

Abstract
Epidermal growth factor (EGF) stimulates the homodimerization of EGF receptor (EGFR) and the heterodimerization of EGFR and ErbB2. The EGFR homodimers are quickly endocytosed after EGF stimulation as a means of down-regulation. However, the results from experiments on the ability of ErbB2 to undergo ligand-induced endocytosis are very controversial. It is unclear how the EGFR–ErbB2 heterodimers might behave. In this research, we showed by subcellular fractionation, immunoprecipitation, Western blotting, indirect immunofluorescence, and microinjection that, in the four breast cancer cell lines MDA453, SKBR3, BT474, and BT20, the EGFR–ErbB2 heterodimerization levels were positively correlated with the ratio of ErbB2/EGFR expression levels. ErbB2 was not endocytosed in response to EGF stimulation. Moreover, in MDA453, SKBR3, and BT474 cells, which have very high levels of EGFR–ErbB2 heterodimerization, EGF-induced EGFR endocytosis was greatly inhibited compared with that in BT20 cells, which have a very low level of EGFR–ErbB2 heterodimerization. Microinjection of an ErbB2 expression plasmid into BT20 cells significantly inhibited EGF-stimulated EGFR endocytosis. Coexpression of ErbB2 with EGFR in 293T cells also significantly inhibited EGF-stimulated EGFR endocytosis. EGF did not stimulate the endocytosis of ectopically expressed ErbB2 in BT20 and 293T cells. These results indicate that ErbB2 and the EGFR–ErbB2 heterodimers are impaired in EGF-induced endocytosis. Moreover, when expressed in BT20 cells by microinjection, a chimeric receptor composed of the ErbB2 extracellular domain and the EGFR intracellular domain underwent normal endocytosis in response to EGF, and this chimera did not block EGF-induced EGFR endocytosis. Thus, the endocytosis deficiency of ErbB2 is due to the sequence of its intracellular domain.
INTRODUCTION
The receptor for epidermal growth factor (EGF) is the prototype for a subfamily of structurally related proteins (termed the class I/ErbB receptors; Schlessinger and Ullrich, 1992) that mediate the proliferation and differentiation of normal cells (Carraway and Cantley, 1994). The other three members of the ErbB receptor family include ErbB2/Her2/neu (Bargmann et al., 1986; Yamamoto et al., 1986), ErbB3/Her3 (Kraus et al., 1989), and ErbB4/Her4 (Plowman et al., 1993). It has been suggested that the aberrant activation of their kinase activities contributes to tumorigenesis or cancer progression (Peles and Yarden, 1993). In particular, amplification or overexpression of the ErbB2 gene has been found in ∼25–30% of human breast cancers (Slamon et al., 1987, 1989).
Receptor tyrosine kinases (RTKs) are activated after homodimerization or after heterodimerization (Heldin, 1995). The EGF receptor (EGFR) was the first RTK shown to dimerize after ligand binding (Yarden and Schlessinger, 1987). However, within the same subfamily of RTKs, heterodimerization of receptors has also been observed. Heregulin (HRG), which is structurally related to EGF, was found to induce heterodimeric complexes between ErbB2 and ErbB3 or ErbB4 (Peles and Yarden, 1993; Plowman et al., 1993; Sliwkowski et al., 1994; Karunagaran et al., 1996; Pinkas-Kramarski et al., 1996; Tzahar et al., 1996; Graus-Porta et al., 1997). Also, EGF itself can induce the heterodimerization of EGFR and ErbB2 (Goldman et al., 1990; Wada et al., 1990; Soltoff et al., 1994). In fact, heterodimerization is preferred in cells that coexpress both EGFR and ErbB2 (Qian et al., 1994; Karunagaran et al., 1996; Pinkas-Kramarski et al., 1996; Tzahar et al., 1996; Graus-Porta et al., 1997). The interaction of EGFR with ErbB2 may, in fact, be crucial. The expression of a kinase-negative ErbB2 mutant is capable of suppressing normal EGFR signaling after EGF stimulation in a dominant negative manner (Qian et al., 1994). Single-chain intracellular retention of ErbB2 in T47D human breast cancer cells (which express all four class I RTKs at moderate levels) markedly impairs signaling induced by EGF and HRG (Graus-Porta et al., 1995).
Dimerization of RTKs is followed by receptor “autophosphorylation,” which occurs when one receptor molecule phosphorylates the other in the dimer (Ullrich and Schlessinger, 1990). Signal transduction by the ErbB family receptors absolutely requires tyrosine kinase activity and tyrosine autophosphorylation (Ullrich and Schlessinger, 1990). Many downstream signaling molecules complex with activated RTKs via the Src homology region 2 domains that bind to phosphotyrosine (pTyr) residues present in specific amino acid sequences in the RTKs (Anderson et al., 1990; Moran et al., 1990; Koch et al., 1991; Pawson, 1995). The formation of the protein complexes then activates several signaling pathways, including the best-elucidated Ras pathway.
Binding of EGF to the EGFR rapidly induces the clustering of ligand–receptor complexes in coated pits, internalization of the complexes, and ultimately lysosomal degradation of both EGF and its receptor (Carpenter, 1987). The endocytic pathway, therefore, is a mechanism for the gradual attenuation of plasma membrane (PM) signaling complexes. Although a molecular mechanism for the rapid endocytosis of growth factor–receptor complexes has not been established, recent evidence suggests that normal endocytosis and down-regulation of EGFR require the activation of intrinsic tyrosine kinase activity and autophosphorylation (Chen et al., 1987; Honegger et al., 1987; Helin and Beguinot, 1991; Sorkin et al., 1991, 1992). Whether the EGFR tyrosine kinase activity is directly required for its internalization remains disputed (Glenney, et al., 1988; Chen et al., 1989; Felder et al., 1990, 1992; Honegger et al., 1990; Wiley et al., 1991; Sorkin et al., 1993). EGFR mutants truncated from C termini to residue 991 (Chang et al., 1993) or to residue 973 (Decker et al., 1992) were internalized inefficiently, and a mutant truncated at residue 958 was not internalized (Chang et al., 1993). Simultaneous point mutation of five tyrosine residues (Tyr-992, Tyr-1068, Tyr-1086, Tyr-1148, and Tyr-1173) to phenylalanine reduced the internalization rate to a minimum (one-quarter of the wild-type EGFRs) (Sorkin et al., 1992). Our recent finding that the binding of GRB2 to EGFR is required for the normal endocytosis and down-regulation of EGFR (Wang and Moran, 1996) has further supported the model that depicts tyrosine kinase activity and autophosphorylation of EGFR as being required for EGFR endocytosis and down-regulation.
However, the results have been very controversial regarding the endocytosis of the other three members of the ErbB receptor family, ErbB2, ErbB3, and ErbB4. Some studies suggest that ErbB2, ErbB3, and ErbB4 are impaired in endocytosis (Sorkin et al., 1993; Baulida et al., 1996). Using a chimeric receptor composed of the EGFR extracellular domain and the ErbB2 cytoplasmic domain, one study showed that EGFR/ErbB2 chimeras internalize 125I-EGF severalfold more slowly than the EGFR (Sorkin et al., 1993). This study also indicated that the EGFR/ErbB2 chimeras were activated by EGF, and the impaired internalization capacity of this receptor was due to the sequences in the ErbB2 C-terminal domain. More recently, studies with EGF-responsive chimeric receptors containing the EGFR extracellular domain and different ErbB cytoplasmic domains (EGFR/ErbB) have indicated that all EGFR/ErbB receptors show impaired ligand-induced internalization, down-regulation, and degradation (Baulida et al., 1996). Moreover, HRG-responsive, wild-type ErbB4 does not mediate the rapid internalization of 125I-HRG (Baulida et al., 1996). In contrast, it has been shown that upon binding of certain mAbs, ErbB2 undergoes internalization using a pathway shared by other growth factor receptors when induced by ligand and antibodies (Drebin et al., 1985; Klapper et al., 1997). The intrinsic abilities of the mAbs to induce the endocytic degradation of ErbB2 are strictly dependent on antibody bivalency, implying that their association is with the ErbB2 homodimers (Gilboa et al., 1995; Hurwitz et al., 1995; Klapper et al., 1997). A point mutation in the transmembrane domain of the rat ErbB2 (Val-664 replaced by Glu) results in a constitutively dimerized and permanently active receptor (Bargmann et al., 1986; Stern et al., 1988; Weiner et al., 1989), and this activated ErbB2 homodimer is internalized like EGFR (Gilboa et al., 1995). Recently, it has been reported that the addition of EGF results in the endocytosis and down-regulation of ErbB2 in nontransformed epithelial cells (Worthylake and Wiley, 1997).
These controversial results raise several questions. Does the activated ErbB2 receptor contain all of the necessary signals to mediate its endocytosis? Does ErbB2 undergo endocytosis in response to EGF stimulation? What is the molecular mechanism that regulates ErbB2 encocytosis in response to EGF? So far, no ligand has been found to directly bind ErbB2. ErbB2 is activated by both EGF and HRG indirectly through heterodimerization with EGFR, ErbB3, and ErbB4. It has been shown that in response to EGF stimulation, EGFR forms both homodimers with itself and heterodimers with ErbB2 (Yarden and Schlessinger, 1987; Goldman et al., 1990; Wada et al., 1990; Spivak-Kroizman et al., 1992; Carraway and Cantley, 1994; Qian et al., 1994; Soltoff et al., 1994). In fact, heterodimerization is preferred in cells that express both EGFR and ErbB2 (Qian et al., 1994). Therefore, in addition to using chimeric receptors, another approach to studying the ability of ErbB2 to undergo ligand-induced endocytosis is to examine the EGF-induced endocytosis of the EGFR–ErbB2 heterodimers. In this report, we studied EGF-induced endocytosis of the EGFR–ErbB2 heterodimers in several breast cancer cell lines that express EGFR and ErbB2 at different levels and in 293T cells that were transiently transfected with EGFR and/or ErbB2. We demonstrate that ErbB2 and the EGFR–ErbB2 heterodimers are endocytosis deficient in response to EGF stimulation.
MATERIALS AND METHODS
Cells
MDA453, SKBR3, BT474, BT20, and 293T cells were grown at 37°C in Dulbecco’s modified Eagle’s medium containing 10% FBS, penicillin, and streptomycin and were maintained in a 5% CO2 atmosphere.
Antibodies and Chemicals
All of the fluorescent-conjugated secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). HRP-conjugated secondary antibodies were purchased from Bio-Rad (Hercules, CA). Unless otherwise specified, all the chemicals were purchased from Sigma (St. Louis, MO).
Subcellular Fractionation
The isolation of PM and endosomal (EN) fractions was carried out by a method modified from those of Wang et al. (1996) and Di Guglielmo et al. (1994). Three 150-mm-diameter plates of cells were used for each condition. At 90% confluence, cells were serum starved by incubation in serum-free medium for 24 h. Cells were then treated with EGF (Upstate Biotechnology, Lake Placid, NY) at a concentration of 100 ng/ml for 60 min at 4°C, which was referred to as 0 min. Some cells were further incubated at 37°C for 15, 30, and 60 min. All of the following procedures were performed at 0–4°C. Cell monolayers in three plates were scraped into 4.5 ml of homogenization buffer [0.25 M sucrose, 20 mM Tris-HCl, 1 mM MgCl2, 4 mM NaF, 0.5 mM Na3VO4, 0.02% NaN3, 0.1 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride, 10 μg/ml aprotinin, 1 μM pepstatin A, pH 7] and homogenized with a glass Potter-type homogenizer. The homogenates were centrifuged at 280 × g for 5 min to remove the cell debris and nuclei (pellet 1 [P1]). Supernatant 1 (S1) was then centrifuged at 1500 × g for 10 min to yield a supernatant (S2), which was used to isolate the EN fraction, and a pellet (P2), which was used to isolate the PM fraction. Next, P2 was resuspended in homogenization buffer, and the sucrose concentration was adjusted to 1.42 M. This homogenate was overlaid with 0.25 M sucrose and centrifuged at 82,000 × g for 1 h. The pellicule at the 0.25–1.42 M interface was also collected, and the sucrose concentration was then adjusted to 0.39 M and centrifuged at 1500 × g for 10 min to obtain the PM fraction. The PM fraction was resuspended in homogenization buffer. The S2 fraction was centrifuged at 200,000 × g for 30 min to yield a cytosolic (Cyt) fraction and a microsomal pellet, which was resuspended to 1.15 M sucrose in homogenization buffer. This resuspension was overlaid with 1.00 and 0.25 M sucrose cushions and centrifuged at 200,000 × g for 1.5 h. The EN fraction was collected at the 0.25–1.00 M sucrose interface.
Immunoprecipitation
The cells were lysed with immunoprecipitation buffer (20 mM Tris, 150 mM NaCl, 1% NP40, 0.1% sodium deoxycholate, 100 mM NaF, 0.5 mM Na3VO4, 0.02% NaN3, 0.1 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride, 10 μg/ml aprotinin, 1 μM pepstatin A, pH 7.5) overnight at 4°C. The cell lysates were then centrifuged at 100,000 × g for 1 h to remove unsolubilized debris. The supernatants, containing 1 mg of total proteins, were then incubated with 1 μg of mouse anti-ErbB2 antibody (immunoglobulin G1 [IgG1]) 9G6 (Santa Cruz Biotechnology, Santa Cruz, CA) for 2 h with gentle mixing by inverting. After that, goat anti-mouse IgG conjugated with agarose was added to each fraction and incubated for 2 h with agitation. Finally, both the agarose beads and the nonprecipitated supernatant were collected by centrifugation. The agarose beads were washed twice with immunoprecipitation buffer. For the control experiments, mouse anti-ErbB2 IgG1 was substituted with normal mouse IgG1 (Sigma), and no ErbB2 and EGFR were precipitated with this normal mouse IgG1.
Immunoblotting
For the detection of EGFR and ErbB2 in the total lysates of MDA453, SKBR3, BT474, and BT20 cells, aliquots containing 20 μg of protein from each cell lysate were used. For the detection of EGFR and pTyr in both the anti-ErbB2 immunoprecipitates and nonprecipitated supernatant, 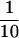 of the immunoprecipitate and the nonprecipitated supernatant from each lysate was used. To examine EGFR and ErbB2 in each subcellular fraction of the various cell lines, aliquots containing 10 μg of protein from each fraction were used. The protein samples were separated by electrophoresis through 10% polyacrylamide SDS-containing gels and electrophoretically transferred onto nitrocellulose filter paper. Filters were probed with polyclonal rabbit anti-ErbB2 C18 (Santa Cruz), polyclonal rabbit anti-EGFR 1005 (Santa Cruz), or monoclonal mouse anti-pTyr antibody PY20 (Santa Cruz). The primary antibodies were detected with a polyclonal goat anti-rabbit Ig coupled to HRP or a polyclonal goat anti-mouse Ig coupled to HRP followed by enhanced chemiluminescence development (Pierce Chemical, Rockford, IL) and light detection with Eastman Kodak (Rochester, NY) RP film.
of the immunoprecipitate and the nonprecipitated supernatant from each lysate was used. To examine EGFR and ErbB2 in each subcellular fraction of the various cell lines, aliquots containing 10 μg of protein from each fraction were used. The protein samples were separated by electrophoresis through 10% polyacrylamide SDS-containing gels and electrophoretically transferred onto nitrocellulose filter paper. Filters were probed with polyclonal rabbit anti-ErbB2 C18 (Santa Cruz), polyclonal rabbit anti-EGFR 1005 (Santa Cruz), or monoclonal mouse anti-pTyr antibody PY20 (Santa Cruz). The primary antibodies were detected with a polyclonal goat anti-rabbit Ig coupled to HRP or a polyclonal goat anti-mouse Ig coupled to HRP followed by enhanced chemiluminescence development (Pierce Chemical, Rockford, IL) and light detection with Eastman Kodak (Rochester, NY) RP film.
Immunofluorescence
Cells were grown on glass coverslips to subconfluence and serum starved for 24 h. After treatment with EGF (100 ng/ml) for the indicated time, the cells were fixed by immersion in −20°C methanol for 5 min. After removal of the methanol and washing with PBS, the cells were permeabilized with 0.2% Triton X-100 for 10 min and blocked with 3% BSA for 30 min. Next, for single immunofluorescent labeling, the cells were incubated with monoclonal mouse anti-EGFR antibody (1:10; PharMingen, San Diego, CA) or mouse monoclonal anti-ErbB2 antibody 9G6 (1:20) at room temperature for 1 h. After three washes with PBS, the cells were incubated with donkey anti-mouse IgG conjugated with FITC (1:50). For double immunofluorescent labeling, the cells were incubated with both monoclonal mouse anti-ErbB2 antibody 9G6 (1:20) and polyclonal sheep anti-EGFR antibody (1:40; Upstate Biotechnology) at room temperature for 1 h. After three washes with PBS, the cells were incubated with donkey anti-mouse IgG conjugated with FITC (1:50) and donkey anti-sheep IgG conjugated with TRITC (1:50). Samples were visualized by using a fluorescence microscope and a Zeiss (Thornwood, NY) oil immersion lens. In control experiments, polyclonal or monoclonal antibodies were substituted with normal rabbit serum or mouse ascites fluids, respectively, and no specific staining was observed.
Internalization of Texas Red–conjugated EGF
Cells were grown on glass coverslips to subconfluence and then serum starved for 24 h. After treatment with Texas Red–conjugated EGF (TR-EGF, 100 ng/ml; Molecular Probes, Eugene, OR) for the indicated time, the cells were fixed by immersion in −20°C methanol for 5 min. After removal of methanol and washing with PBS, the cells were visualized by using a fluorescence microscope and a Zeiss oil immersion lens.
Chimeric Receptor Construct
The chimeric ErbB2/EGFR receptor was engineered by joining the ErbB2 extracellular domain (corresponding to amino acid positions 1–655 of Coussens et al., 1985) and EGFR transmembrane and intracellular domains (corresponding to amino acid positions 623-1210 according to Ullrich et al., 1984). Briefly, a KpnI site was introduced into the 5′ end and a SalI site was introduced into the 3′ end of the ErbB2 extracellular domain, and a SalI site was introduced into the 5′ end and a KpnI site was introduced into the 3′ end of EGFR transmembrane and intracellular domains, by PCR. The PCR products were subcloned into pCR-XL-TOPO vector by the TOPO XL PCR cloning kit (Invitrogen, San Diego, CA) according to the manufacturer’s instructions. After digestion with KpnI and SalI, the ErbB2 extracellular domain and EGFR transmembrane plus intracellular membrane domain were ligated and inserted in frame into pcDNA3.1(−)/Myc-His mammalian expression vector [pcDNA3.1(−)/ErbB2/EGFR/Myc-His].
Transient Expression of EGFR and ErbB2 in 293T Cells
293T cells were transiently transfected with an EGFR expression plasmid, pcDNA3.1(−)/EGFR, or an ErbB2 expression plasmid, pcDNA3.1(−)/ErbB2, or cotransfected with both pcDNA3.1(−)/EGFR and pcDNA3.1(−)/ErbB2 by calcium phosphate precipitation. Thirty-six hours after transfection, the cells were used for immunofluorescence analysis and subcellular fractionation.
Microinjection
The microinjection experiments were carried out by a method described previously (Wang and Moran, 1996; Wang et al., 1998). BT20 cells were grown on glass coverslips to subconfluence. The ErbB2 expression plasmid pcDNA3.1(−)/ErbB2 (50 μg/ml), the chimeric ErbB2/EGFR expression plasmid pcDNA3.1(−)/ErbB2/EGFR/Myc-His, or a control plasmid, pcDNA3.1(−)/Myc-His/LacZ (50 μg/ml; Invitrogen) in microinjection buffer (50 mM HEPES, pH 7.2, 100 mM KCl, 5 mM Na2HPO4) was injected into the nuclei of the cells. After microinjection, cells were returned to the cell culture incubator for 12 h and then serum starved for another 24 h. After treatment with EGF (100 ng/ml) at 4°C for 60 min and further incubation with serum-free medium at 37°C for 30 min, the cells were fixed with methanol. Cells microinjected with pcDNA3.1(−)/ErbB2 were incubated with both polyclonal sheep anti-EGFR antibody and mouse anti-ErbB2 antibody 9G6. Cells microinjected with pcDNA3.1(−)/ErbB2/EGFR/Myc-His or the control plasmid were incubated with both polyclonal sheep anti-EGFR antibody (Upstate Biotechnology) and monoclonal mouse anti-myc antibody (Santa Cruz). Finally, the cells were incubated with donkey anti-sheep IgG conjugated with TRITC (1:50) and donkey anti-mouse IgG conjugated with FITC (1:50). For the analysis of the endocytosis of TR-EGF, cells were stained with either mouse anti-ErbB2 antibody 9G6 (for ErbB2) or mouse anti-myc antibody (for LacZ), followed by FITC-conjugated donkey anti-mouse IgG. For quantification of the inhibition of EGFR endocytosis, the percentage of injected cells in which EGFR internalization was disrupted was determined by multiplying the number of microinjected cells in which EGFR internalization was blocked by 100 and dividing by the total number of injected cells. For each experiment, 200–300 cells were microinjected with a given solution.
RESULTS
EGF-stimulated Heterodimerization and Phosphorylation of EGFR and ErbB2 in Various Breast Cancer Cell Lines
We have selected several breast cancer cell lines, including MDA453, SKBR3, BT474, and BT20, that have been shown to contain different copies of the EGFR and ErbB2 genes (Kraus et al., 1987; Miller and Hung, 1995). The expression levels of EGFR and ErbB2 were assayed by immunoblotting with anti-EGFR and anti-ErbB2 antibodies (Figure 1A). EGFR was highly expressed in BT20 cells, moderately expressed in SKBR3 and BT474 cells, but expressed at a very low level in MDA453 cells. ErbB2 was highly expressed in BT474, SKBR3, and MDA453 cells but was expressed at a very low level in BT20 cells. The protein levels were consistent with the gene amplification and mRNA transcription levels reported previously (Kraus et al., 1987; Miller and Hung, 1995).
Figure 1.

EGFR and ErbB2 concentrations, EGF-stimulated heterodimerization, and phosphorylation of EGFR and ErbB2 in various breast cancer cell lines. (A) MDA453, SKBR3, BT474, and BT20 cells were lysed with radioimmunoprecipitation assay buffer, and 20 μg of protein from each lysate were separated by 10% SDS-PAGE, transferred onto a nitrocellulose filter, and incubated with rabbit polyclonal anti-ErbB2 or anti-EGFR antibody. (B) MDA453, SKBR3, BT474, and BT20 cells were serum starved or stimulated with EGF (100 ng/ml) at 4°C for 60 min and then lysed with immunoprecipitation buffer. The total lysates were immunoprecipitated with monoclonal anti-ErbB2 antibody. Then, 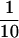 of the total protein from both the anti-ErbB2 immunoprecipitates and the nonprecipitated supernatants was separated by 10% SDS-PAGE, transferred onto a nitrocellulose filter, and incubated with a rabbit polyclonal anti-EGFR antibody or a mouse monoclonal anti-pTyr antibody. Bands were visualized on x-ray film by using an HRP-conjugated secondary antibody and chemiluminescence reagents.
of the total protein from both the anti-ErbB2 immunoprecipitates and the nonprecipitated supernatants was separated by 10% SDS-PAGE, transferred onto a nitrocellulose filter, and incubated with a rabbit polyclonal anti-EGFR antibody or a mouse monoclonal anti-pTyr antibody. Bands were visualized on x-ray film by using an HRP-conjugated secondary antibody and chemiluminescence reagents.
To determine whether EGF stimulates the formation of the EGFR–ErbB2 heterodimers and to examine EGF-stimulated tyrosine phosphorylation of EGFR and ErbB2, MDA453, SKBR3, BT474, and BT20 cells were lysed, and the total lysates were immunoprecipitated with a monoclonal anti-ErbB2 antibody. Both the anti-ErbB2 immunoprecipitates and the nonprecipitated supernatants were immunoblotted with a polyclonal anti-EGFR antibody and a monoclonal anti-pTyr antibody (Figure 1B). The EGFR that coimmunoprecipitated with ErbB2 represented the portion of EGFR that heterodimerized with ErbB2, and the EGFR that remained in the supernatant represented EGFR homodimers and EGFR monomers. As shown in Figure 2, EGF stimulated formation of the EGFR–ErbB2 heterodimers in all four cell lines to a significant extent. After EGF stimulation, two bands corresponding to EGFR and ErbB2 were detected by anti-pTyr antibody in anti-ErbB2 immunoprecipitates in all of the four cell lines, which indicated that EGF stimulated the phosphorylation of both EGFR and ErbB2, and the EGFR that heterodimerized with ErbB2 was phosphorylated. An immunoblot of the nonprecipitated supernatants with anti-ErbB2 antibody showed that ErbB2 was completely precipitated by the ErbB2 antibody (our unpublished results).
Figure 2.

Determination by immunofluorescence analysis of ErbB2 and EGFR endocytosis after EGF stimulation. MDA453, SKBR3, BT474, and BT20 cells on glass coverslips were serum starved or stimulated with EGF (100 ng/ml) at 4°C for 60 min followed by further incubation at 37°C for 30 min. The cells were fixed with methanol and stained with mouse monoclonal anti-ErbB2 or anti-EGFR antibody followed with FITC-conjugated secondary antibody, as described in MATERIALS AND METHODS. Magnification, 200×.
The quantification of the results was made by densitometric analysis of autoradiographs and correcting for the amount of starting material loaded by using the Image Master VDS video documentation system for gel electrophoresis (Pharmacia Biotech, Piscataway, NJ). Among MDA453, SKBR3, BT474, and BT20 cells, MDA453 had the lowest EGFR concentration, and BT20 had the lowest ErbB2 concentration. We assumed the lowest concentration as 1.0, and the relative concentrations of EGFR and ErbB2 for other cell lines were calculated (Table 1). As shown in Table 1, MDA453 cells had the highest ErbB2/EGFR ratio, followed by BT474 and SKBR3 cells, and BT20 cells had the lowest ErbB2/EGFR ratio. After EGF stimulation, EGFR primarily formed heterodimers with ErbB2 in MDA453 and BT474 cells that had very high ErbB2/EGFR ratios. EGFR significantly formed heterodimers in SKBR3 that had a relative high ErbB2/EGFR ratio, whereas it only slightly formed heterodimers in BT20 cells that had a very low ErbB2/EGFR ratio (Table 1). The EGFR–ErbB2 heterodimerization level was calculated as the ratio between the EGFR that coimmunoprecipitated with ErbB2 and the EGFR that remained in the nonprecipitated supernatant. EGFR–ErbB2 heterodimerization levels were positively correlated with the ratio of ErbB2/EGFR in the four breast cancer cell lines.
Table 1.
Relative ErbB2 and EGFR concentrations, the EGFR–ErbB2 dimerization level, and EGFR endocytosis level in various breast cancer cell lines
| MDA453 | SKBR3 | BT474 | BT20 | |
|---|---|---|---|---|
| ErbB2 levela | 2.90 ± 0.45 | 4.26 ± 0.52 | 4.35 ± 0.42 | 1 |
| EGFR levelb | 1 | 4.24 ± 0.38 | 2.90 ± 0.35 | 8.59 ± 0.64 |
| ErbB2/EGFR ratio | 2.90 | 1.00 | 1.55 | 0.12 |
| EGFR–ErbB2 dimerization levelc | 2.00 ± 0.24 | 0.80 ± 0.12 | 1.4 ± 0.11 | 0.10 ± 0.02 |
| EGFR endocytosis leveld (%) | 0 | 15.0 ± 2.1 | 7.0 ± 1.2 | 25.5 ± 4.0 |
Results were quantified by densitometric analysis of autoradiographs and corrected for the amount of starting material loaded by using the Image Master VDS video documentation system for gel electrophoresis (Pharmacia Biotech).
The ErbB2 level was calculated from Figure 1A. The BT20 cell line had the lowest ErbB2 concentration and was set at 1, and the concentrations of ErbB2 for other cell lines were calculated relative to it. Data represent the mean ± SE of three experiments.
The EGFR level was calculated from Figure 1A. The MDA453 cell line had the lowest EGFR concentration and was set at 1, and the concentrations of EGFR for the other cell lines were calculated relative to it. Data represent the mean ± SE of three experiments.
The EGFR–ErbB2 dimerization level was calculated from Figure 1B as the ratio between EGFR which coimmunoprecipitated with ErbB2 and EGFR that was not coimmunoprecipitated with ErbB2. Data represent the mean ± SE of three experiments.
The EGFR endocytosis level was calculated from Figure 4 as the percentage of total EGFR accumulated in the endosome fraction after EGF stimulation for 30 min at 37°C. Data represent the mean ± SE of three experiments.
Impaired Endocytosis of the EGFR–ErbB2 Heterodimers in Response to EGF Stimulation
We tested whether the EGFR–ErbB2 heterodimers were impaired in EGF-induced endocytosis both by immunofluorescent microscopy and by subcellular fractionation combined with immunoblotting. MDA453, SKBR3, BT474, and BT20 cells were cultured in serum-free medium for 24 h and then stimulated with or without EGF at 4°C for 60 min followed by a further incubation at 37°C for 30 min. Immunofluorescence analysis using an anti-ErbB2 antibody indicated that with or without EGF stimulation, ErbB2 was localized at the PM, and no endosome association of ErbB2 was detected in the four cell lines (Figure 2). Because a significant portion of ErbB2 heterodimerized with EGFR in response to EGF stimulation as shown above (Figure 1B), these results suggested that the EGFR–ErbB2 heterodimers were not endocytosed in response to EGF stimulation.
To confirm that the EGFR–ErbB2 heterodimers were endocytosis deficient, we examined EGF-induced EGFR endocytosis (Figure 2). If the EGFR–ErbB2 heterodimers are impaired in endocytosis, the EGFR that heterodimerizes with ErbB2 should be retained in the PM instead of being endocytosed into endosomes after EGF stimulation. As shown in Figure 2, after EGF stimulation, no detectable EGFR was present in endosomes in MDA453 cells, a weak endosome localization of EGFR was observed in BT474, and a significant portion of EGFR was detected in endosomes in SKBR3 cells, whereas EGFR primarily translocated from the PM to the endosomes in BT20 cells. These results indeed suggested that cells with higher EGFR–ErbB2 heterodimerization levels showed weak EGFR endocytosis.
It is very interesting to note that in SKBR3 cells both EGFR and ErbB2 were localized in membrane ruffle after EGF stimulation (Figure 2).
We further analyzed the EGF-stimulated endocytosis of EGFR and ErbB2 by subcellular fractionation combined with immunoblotting. To exclude the possibility that EGF-induced endocytosis of the EGFR–ErbB2 heterodimers is simply delayed or replaced by receptor recycling, EGF-induced translocation of EGFR and ErbB2 was analyzed at several time points ranging from 0 to 60 min. As shown in Figure 3, no EGF-induced endocytosis of ErbB2 was detected in any of the four cell lines up to 60 min, whereas EGFR endocytosis levels were negatively correlated with the levels of EGF-stimulated EGFR–ErbB2 heterodimerization among the four cell lines. Quantification of these results showed that MDA453 cells had the highest EGFR–ErbB2 heterodimerization level and the lowest EGFR endocytosis level, whereas BT20 cells had the lowest EGFR–ErbB2 heterodimerization level and the highest EGFR endocytosis level (Table 1). In SKBR3 and BT474 cells, a significant portion of the EGFR formed heterodimers with ErbB2, and EGFR was weakly endocytosed into endosomes (Table 1).
Figure 3.

Analysis by subcellular fractionation and immunoblotting of EGFR and ErbB2 endocytosis after EGF stimulation. MDA453, SKBR3, BT474, and BT20 cells were cultured in serum-free medium or incubated with EGF (100 ng/ml) at 4°C for 60 min, followed by further incubation with EGF at 37°C for 15, 30, and 60 min as indicated. The cells were fractionated into PM, EN, and Cyt fractions as described in MATERIALS AND METHODS. Proteins (10 μg) from each fraction were separated by SDS-PAGE, transferred onto a nitrocellulose filter, and incubated with rabbit polyclonal anti-ErbB2 or anti-EGFR antibody. Bands were visualized on x-ray film by using an HRP-conjugated secondary antibody and chemiluminescence reagents.
To eliminate the possibility that the observed inhibition of the endocytosis of EGFR–ErbB2 is due to receptor recycling instead of impaired internalization, the endocytosis of TR-EGF was studied in MDA453, SKBR3, BT474, and BT20 cells (Figure 4). If the EGFR–ErbB2 heterodimers are internalized into sorting endosomes and then recycled back to the PM, the TR-EGF should dissociate with the EGFR–ErbB2 heterodimers in the sorting endosomes and further traffick to the late endosomes and lysosomes. In other words, we would observe a strong endosome association but not a PM association of TR-EGF in all four cell lines regardless of the EGFR–ErbB2 heterodimerization levels. The cells were cultured in serum-free medium for 24 h and then incubated with 100 ng/ml TR-EGF at 4°C for 60 min. The cells were either fixed (referred to as 0 min) or further incubated in serum-free medium for 30 min at 37°C. As shown in Figure 4, in all of the cell lines, TR-EGF was localized at the PM at 0 min. The discontinuous membrane distribution of TR-EGF suggested that TR-EGF clustered in the coated pits. After incubation at 37°C for 30 min, TR-EGF was still only detected in the PM of MDA453 and BT474 cells, and only weak TR-EGF was detected in the endosomes of SKBR3 cells. TR-EGF was primarily internalized and localized in endosomes in BT20 cells because of the internalization of the EGFR homodimers. These results further suggested that the EGFR–ErbB2 heterodimers are impaired in EGF-induced endocytosis.
Figure 4.

Determination by immunofluorescence analysis of TR-EGF internalization. MDA453, SKBR3, BT474, and BT20 cells grown on glass coverslips were incubated with TR-EGF (100 ng/ml) at 4°C for 60 min (referred as 0 min; A, C, E, and G). Some coverslips were further incubated at 37°C for 30 min (B, D, F, and H). The cells were then fixed with methanol and examined under the microscope. Magnification, 180×.
Inhibition of EGFR Endocytosis by Overexpressing ErbB2 in BT20 Cells
To determine whether differences in EGFR endocytosis in various cell lines are due to the differences in ErbB2 expression levels or due to the differences in cellular context, we overexpressed ErbB2 in BT20 cells. BT20 cells were microinjected with pcDNA3.1(−)/ErbB2. After incubation at 37°C for 16 h to allow ErbB2 to be expressed, cells were stimulated with EGF at 4°C for 60 min followed by a further incubation at 37°C for 30 min. Immunofluorescence analysis clearly showed that in the cells microinjected with pcDNA3.1(−)/ErbB2, EGFR endocytosis was significantly inhibited compared with the nonmicroinjected cells and the cells microinjected with control plasmid pcDNA3.1(−)/Myc-His/LacZ (Figure 5A). Quantification of the results showed that EGFR endocytosis was inhibited in 87% of the BT20 cells microinjected with pcDNA3.1(−)/ErbB2, whereas EGFR endocytosis was inhibited only in ∼8% of the BT20 cells microinjected with pcDNA3.1(−)/Myc-His/LacZ (Figure 5B).
Figure 5.

Inhibition of EGFR endocytosis in BT20 cells after microinjection of the ErbB2 expression plasmid. (A) BT20 cells were microinjected with the ErbB2 expression plasmid pcDNA3.1(−)/ErbB2 (A–D) or the control plasmid pcDNA3.1(−)/Myc-His/LacZ (E–H) and incubated at 37°C for 24 h. After treatment with EGF (100 ng/ml; (A–C and E–G) or TR-EGF (100 ng/ml; D and H), the cells were fixed with methanol and stained by immunofluorescence. ErbB2 and LacZ expression-positive cells were identified with a mouse anti-ErbB2 (A, C, and D) or mouse anti-myc (B, G, and H) antibody followed by FITC-conjugated anti-mouse IgG. EGFR endocytosis was assayed with sheep polyclonal anti-EGFR antibody followed by a TRITC-conjugated anti-sheep antibody (B, C, F, and G). The endocytosis of TR-EGF was directly examined under fluorescent microscope (D and H). Magnification, 200×. (B) Quantification of inhibition of EGFR and TR-EGF endocytosis after microinjection of ErbB2 expression plasmid. Data are means ± SE of three independent experiments.
TR-EGF was internalized by receptor-mediated endocytosis and concentrated in endosomes in nonmicroinjected BT20 cells, but not in the cells microinjected with pcDNA3.1(−)/ErbB2 (Figure 5). These data and analysis of EGFR localization at several time points ranging from 0 to 60 min after EGF treatment (our unpublished results) indicated that receptor-mediated endocytosis of EGF is blocked, and not simply delayed or replaced by receptor recycling, in cells injected with pcDNA3.1(−)/ErbB2.
EGF-induced Endocytosis of EGFR and ErbB2 in 293T Cells Transfected with EGFR, ErbB2, or Both
To further eliminate the possibility that the observed differences in EGFR endocytosis among the four cell lines were due to the differences in cellular context, 293T cells were transiently transfected with pcDNA3.1(−)/EGFR, pcDNA3.1(−)/ErbB2, or both. After transfection for 24 h, the 293T cells were cultured in serum-free medium for 12 h, with or without additional EGF stimulation at 4°C for 60 min. After further incubation at 37°C for 30 min, cells were fractionated by differential centrifugation and gradient centrifugation into PM, EN, and Cyt fractions. Immunoblotting of nitrocellular-bound SDS-PAGE–resolved samples with an anti-ErbB2 antibody indicated that, with or without EGF stimulation, ErbB2 was localized at the PM in the cells transfected with pcDNA3.1(−)/ErbB2 or cotransfected with both pcDNA3.1(−)/ErbB2 and pcDNA3.1(−)/EGFR (Figure 6). EGFR was localized at the PM without EGF stimulation and endocytosed to endosomes after EGF stimulation in the cells transfected with pcDNA3.1(−)/EGFR alone. However, EGF-stimulated endocytosis of EGFR was significantly inhibited in the cells that were cotransfected with both pcDNA3.1(−)/ErbB2 and pcDNA3.1(−)/EGFR (Figure 6).
Figure 6.

Analysis by subcellular fractionation and immunoblotting of EGFR and ErbB2 endocytosis after EGF stimulation in the transiently transfected 293T cells. (A) Western blot analysis of EGFR and ErbB2 expression in 293T cells transiently transfected with pcDNA3.1(−)/EGFR and/or pcDNA3.1(−)/ErbB2. (B) Determination of EGF-induced EGFR and ErbB2 endocytosis in 293T cells transfected with pcDNA3.1(−)/EGFR and/or pcDNA3.1(−)/ErbB2. 293T cells were cultured in serum-free medium or stimulated with EGF at 4°C for 60 min followed by further incubation in serum-free medium for 15 or 30 min. The cells were subcellular fractionated and immunoblotted for EGFR and/or ErbB2 as described in MATERIALS AND METHODS.
EGF-induced endocytosis of ErbB2 and EGFR in 293T cells transfected with pcDNA3.1(−)/EGFR, pcDNA3.1(−)/ErbB2, or both was also analyzed by immunofluorescent microscopy (Figure 7). The results showed that EGF did not stimulate the endocytosis of ErbB2 in the cells transfected with pcDNA3.1(−)/ErbB2 or both pcDNA3.1(−)/ErbB2 and pcDNA3.1(−)/EGFR, whereas EGF stimulated the endocytosis of EGFR in the cells transfected with pcDNA3.1(−)/EGFR. However, the cotransfection of pcDNA3.1(−)/ErbB2 together with pcDNA3.1(−)/EGFR significantly inhibited EGF-induced EGFR endocytosis (Figure 7).
Figure 7.

Determination by immunofluorescence analysis of EGF-induced EGFR and ErbB2 endocytosis in the transiently transfected 293T cells. 293T were transiently transfected with pcDNA3.1(−)/ErbB2 (A and B), pcDNA3.1(−)/EGFR (C and D), or both (E–H). After the transfection, the cells were cultured in serum-free medium (A, C, E, and G) or incubated with EGF (100 ng/ml) at 4°C for 60 min followed by further incubation for 30 min at 37°C (B, D, F, and H). The cells were then single immunofluorescent stained for ErbB2 (A and B) and EGFR (C and D) or double immunofluorescent stained for both ErbB2 (E and F) and EGFR (G and H), as described in MATERIALS AND METHODS. Magnification, 200×.
Endocytosis of ErbB2/EGFR Chimera in Response to EGF Stimulation
A chimeric receptor composed of the EGFR extracellular domain and the ErbB2 intracellular domain is impaired in EGF-induced endocytosis (Sorkin et al., 1993; Baulida et al., 1996). However, the impaired endocytosis of the chimera may be due to the inappropriate three-dimensional structure that results from the construction of the chimera. To exclude this possibility, we constructed an ErbB2/EGFR chimera composed of the ErbB2 extracellular domain and the EGFR intracellular domain. This ErbB2/EGFR chimera was expressed in BT20 cells by microinjection. Indirect immunofluorescence showed that, after the addition of EGF at 37°C for 30 min, both the chimera and EGFR were internalized into the endosomes (Figure 8). The ability to restore the endocytosis of ErbB2 using the intracellular domain of EGFR further suggests that the impaired endocytosis of ErbB2 may result from its intracellular domain.
Figure 8.

Determination by immunofluorescence analysis of EGF-induced endocytosis of an ErbB2/EGFR chimera. BT20 cells were microinjected with the chimeric ErbB2/EGFR expression plasmid pcDNA3.1(−)/ErbB2/EGFR/Myc/His and incubated at 37°C for 24 h. The cells were either untreated (A and B) or treated with EGF (100 ng/ml) at 37°C for 30 min (C and D). The cells were then fixed with methanol and stained by immunofluorescence. The endocytosis of the chimeric ErbB2/EGFR was assayed by mouse anti-myc antibody (B, G, and H) followed by FITC-conjugated anti-mouse IgG (A and C). The EGFR endocytosis was assayed with sheep polyclonal anti-EGFR antibody followed by a TRITC-conjugated anti-sheep antibody (B and D). Magnification, 200×.
DISCUSSION
EGFR is rapidly endocytosed in response to EGF stimulation (Carpenter, 1987). However, the reported results regarding the ability of ErbB2 to undergo ligand-induced internalization are controversial (Drebin et al., 1985; Sorkin et al., 1993; Baulida et al., 1996; Klapper et al., 1997; Worthylake and Wiley, 1997). It is unclear how the EGFR–ErbB2 heterodimers might behave. In this research, we studied the EGF-induced endocytosis of EGFR–ErbB2 heterodimers in four breast cancer cell lines that express ErbB2 and EGFR at different levels and in 293T cells transfected with EGFR, ErbB2, or both. Consistent with previous reports (Goldman et al., 1990; Wada et al., 1990; Soltoff et al., 1994), our results indicated that in the breast cancer cells that overexpress ErbB2, EGFR primarily heterodimerized with ErbB2 (Figure 1). The heterodimerization levels were positively correlated with the ratio between ErbB2 and EGFR concentrations (Table 1). In all these cell lines, ErbB2 was not endocytosed (Figures 2 and 3), which suggests that ErbB2 and the EGFR–ErbB2 heterodimers are impaired in EGF-induced endocytosis. Furthermore, in MDA453, SKBR3, and BT474 cells that have very high levels of the EGFR–ErbB2 heterodimers, EGF-induced EGFR endocytosis was significantly inhibited compared with that in BT20 cells, which have a very low level of the EGFR–ErbB2 heterodimers (Figures 2 and 3, and Table 1). These results further suggest that the EGFR–ErbB2 heterodimers are not internalized in response to EGF.
To demonstrate that the correlative results obtained from the four breast cancer cell lines did indeed result from the different levels of ErbB2 expression instead of from different cellular contexts, we overexpressed ErbB2 in BT20 cells by microinjection of pcDNA3.1(−)/ErbB2, and we transfected 293T cells with pcDNA3.1(−)/EGFR and/or pcDNA3.1(−)/ErbB2. Our results indicated that ectopically expressed ErbB2 in BT20 cells and 293T cells was not endocytosed in response to EGF stimulation (Figures 5–7). Moreover, overexpression of ErbB2 in BT20 cells significantly inhibited EGF-induced EGFR endocytosis (Figure 5). Coexpression of ErbB2 with EGFR in 293T cells also significantly inhibited EGF-induced EGFR endocytosis (Figures 6 and 7).
We have tested the possibility that the EGFR–ErbB2 heterodimers are simply recycling. If the EGFR–ErbB2 heterodimers are internalized into sorting endosomes and then are recycled back to the PM, it is very likely that the TR-EGF would dissociate from the EGFR–ErbB2 heterodimers in the sorting endosomes and further traffick to the late endosomes and lysosomes. In other words, after stimulation, we would always observe a strong endosome association but not a PM association of TR-EGF in the cells regardless of the EGFR–ErbB2 heterodimerization levels. Our results showed that TR-EGF was internalized by receptor-mediated endocytosis and concentrated in endosomes in BT20 cells but not in BT20 cells microinjected with pcDNA3.1(−)/ErbB2 (Figure 5) and not in MDA453 and BT474 cells (Figure 4). These results suggest that EGF-induced endocytosis of the EGFR–ErbB2 heterodimers is inhibited and not simply replaced by receptor recycling. However, we cannot exclude the possibility that the compartments to which EGFR–ErbB2 complexes are delivered are not of a sufficiently low pH to dissociate the growth factor from the heterodimers before routing back to the PM.
It is interesting to note that the EGFR–ErbB2 heterodimers are present in MDA453, SKBR3, and BT474 cells in the absence of EGF stimulation (Figure 1). To eliminate the possibility that factors, secreted by these cells during the 24-h serum starvation, stimulate the formation of the EGFR–ErbB2 heterodimers, the serum-free medium was replaced every hour to keep secreted factors at very low levels. We observed similar EGFR–ErbB2 heterodimerization in MDA453, SKBR3, and BT474 cells under these conditions (our unpublished results). Therefore, spontaneous dimerization occurs in the cells that overexpress ErbB2. Because of the spontaneous EGFR–ErbB2 heterodimerization, the calculated EGFR–ErbB2 heterodimerization levels after EGF stimulation actually reflected both spontaneous and EGF-stimulated heterodimerization.
The relative EGFR endocytosis levels for the four breast cancer cell lines were calculated as the percentage of total EGFR that accumulated in the endosome fraction (Table 1). The EGFR that accumulated in the endosome fraction only represents a portion of the endocytosed EGFR. During endocytosis, EGFR may be located in other endocytic compartments, such as in coated vesicles and lysosomes. Our subcellular fractionation method was designed to maximize the purity of the endosome fraction, possibly underestimating recovery.
Very recently, it has been reported that EGF induces ErbB2 internalization in mouse B82 fibroblasts. Immunofluorescence analysis with rabbit polyclonal anti-ErbB2 antibody C18 (Santa Cruz) showed that, after EGF treatment for 4 h at 37°C with 100 ng/ml EGF, ErbB2 was colocalized with EGFR in the lysosomes (Worthylake and Wiley, 1997). In our study, we used the same anti-ErbB2 antibody (C18) to analyze EGF-induced endocytosis of ErbB2 in BT20 cells by immunofluorescent microscopy, and the results showed that ErbB2 was colocalized with EGFR in endosomes after EGF stimulation for 30 min at 37°C (our unpublished results). However, when we used monoclonal anti-ErbB2 antibody 9G6 (Santa Cruz), ErbB2 was localized at the PM with or without EGF stimulation (Figure 3). It is very likely that the reported EGF-induced endosome and lysosome localization of ErbB2 in mouse B82 fibroblasts by rabbit polyclonal C18 anti-ErbB2 antibody is due to a cross-reaction with EGFR.
Among the EGFR family receptors, only the endocytosis of EGFR has been extensively studied. Although a molecular mechanism for the rapid endocytosis of growth factor–receptor complexes has not been established, recent evidence suggests that normal endocytosis and down-regulation of EGFR require the activation of intrinsic tyrosine kinase activity and autophosphorylation (Chen et al., 1987; Honegger et al., 1987; Helin and Beguinot, 1991; Sorkin et al., 1991, 1992). EGFR mutants truncated from the C termini to residue 991 (Chang et al., 1993) or residue 973 (Decker et al., 1992) were internalized inefficiently, and a mutant truncated at residue 958 was not internalized (Chang et al., 1993). Simultaneous point mutations of five tyrosine residues (Tyr-992, Tyr-1068, Tyr-1086, Tyr-1148, and Tyr-1173) to phenylalanine reduced the internalization rate to a minimum (Sorkin et al., 1992). Although a few studies about the endocytosis of ErbB2 have been published, the results are very controversial (Drebin et al., 1985; Sorkin et al., 1993; Baulida et al., 1996; Klapper et al., 1997). Using a chimeric receptor composed of the extracellular EGFR binding domain and the cytoplasmic domain of the ErbB2 molecule, it was shown that the EGFR/ErbB2 chimeras internalized 125I-EGF severalfold more slowly than the EGFR (Sorkin et al., 1993; Baulida et al., 1996). This study also indicated that the EGFR/ErbB2 chimeras were activated by EGF and that the impaired internalization capacity of this receptor was due to sequences in the ErbB2 C-terminal domain. This suggests that ErbB2 may lack required internalization signals in its C terminus (Sorkin et al., 1993). However, it is possible that the construction of the chimeras altered the three-dimensional structures, and this alteration resulted in the inhibition of EGF-induced endocytosis. To exclude this possibility, we constructed a chimera composed of the ErbB2 extracellular domain and the EGFR intracellular domain. We showed that, after being expressed in BT20 cells, in response to EGF, this chimera underwent normal endocytosis, as does endogenous EGFR (Figure 8). These results suggest that the intracellular domain of ErbB2 is responsible for the endocytosis deficiency of EGFR–ErbB2 heterodimers.
In contrast, it has been shown that upon binding of certain mAbs, ErbB2 undergoes internalization in a pathway shared by other growth factor receptors in response to ligand or antibody (Drebin et al., 1985; Klapper et al., 1997). The ability of the mAbs to induce endocytic degradation of ErbB2 was strictly dependent on antibody bivalency, implying their association with the ErbB2 homodimers (Gilboa et al., 1995; Hurwitz et al., 1995; Klapper et al., 1997). A point mutation in the transmembrane domain of the rat ErbB2 (Val-664 replaced by Glu) results in a constitutively dimerized and permanently active receptor (Bargmann et al., 1986; Stern et al., 1988; Weiner et al., 1989), and this activated ErbB2 homodimer was internalized like EGFR (Gilboa et al., 1995). These results suggest that ErbB2 contains internalization signals and ErbB2 internalization is dependent on its dimerization.
Our results indicate that ErbB2 and the EGFR–ErbB2 heterodimers are impaired in EGF-induced endocytosis. Because the EGFR in EGFR–ErbB2 heterodimers is phosphorylated in response to EGF stimulation (Figure 1), its internalization signals are likely activated. If ErbB2 does not contain internalization signals as suggested by the studies with chimeric receptors, our results may suggest that either ErbB2 contains inhibitory signals for endocytosis or else that a pair of internalization signals are required for the endocytosis of the EGFR–ErbB2 dimers. On the other hand, if ErbB2 does contain internalization signals, our results may suggest that the pair of internalization signals must be identical to allow internalization. The requirement for the paired internalization signals in the dimer of the receptors may also suggest that the downstream proteins that regulate receptor endocytosis are present in a dimeric form and need to bind to a pair of internalization signals simultaneously. Recently, distinct endocytic responses of heteromeric and homomeric transforming growth factor β receptors have been reported (Anders et al., 1997).
The ErbB2 gene is amplified and/or overexpressed in 25–30% of human breast and ovarian cancers, and overexpression of the receptor is associated with a poor prognosis (Slamon et al., 1987, 1989). Consistent with these clinical observations, the overexpression of ErbB2 in human breast and ovarian cancer cell lines has been shown to increase DNA synthesis, promote cell growth, improve soft agar cloning efficiency and increase tumorigenicity in nude mouse xenograft models (Di Fiore et al., 1987; Hudziak et al., 1987; Pietras et al., 1995; Reese and Slamon, 1997). Because ErbB2 is activated by both EGF and HRG indirectly through heterodimerization with EGFR, ErbB3, or ErbB4, it is not surprising that overexpression of ErbB2 enhances cell signaling. However, the selective overexpression of ErbB2, instead of EGFR, ErbB3, or ErbB4, in breast cancer and various other cancers suggests that unique properties of ErbB2 may contribute to this selection. Our results may suggest a mechanism by which overexpression of ErbB2 contributes to cancer development. It is possible that in the breast cancer cells that overexpress ErbB2, EGFR primarily forms heterodimers with ErbB2. The EGFR–ErbB2 heterodimers are impaired in EGF-induced endocytosis and down-regulation. The impaired endocytosis leads to sustained signaling in response to EGF and subsequently stimulates the overproliferation and transformation of breast cancer cells. Indeed, a mutant EGFR that avoids internalization transmits a growth signal at a lower EGF concentration and is capable of transforming cells in a ligand-dependent manner (Wells et al., 1990). In addition, blocking clathrin-mediated endocytosis by expressing mutant dynamin also enhanced EGF-induced cell proliferation (Vieira et al., 1996). Therefore, this present study provides evidence to favor a mechanistic link between ErbB2 overexpression and cell transformation.
ACKNOWLEDGMENTS
We thank Dr. A. Ullrich for providing EGFR and ErbB2 plasmids. This work was supported in part by funds from the Medical Research Council of Canada (to Z.W.), Cancer Care Ontario (to Z.W.), and the Northern Cancer Research Foundation (to Z.W. and T.Y.). Z.W. is a Medical Research Council of Canada Scholar.
REFERENCES
- Anders RA, Arline SL, Dore JJ, Leof EB. Distinct endocytic responses of heteromeric and homomeric transforming growth factor beta receptors. Mol Biol Cell. 1997;8:2133–2143. doi: 10.1091/mbc.8.11.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D, Koch CA, Gray L, Ellis C, Moran MF, Pawson T. Binding of SH2 domains of phospholipase C gamma 1, GAP, and Src to activated growth factor receptors. Science. 1990;250:979–982. doi: 10.1126/science.2173144. [DOI] [PubMed] [Google Scholar]
- Bargmann CI, Hung MC, Weinberg RA. Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell. 1986;45:649–657. doi: 10.1016/0092-8674(86)90779-8. [DOI] [PubMed] [Google Scholar]
- Baulida J, Kraus MH, Alimandi M, Di Fiore PP, Carpenter G. All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J Biol Chem. 1996;271:5251–5257. doi: 10.1074/jbc.271.9.5251. [DOI] [PubMed] [Google Scholar]
- Carpenter G. Receptors for epidermal growth factor and other polypeptide mitogens. Annu Rev Biochem. 1987;56:881–914. doi: 10.1146/annurev.bi.56.070187.004313. [DOI] [PubMed] [Google Scholar]
- Carraway KL, III, Cantley LC. A neu acquaintance for erbB3 and erbB4: a role for receptor heterodimerization in growth signaling. Cell. 1994;78:5–8. doi: 10.1016/0092-8674(94)90564-9. [DOI] [PubMed] [Google Scholar]
- Chang CP, Lazar CS, Walsh BJ, Komuro M, Collawn JF, Kuhn LA, Tainer JA, Trowbridge IS, Farquhar MG, Rosenfeld MG. Ligand-induced internalization of the epidermal growth factor receptor is mediated by multiple endocytic codes analogous to the tyrosine motif found in constitutively internalized receptors. J Biol Chem. 1993;268:19312–19320. [PubMed] [Google Scholar]
- Chen WS, Lazar CS, Lund KA, Welsh JB, Chang CP, Walton GM, Der CJ, Wiley HS, Gill GN, Rosenfeld MG. Functional independence of the epidermal growth factor receptor from a domain required for ligand-induced internalization and calcium regulation. Cell. 1989;59:33–43. doi: 10.1016/0092-8674(89)90867-2. [DOI] [PubMed] [Google Scholar]
- Chen WS, Lazar CS, Poenie M, Tsien RY, Gill GN, Rosenfeld MG. Requirement for intrinsic protein tyrosine kinase in the immediate and late actions of the EGF receptor. Nature. 1987;328:820–823. doi: 10.1038/328820a0. [DOI] [PubMed] [Google Scholar]
- Coussens L, Yang-Feng TL, Liao YC, Chen E, Gray A, McGrath J, Seeburg PH, Libermann TA, Schlessinger J, Francke U. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science. 1985;230:1132–1139. doi: 10.1126/science.2999974. [DOI] [PubMed] [Google Scholar]
- Decker SJ, Alexander C, Habib T. Epidermal growth factor (EGF)-stimulated tyrosine phosphorylation and EGF receptor degradation in cells expressing EGF receptors truncated at residue 973. J Biol Chem. 1992;267:1104–1108. [PubMed] [Google Scholar]
- Di Fiore PP, Pierce JH, Fleming TP, Hazan R, Ullrich A, King CR, Schlessinger J, Aaronson SA. Overexpression of the human EGF receptor confers an EGF- dependent transformed phenotype to NIH 3T3 cells. Cell. 1987;51:1063–1070. doi: 10.1016/0092-8674(87)90592-7. [DOI] [PubMed] [Google Scholar]
- Di Guglielmo GM, Baass PC, Ou WJ, Posner BI, Bergeron JJ. Compartmentalization of SHC, GRB2 and mSOS, and hyperphosphorylation of Raf-1 by EGF but not insulin in liver parenchyma. EMBO J. 1994;13:4269–4277. doi: 10.1002/j.1460-2075.1994.tb06747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drebin JA, Link VC, Stern DF, Weinberg RA, Greene MI. Down-modulation of an oncogene protein product and reversion of the transformed phenotype by monoclonal antibodies. Cell. 1985;41:697–706. doi: 10.1016/s0092-8674(85)80050-7. [DOI] [PubMed] [Google Scholar]
- Felder S, LaVin J, Ullrich A, Schlessinger J. Kinetics of binding, endocytosis, and recycling of EGF receptor mutants. J Cell Biol. 1992;117:203–212. doi: 10.1083/jcb.117.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder S, Miller K, Moehren G, Ullrich A, Schlessinger J, Hopkins CR. Kinase activity controls the sorting of the epidermal growth factor receptor within the multivesicular body. Cell. 1990;61:623–634. doi: 10.1016/0092-8674(90)90474-s. [DOI] [PubMed] [Google Scholar]
- Gilboa L, Ben-Levy R, Yarden Y, Henis YI. Roles for a cytoplasmic tyrosine and tyrosine kinase activity in the interactions of Neu receptors with coated pits. J Biol Chem. 1995;270:7061–7067. doi: 10.1074/jbc.270.13.7061. [DOI] [PubMed] [Google Scholar]
- Glenney JR, Jr, Chen WS, Lazar CS, Walton GM, Zokas LM, Rosenfeld MG, Gill GN. Ligand-induced endocytosis of the EGF receptor is blocked by mutational inactivation and by microinjection of anti-phosphotyrosine antibodies. Cell. 1988;52:675–684. doi: 10.1016/0092-8674(88)90405-9. [DOI] [PubMed] [Google Scholar]
- Goldman R, Levy RB, Peles E, Yarden Y. Heterodimerization of the erbB-1 and erbB-2 receptors in human breast carcinoma cells: a mechanism for receptor transregulation. Biochemistry. 1990;29:11024–11028. doi: 10.1021/bi00502a002. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997;16:1647–1655. doi: 10.1093/emboj/16.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus-Porta D, Beerli RR, Hynes NE. Single-chain antibody-mediated intracellular retention of ErbB-2 impairs Neu differentiation factor and epidermal growth factor signaling. Mol Cell Biol. 1995;15:1182–1191. doi: 10.1128/mcb.15.3.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin CH. Dimerization of cell surface receptors in signal transduction. Cell. 1995;80:213–223. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- Helin K, Beguinot L. Internalization and down-regulation of the human epidermal growth factor receptor are regulated by the carboxyl-terminal tyrosines. J Biol Chem. 1991;266:8363–8368. [PubMed] [Google Scholar]
- Honegger AM, Schmidt A, Ullrich A, Schlessinger J. Separate endocytic pathways of kinase-defective and -active EGF receptor mutants expressed in same cells. J Cell Biol. 1990;110:1541–1548. doi: 10.1083/jcb.110.5.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honegger AM, Szapary D, Schmidt A, Lyall R, Van Obberghen E, Dull TJ, Ullrich A, Schlessinger J. A mutant epidermal growth factor receptor with defective protein tyrosine kinase is unable to stimulate proto-oncogene expression and DNA synthesis. Mol Cell Biol. 1987;7:4568–4571. doi: 10.1128/mcb.7.12.4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudziak RM, Schlessinger J, Ullrich A. Increased expression of the putative growth factor receptor p185HER2 causes transformation and tumorigenesis of NIH 3T3 cells. Proc Natl Acad Sci USA. 1987;84:7159–7163. doi: 10.1073/pnas.84.20.7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz E, Stancovski I, Sela M, Yarden Y. Suppression and promotion of tumor growth by monoclonal antibodies to ErbB-2 differentially correlate with cellular uptake. Proc Natl Acad Sci USA. 1995;92:3353–3357. doi: 10.1073/pnas.92.8.3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunagaran D, Tzahar E, Beerli RR, Chen X, Graus-Porta D, Ratzkin BJ, Seger R, Hynes NE, Yarden Y. ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: implications for breast cancer. EMBO J. 1996;15:254–264. [PMC free article] [PubMed] [Google Scholar]
- Klapper LN, Vaisman N, Hurwitz E, Pinkas-Kramarski R, Yarden Y, Sela M. A subclass of tumor-inhibitory monoclonal antibodies to ErbB- 2/HER2 blocks cross-talk with growth factor receptors. Oncogene. 1997;14:2099–2109. doi: 10.1038/sj.onc.1201029. [DOI] [PubMed] [Google Scholar]
- Koch CA, Anderson D, Moran MF, Ellis C, Pawson T. SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science. 1991;252:668–674. doi: 10.1126/science.1708916. [DOI] [PubMed] [Google Scholar]
- Kraus MH, Issing W, Miki T, Popescu NC, Aaronson SA. Isolation and characterization of ERBB3, a third member of the ERBB/epidermal growth factor receptor family: evidence for overexpression in a subset of human mammary tumors. Proc Natl Acad Sci USA. 1989;86:9193–9197. doi: 10.1073/pnas.86.23.9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus MH, Popescu NC, Amsbaugh SC, King CR. Overexpression of the EGF receptor-related proto-oncogene erbB-2 in human mammary tumor cell lines by different molecular mechanisms. EMBO J. 1987;6:605–610. doi: 10.1002/j.1460-2075.1987.tb04797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SA, Hung M-C. Regulation of Her2/neu gene expression (review) Oncol Rep. 1995;2:497–503. doi: 10.3892/or.2.4.497. [DOI] [PubMed] [Google Scholar]
- Moran MF, Koch CA, Anderson D, Ellis C, England L, Martin GS, Pawson T. Src homology region 2 domains direct protein-protein interactions in signal transduction. Proc Natl Acad Sci USA. 1990;87:8622–8626. doi: 10.1073/pnas.87.21.8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T. Protein modules and signaling networks. Nature. 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- Peles E, Yarden Y. Neu and its ligands: from an oncogene to neural factors. Bioessays. 1993;15:815–824. doi: 10.1002/bies.950151207. [DOI] [PubMed] [Google Scholar]
- Pietras RJ, Arboleda J, Reese DM, Wongvipat N, Pegram MD, Ramos L, Gorman CM, Parker MG, Sliwkowski MX, Slamon DJ. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene. 1995;10:2435–2446. [PubMed] [Google Scholar]
- Pinkas-Kramarski R, et al. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J. 1996;15:2452–2467. [PMC free article] [PubMed] [Google Scholar]
- Plowman GD, Culouscou JM, Whitney GS, Green JM, Carlton GW, Foy L, Neubauer MG, Shoyab M. Ligand-specific activation of HER4/p180erbB4, a fourth member of the epidermal growth factor receptor family. Proc Natl Acad Sci USA. 1993;90:1746–1750. doi: 10.1073/pnas.90.5.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, LeVea CM, Freeman JK, Dougall WC, Greene MI. Heterodimerization of epidermal growth factor receptor and wild-type or kinase-deficient Neu: a mechanism of interreceptor kinase activation and transphosphorylation. Proc Natl Acad Sci USA. 1994;91:1500–1504. doi: 10.1073/pnas.91.4.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese DM, Slamon DJ. HER-2/neu signal transduction in human breast and ovarian cancer. Stem Cells. 1997;15:1–8. doi: 10.1002/stem.150001. [DOI] [PubMed] [Google Scholar]
- Schlessinger J, Ullrich A. Growth factor signaling by receptor tyrosine kinases. Neuron. 1992;9:383–391. doi: 10.1016/0896-6273(92)90177-f. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- Sliwkowski MX, Schaefer G, Akita RW, Lofgren JA, Fitzpatrick VD, Nuijens A, Fendly BM, Cerione RA, Vandlen RL, Carraway KL., III Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinity receptor for heregulin. J Biol Chem. 1994;269:14661–14665. [PubMed] [Google Scholar]
- Soltoff SP, Carraway KL, III, Prigent SA, Gullick WG, Cantley LC. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol. 1994;14:3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin A, Di Fiore PP, Carpenter G. The carboxyl terminus of epidermal growth factor receptor/erbB-2 chimerae is internalization impaired. Oncogene. 1993;8:3021–3028. [PubMed] [Google Scholar]
- Sorkin A, Helin K, Waters CM, Carpenter G, Beguinot L. Multiple autophosphorylation sites of the epidermal growth factor receptor are essential for receptor kinase activity and internalization. Contrasting significance of tyrosine 992 in the native and truncated receptors. J Biol Chem. 1992;267:8672–8678. [PubMed] [Google Scholar]
- Sorkin A, Waters C, Overholser KA, Carpenter G. Multiple autophosphorylation site mutations of the epidermal growth factor receptor. Analysis of kinase activity and endocytosis. J Biol Chem. 1991;266:8355–8362. [PubMed] [Google Scholar]
- Spivak-Kroizman T, Rotin D, Pinchasi D, Ullrich A, Schlessinger J, Lax I. Heterodimerization of c-erbB2 with different epidermal growth factor receptor mutants elicits stimulatory or inhibitory responses. J Biol Chem. 1992;267:8056–8063. [PubMed] [Google Scholar]
- Stern DF, Kamps MP, Cao H. Oncogenic activation of p185neu stimulates tyrosine phosphorylation in vivo. Mol Cell Biol. 1988;8:3969–3973. doi: 10.1128/mcb.8.9.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D, Lavi S, Ratzkin BJ, Yarden Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol. 1996;16:5276–5287. doi: 10.1128/mcb.16.10.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich A, Coussens L, Hayflick JS, Dull TJ, Gray A, Tam AW, Lee J, Yarden Y, Libermann TA, Schlessinger J. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature. 1984;309:418–425. doi: 10.1038/309418a0. [DOI] [PubMed] [Google Scholar]
- Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- Vieira AV, Lamaze C, Schmid SL. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science. 1996;274:2086–2089. doi: 10.1126/science.274.5295.2086. [DOI] [PubMed] [Google Scholar]
- Wada T, Qian XL, Greene MI. Intermolecular association of the p185neu protein and EGF receptor modulates EGF receptor function. Cell. 1990;61:1339–1347. doi: 10.1016/0092-8674(90)90697-d. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gluck S, Zhang L, Moran M. Requirement for Phospholipase C-gamma1 enzymatic activity in growth factor-induced mitogenesis. Mol Cell Biol. 1998;18:590–597. doi: 10.1128/mcb.18.1.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Moran MF. Requirement for the adapter protein GRB2 in EGF receptor endocytosis. Science. 1996;272:1935–1939. doi: 10.1126/science.272.5270.1935. [DOI] [PubMed] [Google Scholar]
- Wang Z, Tung PS, Moran MF. Association of p120 ras GAP with endocytic components and colocalization with epidermal growth factor (EGF) receptor in response to EGF stimulation. Cell Growth & Differ. 1996;7:123–133. [PubMed] [Google Scholar]
- Weiner DB, Liu J, Cohen JA, Williams WV, Greene MI. A point mutation in the neu oncogene mimics ligand induction of receptor aggregation. Nature. 1989;339:230–231. doi: 10.1038/339230a0. [DOI] [PubMed] [Google Scholar]
- Wells A, Welsh JB, Lazar CS, Wiley HS, Gill GN, Rosenfeld MG. Ligand-induced transformation by a noninternalizing epidermal growth factor receptor. Science. 1990;247:962–964. doi: 10.1126/science.2305263. [DOI] [PubMed] [Google Scholar]
- Wiley HS, Herbst JJ, Walsh BJ, Lauffenburger DA, Rosenfeld MG, Gill GN. The role of tyrosine kinase activity in endocytosis, compartmentation, and down-regulation of the epidermal growth factor receptor. J Biol Chem. 1991;266:11083–11094. [PubMed] [Google Scholar]
- Worthylake R, Wiley HS. Structural aspects of the epidermal growth factor receptor required for transmodulation of erbB2/neu. J Biol Chem. 1997;272:8594–8601. doi: 10.1074/jbc.272.13.8594. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Ikawa S, Akiyama T, Nomura N, Miyajima N, Saito T, Toyoshima K. Similarity of protein encoded by the human c-erb-B-2 gene to epidermal growth factor receptor. Nature. 1986;319:230–234. doi: 10.1038/319230a0. [DOI] [PubMed] [Google Scholar]
- Yarden Y, Schlessinger J. Self-phosphorylation of epidermal growth factor receptor: evidence for a model of intermolecular allosteric activation. Biochemistry. 1987;26:1434–1442. doi: 10.1021/bi00379a034. [DOI] [PubMed] [Google Scholar]