes, respectively. Rfree was calculated with a random 7% subset from all reflections which was not included in the refinement. Rwork was calculated with remaining reflections.
Superposition of structures and calculation of r.m.s.d were performed by the align algorithm of the program PyMOL (The PyMOL Molecular Graphics System, Schrödinger, LLC, New York, NY). Interaction between MPD and α-hemolysin was calculated based on the proximity of atoms using the program CONTACT in the ccp4 program package.29
Estimation of oligomer formation in aqueous solution by size exclusion chromatography—Purified monomeric α-hemolysin was stored until use at a concentration of 4.9 mg mL−1 in 20 mM Tris-HCl (pH 8.0), 200 mM NaCl at 4°C. After 1 day, 2 weeks, 4 weeks, 10 weeks, and 1 year, 2 mL of the stored α-hemolysin was loaded onto a HiLoad 26/60 Superdex 200-pg column (GE Healthcare Biosciences AB) pre-equilibrated with 20 mM Tris-HCl (pH 8.0), 200 mM NaCl.
Evaluation of the effects of the components in the crystallization buffer on oligomer formation—MPD and ammonium acetate were added to the purified monomeric α-hemolysin to final concentrations of 20% and 0.2 M, respectively, and then incubated at 25°C for 24 h. An equal volume of buffer containing SDS [62.5 mM Tris-HCl (pH 6.8), 2.3% (w/v) SDS, 8% (w/v) glycerol, 1.4 M β-mercaptoethanol, 0.1% (w/v) bromophenol blue] was added. Each solution was subjected to electrophoresis on 10% SDS-polyacrylamide gels. The gels were stained with Coomassie brilliant blue R-250. To evaluate the effects of MPD concentration on oligomer formation, 2.5, 5, 7.5, 10, 20, 30, or 40% MPD was added to monomeric α-hemolysin, and the generated oligomer was assayed by SDS-PAGE as described earlier.
Acknowledgments
The authors thank Mr. K. Yamashita and Mr. H. Kawauchi of Hokkaido University for their help with structure analysis. They also thank Dr. S. Galdiero of the University of Naples, Italy, for providing PDB files of heptameric α-hemolysin in complex with DiC3PC and GPC.
REFERENCES
- 1.Gouaux E. alpha-Hemolysin from Staphylococcus aureus: an archetype of beta-barrel, channel-forming toxins. J Struct Biol. 1998;121:110–122. doi: 10.1006/jsbi.1998.3959. [DOI] [PubMed] [Google Scholar]
- 2.Walker B, Krishnasastry M, Zorn L, Bayley H. Assembly of the oligomeric membrane pore formed by Staphylococcal alpha-hemolysin examined by truncation mutagenesis. J Biol Chem. 1992;267:21782–21786. [PubMed] [Google Scholar]
- 3.Walker B, Braha O, Cheley S, Bayley H. An intermediate in the assembly of a pore-forming protein trapped with a genetically-engineered switch. Chem Biol. 1995;2:99–105. doi: 10.1016/1074-5521(95)90282-1. [DOI] [PubMed] [Google Scholar]
- 4.Montoya M, Gouaux E. Beta-barrel membrane protein folding and structure viewed through the lens of alpha-hemolysin. Biochim Biophys Acta. 2003;1609:19–27. doi: 10.1016/s0005-2736(02)00663-6. [DOI] [PubMed] [Google Scholar]
- 5.Bayley H, Jayasinghe L, Wallace M. Prepore for a breakthrough. Nat Struct Mol Biol. 2005;12:385–386. doi: 10.1038/nsmb0505-385. [DOI] [PubMed] [Google Scholar]
- 6.Iacovache I, van der Goot FG, Pernot L. Pore formation: an ancient yet complex form of attack. Biochim Biophys Acta. 2008;1778:1611–1623. doi: 10.1016/j.bbamem.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 7.Banerjee A, Mikhailova E, Cheley S, Gu LQ, Montoya M, Nagaoka Y, Gouaux E, Bayley H. Molecular bases of cyclodextrin adapter interactions with engineered protein nanopores. Proc Natl Acad Sci USA. 2010;107:8165–8170. doi: 10.1073/pnas.0914229107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 9.Galdiero S, Gouaux E. High resolution crystallographic studies of alpha-hemolysin-phospholipid complexes define heptamer-lipid head group interactions: implication for understanding protein-lipid interactions. Protein Sci. 2004;13:1503–1511. doi: 10.1110/ps.03561104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olson R, Nariya H, Yokota K, Kamio Y, Gouaux E. Crystal structure of staphylococcal LukF delineates conformational changes accompanying formation of a transmembrane channel. Nat Struct Biol. 1999;6:134–140. doi: 10.1038/5821. [DOI] [PubMed] [Google Scholar]
- 11.Pedelacq JD, Maveyraud L, Prevost G, Baba-Moussa L, Gonzalez A, Courcelle E, Shepard W, Monteil H, Samama JP, Mourey L. The structure of a Staphylococcus aureus leucocidin component (LukF-PV) reveals the fold of the water-soluble species of a family of transmembrane pore-forming toxins. Structure. 1999;7:277–287. doi: 10.1016/s0969-2126(99)80038-0. [DOI] [PubMed] [Google Scholar]
- 12.Guillet V, Roblin P, Werner S, Coraiola M, Menestrina G, Monteil H, Prevost G, Mourey L. Crystal structure of leucotoxin S component: new insight into the Staphylococcal beta-barrel pore-forming toxins. J Biol Chem. 2004;279:41028–41037. doi: 10.1074/jbc.M406904200. [DOI] [PubMed] [Google Scholar]
- 13.Meesters C, Brack A, Hellmann N, Decker H. Structural characterization of the alpha-hemolysin monomer from Staphylococcus aureus. Proteins. 2009;75:118–126. doi: 10.1002/prot.22227. [DOI] [PubMed] [Google Scholar]
- 14.Bhakdi S, Fussle R, Tranum-Jensen J. Staphylococcal alpha-toxoligomerization of hydrophilic monomers to form amphiphilic hexamers induced through contact with deoxycholate detergent micelles. Proc Natl Acad Sci USA. 1981;78:5475–5479. doi: 10.1073/pnas.78.9.5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weissmann G, Sessa G, Bernheimer AW. Staphylococcal alpha-toxeffects on artificial lipid spherules. Science. 1966;154:772–774. doi: 10.1126/science.154.3750.772. [DOI] [PubMed] [Google Scholar]
- 16.Freer JH, Arbuthnott JP, Bernheimer AW. Interaction of staphylococcal alpha-toxin with artificial and natural membranes. J Bacteriol. 1968;95:1153–1168. doi: 10.1128/jb.95.3.1153-1168.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watanabe M, Tomita T, Yasuda T. Membrane-damaging action of staphylococcal alpha-toxin on phospholipid-cholesterol liposomes. Biochim Biophys Acta. 1987;898:257–265. doi: 10.1016/0005-2736(87)90065-4. [DOI] [PubMed] [Google Scholar]
- 18.Czajkowsky DM, Sheng S, Shao Z. Staphylococcal alpha-hemolysin can form hexamers in phospholipid bilayers. J Mol Biol. 1998;276:325–330. doi: 10.1006/jmbi.1997.1535. [DOI] [PubMed] [Google Scholar]
- 19.Valeva A, Hellmann N, Walev I, Strand D, Plate M, Boukhallouk F, Brack A, Hanada K, Decker H, Bhakdi S. Evidence that clustered phosphocholine head groups serve as sites for binding and assembly of an oligomeric protein pore. J Biol Chem. 2006;281:26014–26021. doi: 10.1074/jbc.M601960200. [DOI] [PubMed] [Google Scholar]
- 20.Tomita T, Watanabe M, Yasuda T. Influence of membrane fluidity on the assembly of Staphylococcus aureus alpha-toxin, a channel-forming protein, in liposome membrane. J Biol Chem. 1992;267:13391–13397. [PubMed] [Google Scholar]
- 21.Jayasinghe L, Miles G, Bayley H. Role of the amino latch of staphylococcal alpha-hemolysin in pore formation: a co-operative interaction between the N terminus and position 217. J Biol Chem. 2006;281:2195–2204. doi: 10.1074/jbc.M510841200. [DOI] [PubMed] [Google Scholar]
- 22.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 23.Vagin A, Teplyako A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 24.Yao M, Zhou Y, Tanaka I. LAFIRE: software for automating the refinement process of protein-structure analysis. Acta Crystallogr D Biol Crystallogr. 2006;62:189–196. doi: 10.1107/S0907444905038965. [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.Afonine PV, Grosse-Kunstleve RW, Adams PD. CCP4 Newsletter. 2005. The phenix refinement framework. contribution 8. [Google Scholar]
- 27.Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Cryst. 2006;39:109–111. [Google Scholar]
- 28.Lovell SC, Davis IW, Arendall WB, III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 29.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]