

Abstract
Familial haemophagocytic lymphohistiocytosis (FHL) is a rare autosomal recessive disorder of immune dysregulation associated with uncontrolled T cell and macrophage activation and hypercytokinaemia. The incidence of FHL is 0·12/100·000 children born per year, with a male to female ratio of 1:1. The disease is classified into six different types based on genetic linkage analysis and chromosomal localization; five specific genetic defects have been identified, which account for approximately 90% of all patients. Type 1 is due to an as yet unidentified gene defect located on chromosome nine. Type 2 is caused by mutations in the perforin (PRF1) gene, type 3 by mutations in the Munc-13–4 (UNC13D) gene, type 4 by mutations in the syntaxin 11 (STX11) gene and the recently described type 5 due to mutations in the gene encoding syntaxin binding protein 2 (STXBP-2). The incidence of the five types varies in different ethnic groups. The most common presenting features are pyrexia of unknown origin, pronounced hepatosplenomegaly and cytopenias. Neurological features tend to present later and are associated with poor prognosis. Absent or decreased lymphocyte cytotoxicity is the cellular hallmark of FHL. Biochemical features such as hyperferritinaemia, hypertriglyceridaemia and hypofibrinogenaemia are usually present, along with high levels of soluble interleukin 2 receptor in the blood and cerebrospinal fluid. Bone marrow aspirate may demonstrate the characteristic haemophagocytes, but initially is non-diagnostic in two-thirds of patients. Established international clinical, haematological and biochemical criteria now facilitate accurate clinical diagnosis. The disease is fatal unless a haematopoietic stem cell transplant (HSCT) is performed. The introduction of HSCT has dramatically improved the prognosis of the disease. However, the mortality of the disease is still significantly high and a number of challenges remain to be addressed. Active disease at the time of the transplant is the major significant poor prognostic factor. Delayed diagnosis, after irreversible organ damage has occurred, especially neurological damage, disease reoccurrence and pre-transplant mortality, remain a concern.
Keywords: diagnosis, familial, haemophagocytic lymphohistiocytosis, treatment
Introduction
Haemophagocytic lymphohistiocytosis (HLH) describes a clinical and immunological syndrome characterized by an excessive inflammatory response due to hyperactivation of macrophages and cytotoxic T lymphocytes (CTLs), resulting in progressive multi-organ failure if untreated. Increased production of proinflammatory cytokines and tissue infiltration by macrophages/histiocytes and CTLs are cardinal features of the phenotype. HLH is the clinical and immunological manifestation of several different disease entities, including primary or familial haemophagocytic lymphohistiocytosis (FHL) and secondary forms, which are associated with infections, autoimmune diseases and malignancies. Other genetic immunodeficiencies can also develop HLH as part of the disease process, which can make diagnosis of the underlying defect challenging. HLH has also been described in association with inborn errors of metabolism, e.g. lysinuric protein intolerance and multiple sulphatase deficiency [1]. This review will focus on FHL, with additional reference to other primary immunodeficiencies where HLH is a significant disease manifestation.
Epidemiology
Familial haemophagocytic lymphohistiocytosis is a disorder of early childhood. It was first described in 1952 by Farquhar and Claireaux, who identified the disease in two fatal cases involving siblings, with the major findings being a marked increase of histiocytes in the bone marrow at the expense of blood-forming cells [2]. The condition was shown to be more prevalent in consanguineous families, suggesting an autosomal recessive pattern of inheritance [3]. The reported incidence varies among different studies, probably reflecting different prevalence within different ethnic groups and the higher index of suspicion and improved diagnosis in more recent studies. It has been reported to be 0·12 per 100 000 children per year in Sweden [4], whereas it was found to be 0·342/100 000 in Japan [5]. The frequency of the disease among hospitalized patients in Turkey is 7·5 in 10 000, which is due most probably to the high rate of consanguineous marriage in Turkish pedigrees [6].
Pathophysiology
The salient features of FHL are systemic release of proinflammatory cytokines, persistent activation of macrophages/histiocytes and T cells and multi-system inflammation with characteristic clinical and laboratory features of fever, hepatosplenomegaly, cytopenias, coagulopathy and hypertriglyceridaemia.
The pathogenesis of the disease remains controversial. From studies in humans it is clear that the principal underlying defect is impaired T and natural killer (NK) cell cytotoxicity, even though the actual numbers of these effector cells are normal [7]. However, the precise mechanism that links the defects in cytotoxicity and clinical disease is not fully understood. NK cells and CTLs play a major role in the immune response to invading pathogens, predominantly viruses. They secrete soluble mediators such as interferon (IFN)-γ, which enhances immunity, interferes with viral replication and is also a potent macrophage activator. CTLs also mediate direct killing of infected cells and antigen-presenting cells (APCs), including macrophages, predominantly via the release of cytotoxic granules [8]. Studies in the perforin (PRF)-deficient and Unc13d-deficient mouse models have shown that when the cytotoxic function is impaired there is a disproportional expansion of CD8 T cells and excessive production of cytokines, including IFN-γ, and persistent macrophage activation [9–11]. This leads to tissue infiltration by macrophages/histiocytes and increased production of proinflammatory cytokines, giving rise to clinical features resembling those of the human disease. These experiments and observations in human disease gave rise to the hypothesis that granule-dependent cytotoxicity plays a vital role in T lymphocyte homeostasis following infection, possibly through direct killing of CTLs [8]. However, whether this occurs in vivo remains to be proved. The role of hyperactivated tissue infiltrating macrophages in the pathophysiology of the disease and whether they represent the cause or the result of the disease also remains to be elucidated.
During active disease there is a high serum concentration of IFN-γ, tumour necrosis factor (TNF)-α, interleukin (IL)-6, IL-8, IL-10, IL-12, IL-18 and macrophage inflammatory protein (MIP)-1α[12–14]. Cytokines may also infiltrate tissues and lead directly to necrosis and organ failure [15,16]. Fever and cytopenias are prominent in FHL and may be induced by several cytokines, such as TNF, IL-6 and IFN-γ. TNF also has a procoagulant activity, which may account for the hypofibrinogenaemia [17]. The hypertriglyceridaemia is associated with reduced lipoprotein lipase, due to the presence of TNF [18].
Identification of the molecular defects associated with FHL has now given a greater insight into the mechanisms by which the cellular defects arise.
Genetic basis of FHL
FHL has an autosomal recessive trait, and to date five genetic loci have been identified giving rise to five different subtypes of the disease (Table 1). All the genes identified to date encode for proteins that participate in the granule-dependent cytotoxic function of NK and T cells (Fig. 1) and explain the cellular pathogenesis described above. This mechanism is essential for the elimination of infected target cells and control of the immune response [19].
Table 1.
Summary of eight studies examining the percentage of patients with different forms of familial haemophagocytic lymphohistiocytosis (FHL). Column 1 provides the references, column 2 summarizes the patients and their ethnic origin and column 3 provides percentages for the different types of FHL. Seven of the studies used genetic analysis and one protein screening.
| Reference | Numerical breakdown | Percentages |
|---|---|---|
| ‘Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis’[28] | 34 families from various countries 7 families FHL2 mutations | FHL1 = 10% FHL2 = 20% (FHL2 = 30% in Turkish patients) FHL2 = 20–40% (mutations and polymorphism) |
| ‘Characterisation of diverse PRF1 mutations leading to decreased NK cell activity in North American families with haemophagocytic lymphohistiocytosis’[31] | 50 families from North America 43 studied for perforin mutations (the other 7 unlikely to be FHL2) 25 families FHL2 | FHL2 = 50% |
| ‘Genetic subtypes of familial hemophagocytic lymphohistiocytosis: correlations with clinical features and cytotoxic T lymphocyte/natural killer cell functions’[39] | 35 patients with FHL in Japan 11 patients FHL2 8 patients FHL3 16 patients non-FHL2/FH3 | FHL2 = 31% FHL3 = 23% Non-FHL2/3 = 46% |
| ‘Spectrum and clinical implications of syntaxin 11 gene mutations in FHL: association with disease-free remissions and haematopoietic malignancies’[43] | 28 families non-FHL2, various countries of origin 4 families FHL4 (all Turkish) | FHL4 = 14% of non-FHL2 |
| ‘Novel Munc13–4 mutations in children and young adults with haemophagocytic lymphohistiocytosis’[38] | 30 families with FHL, non- FHL2 (26 of Italian origin) 15 families FHL3 | FHL3 = 50% of non-FHL2 cases |
| ‘Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analysis of PRF1, UNC13D, STX11 and RAB27A’[35] | 63 unrelated patients from various ethnic backgrounds 38 patients with mutations 20 patients FHL1 12 patients FHL2 6 patients FHL4 (all Turkish) | FHL2 = 32% FHL3 = 19% FHL4 = 9·5% Gene defect found; 80% of Turkish and 30% of German patients |
| ‘Familial hemophagocytic lymphohistiocytosis: protein screening, genetic analysis and ethnicity’ Walshe D, Peraj R, Maeney C XIIth Meeting of the ESID, 2006 | 88 FHL patients from various ethnic backgrounds underwent protein screening | Perforin = 19% Munc13–4 = 25% SAP = 4% 52% cause not identified |
| ‘Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells’[45] | 80 FHL patients, ethnic background not reported | FHL2 + FHL3 + FHL4 = 80% FHL5 = 10% Unknown = 10% |
Fig. 1.
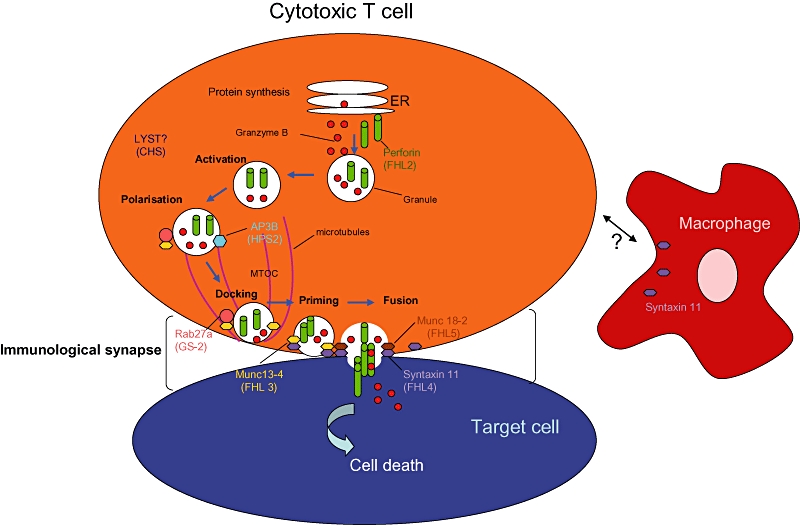
Model of the molecular mechanisms of familial haemophagocytic lymphohistiocytosis. Following cytotoxic T cell activation and the formation of the immunological synapse with the target cell, the microtubule organizing centre (MTOC) moves to the contact site repolarizing the microtubule network. Lytic granules move along the microtubules towards the contact site and they dock to the plasma membrane. Priming and fusion with the membrane follows and the contents of the lytic granules, i.e. perforin and granzymes, are released into the intercellular space, causing rapid death of the target cell. Defects in trafficking [?lysosomal trafficking regulator (LYST)], docking (Rab27a), priming (Munc13-4) fusion [syntaxin (STX)11 and STXB2 or Munc18-2) and target cell entry (perforin) all result in different types of the disease which are indicated. The precise mechanisms and other effectors involved in the process remain to be elucidated.
A genomic region on chromosome 9 (9q21) is linked with FHL1, although the gene responsible and its specific product remain unknown to date [20]. The FHL1 locus is thought to account for 10% of gene defects causing FHL. The genes responsible for four other different types of FHL have now been identified.
In 1999 the gene encoding perforin on chromosome 10q21–22 locus was identified as the cause for FHL2 [21]. Perforin is found together with granzyme B in intracellular lysosomal granules in CTLs and NK cells. When NK cells or CTLs come into contact and engage with target cells, an immunological synapse is formed through reorganization of the microtubule organizing complex (MTOC) [19]. Lytic granules traffic to the contact site along the MTOC, dock and fuse with the plasma membrane and release their contents, which include perforin and granzyme B, into the contact site between effector cell and target cell. Perforin forms pores in the presence of calcium on the surface membrane of the target cell, allowing the entry of granzymes which then activate a cascade that leads eventually to apoptosis of the target cell [22]. In the absence of perforin, granzymes can still enter into the target cell, but adequate toxicity is not produced. Many missense mutations of PRF1 result in a conformational change to the perforin molecule that inhibits proteolytic cleavage of the perforin precursors. As a result, no mature or functional form of the protein is expressed [23].
More than 50 perforin mutations have been described to date [8,23–35]. Data from these studies suggests that there is genotype/phenotype correlation with regard to disease onset and clinical severity. Deleterious non-sense mutations resulting in no perforin production are associated with an early age of disease onset [31], whereas patients with missense mutations (homozygous or compound heterozygous) present at a later age [25,33]. The most common mutation observed in white Caucasians is heterozygosity for C272T causing an A91V substitution and has been proposed as a disease-modifying mutation [36].
Perforin mutations (FHL2) account for 20–40% of all FHL cases, with a somewhat higher prevalence in Turkey [28]. The percentage of perforin mutations also appears to be higher in North American families (approximately 50% of all FHL cases). Notably in African American patients, all individuals carry the same 50delT mutation, suggesting a founder effect in the population [31]. In Japan the most common perforin mutation is the 1090-1091delCT (62·5% of perforin mutations); the second most common mutation is the 207delC (37·5% of perforin mutations) [34], with perforin mutations accounting for 40% of the total FHL cases. Turkish families express the Trp374X perforin mutation in high frequency, which is associated with early disease onset [35], and Italian families express the A91V sequence variant. That certain mutations are unique in some populations suggests that each of the above perforin gene abnormalities occurred in separate common ancestors and may confer a survival advantage in heterozygote carriers [8].
The genetic basis of FHL3 was determined in 2003 [37]. The UNC13D gene is located on chromosome 17q25 and encodes the Munc13-4 protein. Six different mutations were identified originally in 10 patients from seven families. The resulting clinical and immunological phenotype was indistinguishable from that of FHL2 [37]. A further 12 new mutations were identified in a cohort of 30 families with FHL, where perforin mutations had been excluded [38]. Munc13-4 is a member of the UNC13 family of intracellular proteins and is required for vesicle priming. It co-localizes with cytolytic granules near the site of contact between the T cell and the target cell. In FHL3 patients, docking of the lytic granules, containing perforin and granzymes A and B, on the plasma membrane is normal. Priming of the granules, before fusion and subsequent release of cytolytic enzymes, is impaired.
In Japan Munc13-4 mutations were found in six of 16 patients with FHL, suggesting that FHL3 may account for 20–25% of all FHL cases [39].
Syntaxin 11 (STX11) is located on chromosome 6q24 and is the molecular basis of FHL4 [40]. The locus was identified in a large consanguineous Kurdish family with five children with FHL with a deletion in the STX11 gene. Three different mutations have been identified in a total of 10 FHL4 patients from six different families, all of them with a Turkish/Kurdish ethnic background. The product of the STX11 gene is a protein, syntaxin 11, a member of soluble N-ethylmaleimide-sensitive factor attachment protein receptors present on target cell membranes (t-SNARES). Syntaxin 11 is expressed in monocytes, NK cells and cytotoxic T cells and is involved in vesicle priming and membrane fusion [41,42].
Mutations in the STX11 may account for 14% of non-FHL1 cases; the figure is higher (21%) for the Turkish population [43]. Syntaxin 11 mutations have not been identified in FHL patients in Japan [44] or in any other ethnic group, suggesting that mutations in this gene may be limited to a specific ethnic group.
Very recently, a new genetic locus has been identified as a cause of FHL-5 by two separate groups [45,46]. The gene is located on chromosome 19p and encodes for Munc 18-2 (or syntaxin binding protein 2, STXBP2), a protein involved in the regulation of intracellular trafficking and control of SNARE complex assembly and disassembly. Through interaction with various SNARE proteins in different cell types, STXBP2 contributes to the granule exocytosis machinery [47,48]. Its interaction partner in lymphocytes is syntaxin 11, the protein defective in FHL-4. The majority of mutations identified are homozygous missense mutations affecting highly conserved residues at the binding site for syntaxin 11, resulting in reduced expression of both proteins. The less common splice site mutations have less impact as part of the protein function and its interaction with syntaxin 11 is preserved. Degranulation of NK and T cells from STXBP2-deficient patients is impaired, as is cytotoxic function of NK cells. Although the majority of patients are from consanguineous Turkish and Saudi Arabian families, identification of mutations in patients from central Europe implies that the defect is not confined to a specific ethnic group. The frequency of FHL-5 is estimated at 10% of the total FHL cases [45].
Despite genetic heterogeneity, patients with FHL generally have a homogeneous and indistinguishable clinical phenotype, which is independent of the gene responsible for the disease. A study of a Japanese cohort of patients with FHL2 and FHL3 suggested a correlation between the alloantigen-specific CTL-mediated toxicity and the detected amount of perforin expression [39], but these findings remain to be confirmed. Higher serum ferritin and CD25 (sIL-2R) levels have been associated with more severe cases of FHL2 compared with FHL3 [49], but were performed in only one ethnic group. A more recent study established a relationship between ethnicity and genotype, PRF1 being more common in patients from the Middle East, and STX11 presents almost exclusively in Turkish patients [50].
Clinical presentation
Approximately 70–80% of patients with FHL manifest the symptoms of HLH in the first year of life [51], with the peak age of presentation between 1 and 6 months of age. In rare cases, the disease may even develop in utero and be present at birth [52]. Presentation at late age, even in teenagehood, however, can also occur [53]. In addition, several adults (oldest 46 years in our cohort) have been diagnosed with FHL2 and are associated with missense mutations in the perforin gene, which results in partial protein expression and may account for the delayed presentation (K. Gilmour, personal communication). There is a tendency towards a similar age at onset within the same families [3]. These rare cases emphasize the heterogeneity of this disease and the diversity of its clinical presentations [54]. Although the male to female ratio is reported to be 1:1 [4], a review of 121 cases shows a skewing to the male population with a male : female ratio of 1·28:1 [55]. X-linked lymphoproliferative disease (XLP) can present with HLH and the presence of such cases among this cohort may account for this discrepancy.
The most common symptoms of FHL are fever, hepatosplenomegaly and cytopenias. Fever is typically prolonged and hepatosplenomegaly is pronounced and progressive. Lymphadenopathy develops in fewer than half the patients and is not usually remarkable. Oedema may be present. Skin manifestations are common, but non-specific. Transient maculopapular, nodular or purpuric skin rashes may develop and may be associated with high fever, and are present in up to 30–40% of cases [5]. Neurological abnormalities are another prominent feature of the disease and are associated with histiocytic infiltration into the central nervous system (CNS), and are more common later in the disease and may be a major feature of advanced FHL. The younger patient may present with irritability, bulging fontanelle, neck stiffness, hypotonia or hypertonia and convulsions in addition to other systemic symptoms. Cranial nerve (VI–VII) palsy, ataxia, hemiplegia/tetraplegia, blindness, unconsciousness and signs of raised intracranial pressure may develop. The disease is often triggered by infections, most commonly viral, but also bacterial. The most common viral pathogens are Epstein–Barr virus (EBV), cytomegalovirus and parvovirus [56].
Laboratory findings
Cytopenia is one of the most common findings of FHL, and in particular thrombocytopenia. Anaemia and neutropenia are also found at presentation. Reticulocytes are moderately raised despite the severe anaemia. The hallmark of FHL is impaired or absent NK cell and T cell cytotoxicity, although the absolute numbers are usually normal. The NK cell cytotoxicity is also defective in cases of secondary HLH. Function of NK cells and CTLs can be measured by the lysis of K562 erythroleukaemia cells in a chromium release assay. Impaired NK cell cytotoxicity can also be found in asymptomatic parents and siblings of patients, some of whom, but not all, are carriers of the disease. Therefore, NK cell function is not a useful tool in selecting siblings of FHL patients who are at risk of developing the disease [57] and may reflect the poor reliability of the assay.
Additional laboratory findings include hypertriglyceridaemia, hyperferritinaemia and liver dysfunction with elevated lactate dehydrogenase, serum transaminases and bilirubin. Hyperferritinaemia is very remarkable and characteristic and should alarm clinicians of the diagnosis. Patients can have coagulation abnormalities with hypofibrinogenaemia. Low fibrinogen is not usually associated with disseminated intravascular coagulation. Any bleeding tendency, when present in FHL, is usually the result of the low platelets [58]. The lipoprotein pattern was studied in nine children with FHL [18]. Triglycerides were elevated markedly in serum and in low- and very low-density lipoproteins and cholesterol was raised in very low-density lipoproteins. Both cholesterol and triglycerides were extremely low in high-density lipoproteins. All those abnormalities were reversible and normalized when patients went into remission.
Levels of the alpha chain of the IL-2 receptor (IL-2R), also known as CD25, are high in serum and cerebrospinal fluid of FHL patients. Other cytokines, such as IFN-γ, may be elevated. The concentration of CD25 correlates with disease activity and is elevated in children with untreated disease and returns to normal after treatment, and may provide a useful monitoring tool for the activity of the disease and prediction of relapse [59].
The neurological abnormalities observed in FHL can be associated with spinal fluid hyperproteinaemia and a moderate pleiocytosis, predominantly of lymphocytes and monocytes. More than 50% of patients will have these findings in the CSF, even in the absence of clinical symptoms. Caution must be exercised when performing a lumbar puncture in patients with neurological symptoms in case of raised intracranial pressure.
In patients with FHL there is accumulation of activated macrophages, lymphocytes and haemophagocytosis in the bone marrow, spleen, liver, lymph nodes and CNS. Haemophagocytosis may not be found in the initial stages of the disease [60,61]. In two-thirds of patients the bone marrow is non-diagnostic initially. Repeat bone marrow aspirates over time may eventually demonstrate haemophagocytosis. In exceptional cases, and if bone marrow is non-diagnostic, a biopsy from other organs, such as the liver or spleen [62,63], may be useful although the risk of bleeding following splenic biopsy must always be taken into account.
Diagnosis
If FHL is suspected, investigations should include full blood count and blood film, liver function tests, triglycerides, ferritin and coagulation profile. Bone marrow aspiration is performed and, if negative initially, should be repeated at a later stage if FHL is still a possible diagnosis. Lumbar puncture should be considered when there is suspicion of neurological involvement. A consensus set of criteria has been established by the Histiocyte Society and was revised in 2007 (HLH 2004 diagnostic guidelines; Table 2) [61,64]. These criteria have been extremely useful in allowing a clinical and laboratory diagnosis of HLH to be made while more definitive molecular assays are being undertaken. Unless family history or genetic testing is consistent with FLH, patients should present with five of the eight criteria.
Table 2.
Revised haemophagocytic lymphohistiocytosis (HLH) 2004 diagnostic criteria.
| The diagnosis of HLH can be established if one of either 1 or 2 below is fulfilled |
| 1.A molecular diagnosis consistent with HLH |
| 2.Diagnostic criteria for HLH fulfilled (five of eight criteria below) |
| Initial diagnostic criteria |
| 1.Fever |
| 2.Splenomegaly |
| 3.Cytopenia affecting at least two of the three lineages in the peripheral blood |
| Haemoglobin <90 g/l (in neonates haemoglobin <100 g/l) |
| Platelets <100 × 10˄9/l |
| Neutrophils <1·0 × 10˄9/l |
| 4.Hypertriglyceridaemia and/or hypofibrinogenaemia |
| Fasting triglycerides ≥3·0 mmol/l (i.e. ≥265 mg/dl) |
| Fibrinogen ≤1·5 g |
| 5.Haemophagocytosis in bone marrow, spleen or lymph nodes |
| No sigh of malignancy |
| Additional criteria |
| 6.Low/absent natural killer cell activity |
| 7.Hyperferritinaemia |
| Ferritin >500 µg/l |
| 8.High soluble interleukin 2 receptor levels |
| sIL-2R ≥2400 U/ml |
| Additional comments (Table 2) |
| 1.If bone marrow aspirate shows no haemophagocytosis look for evidence in other organs or perform serial marrow aspirates |
| 2.Diagnosis would be supported by: |
| (a)Spinal fluid pleiocytosis (mononuclear cells) and/or elevated spinal fluid protein |
| (b)Liver biopsy showing findings consistent with chronic persistent hepatitis |
| 3.Other clinical and laboratory findings which would point towards the diagnosis of FHL are: cerebromeningeal symptoms, lymph node enlargement, jaundice, oedema, skin rash, hepatic enzyme abnormalities, hypoproteinaemia, hyponatraemia, elevated very low-density lipoproteins (VLDL) and low high-density lipoproteins (HDL) |
If there is any clinical suspicion, screening for primary immunodeficiencies that present with HLH such as X-linked lymphoproliferative disease (XLP), Griscelli syndrome (GS) and Chediak–Higashi syndrome (CHS) should be performed (see also next section). At Great Ormond Street Hospital, rapid screening of perforin by fluorescence activated cell sorter (FACS) flow cytometry has enabled a 2-h screen for FHL2 (Fig. 2) [29,65]. FACS for perforin also identifies heterozygous carriers of FHL2. In addition, a granule release assay (GRA) has been developed that enables rapid screening of the proteins involved in the transport and fusion of cytotoxic granules to the cell membrane, providing an alternative tool for the assessment of the cytotoxic function of both T and NK cells [66–68]. The GRA assay is based on surface staining for CD107a (LAMP 1) on the surface of peripheral blood mononuclear cells (PMBCs) following stimulation with phytohaemagglutinin (PHA) or anti-CD3 (Fig. 3a). CD107a is present on the membrane of secretory granules within T and NK cells. It can be detected on the cell surface only when the secretory granules fuse with the cell membrane. Detection of CD107a on the cell surface following stimulation implies an intact pathway of granule exocytosis and normal function of the proteins in that pathway. Absence of CD107a expression on the cell surface suggests a defect in secretory granule migration, docking, priming or fusion. This screen cannot identify the exact defect. If the GRA is absent or abnormal, Munc13-4 and syntaxin 11 are screened by immunoblot followed by genetic analysis, as indicated (Fig. 3b). The GRA has identified FHL-3, FHL-4, GS and CHS patients as well as one patient with Hermansky–Pudlak syndrome type II. Patients with FHL-5 due to STXBP-2 mutations will also have defects on this assay. Patients with FHL2 have normal GRA (K. Gilmour, personal communication) [68].
Fig. 2.
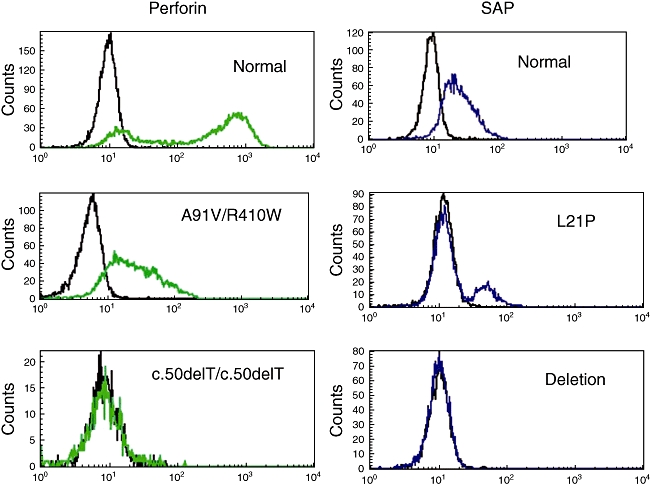
Fluorescence activated cell sorter (FACS) base diagnosis of X-linked lymphoproliferative disease (XLP) and familial haemophagocytic lymphohistiocytosis (FHL)2. FACS plots on the left are gated on natural killer (NK) cells and show a normal individual on top and two individuals with confirmed perforin mutations below. FACS plots of the right gated on CD8+ T cells show a normal individual on top and two confirmed XLP patients below. Mutations are indicated in each plot.
Fig. 3.
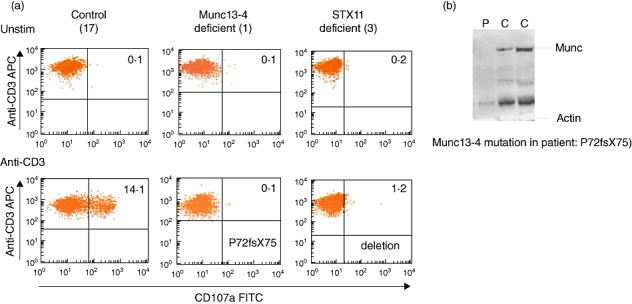
Granule release assay used to screen patients for familial haemophagocytic lymphohistiocytosis (FHL)3 and FHL4. (a) Granule release assay of CD8+ T cells unstimulated or stimulated with anti-CD3. The normal individual is on the left, FHL3 patient in the middle and FHL4 patient is on the right. Mutations are indicated on the plots. (b) Immunoblot showing absent Munc13-4 in a FHL3 patient (P) and normal Munc13-4 expression in two normal controls. A control protein actin is indicated below.
Prenatal and pre-implantation diagnosis is possible by genetic analysis once the gene defect within a family is known. Prenatal diagnosis was first performed in two unrelated Turkish families harbouring a perforin mutation [69].
Differential diagnosis
Four different inherited immunodeficiency disorders can present with HLH, with indistinguishable clinical presentation from FHL.
XLP, also known as Duncan's syndrome, is caused by mutations in the gene SH2D1A [70]. The gene encodes a protein known as SH2D1A or SAP [signalling lymphocyte activating molecule (SLAM)-associated protein]. This protein is important for signal transduction in a variety of immune cells and for activation of cytotoxic function in CTLs and NK cells. In T cells, the protein binds to intracellular motifs of the SLAM. In NK cells, it binds predominantly to 2B4, an NK cell-activating receptor, with a 20–35% homology with SLAM [71]. Mutations in SH2D1A result in dysregulated signalling pathways and impaired cell–cell interactions at multiple levels [72]: decreased IL-10 production and inducible co-stimulatory molecule (ICOS) expression on CD4 T cells result in impaired T cell help and impaired development of memory B cells and long-lived plasma cells. NK T cell development is abrogated. The 2B4-mediated IFN-γ production and cytotoxicity of CD8 T cells and NK cells is impaired. In addition, the polarization of cytotoxic mediators such as perforin to the immunological synapse and the subsequent target cell lysis is also defective in CD8 T cells [73].
Mutations in the SH2D1A gene have been identified in approximately 55–60% of patients with an XLP phenotype. More recently, mutations in X-linked inhibitor of apoptosis (XIAP) have been described as another cause of XLP with similar defects of cytotoxicity [74]. The disease presents typically with EBV-related HLH, but other viral infections can also trigger the clinical syndrome. Approximately 60% of patients will develop fulminant infectious mononucleosis, the majority whom die within 1–2 months [75,76]. In a significant number of cases no infectious triggers have been identified. Other clinical manifestations include hypogammaglobulinaemia and lymphomas. Patients are often completely asymptomatic, sometimes until adulthood, before the clinical phenotype occurs.
In a series of 25 male patients with HLH, mutations in SH2D1A were found in four patients, suggesting that all male patients with HLH should be screened for XLP [77]. Flow cytometric detection of SAP provides a rapid tool for diagnosis [65], and in Great Ormond Street hospital is performed concurrently with screening for FHL2 (Fig. 2). Detection of SAP by Western blot is an alternative method but has now been largely superseded by the FACS-based assays. Patients with XLP have normal GRA (K. Gilmour, personal communication).
GS and CHS are autosomal recessive disorders associated with partial albinism and immunodeficiency due to lysosomal trafficking defects. GS is characterized by pigmentary dilution with silvery grey sheen of hair. Three subtypes of the disease have been described: GS1 is caused by mutations in myosine VA (MYO5a) [78] and GS2 is caused by mutations in Rab27a [79]. Both these genes are located on chromosome 15q21. Recently, a third form of the disease (GS3) has been described due to mutations in the melanophilin gene (MLPH) [80]. Patients with GS1 do not develop HLH, but have severe neurological abnormalities. Patients with GS3 have isolated hypopigmentation. Only GS2 patients with Rab27a mutations develop HLH. Rab27a interacts with Munc13-4 and is involved in cytotoxic granule exocytosis. The Munc13-4/Rab27a complex is essential for docking of the secretory granules with the plasma membrane in haematopoietic cells [81,82].
CHS is caused by mutations in the lysosomal trafficking regulator (LYST) gene, which was mapped to chromosome 1q43 [83]. The function of LYST is not yet clear but its product seems to be involved in the final steps of granule secretion.
In addition to HLH, CHS patients demonstrate hypopigmentation, bleeding tendency, neurological symptoms and frequent pyogenic infections. Approximately 85% of CHS patients develop HLH [84]. GS and CHS can be diagnosed by microscopy on a shaft of hair.
Hermansky–Pudlak syndrome type II (HPS II) is characterized by oculocutaneous albinism, bleeding and neutropenia and is distinguished from other forms of the disease by an increased susceptibility to infections [85]. It is caused by mutations in the cytosolic adaptor protein AP-3 (AP3B1). A single case of HPS II has been described which developed fatal HLH [86]. The patient had impaired NK cell and CTL cytotoxicity.
In addition to the above, well-established causes of familial/primary HLH, other primary immunodeficiencies such as DiGeorge syndrome [87], severe combined immunodeficiency (SCID) and chronic granulomatous disease [88] may occasionally present with features of HLH.
The clinical syndrome of HLH may also be associated with severe infections, most commonly viral infections [predominantly of the herpes group and human immunodeficiency virus (HIV)], but parasitic and bacterial infections have also been reported. Leishmania infection in particular may mimic or cause secondary HLH [89–91]. Visceral leishmaniasis is a systemic disease caused by infection of the reticuloendothelial system by the protozoan Leishmania. It is endemic in the Mediterranean basin, but can also be contracted during short visits. It presents with typical clinical features of HLH, with fevers, pancytopenia and hepatosplenomegaly. There are no discriminating signs from other causes of HLH. Bone marrow examination reveals haemophagocytosis in the majority of cases, but the protozoan species are more difficult to identify and repeated bone marrow aspiration, blood cultures or serology may be necessary [91]. Liposomal amphotericin is the drug of choice. The diagnosis should always be considered in patients coming from endemic areas or those who have travelled to endemic areas.
Haematological abnormalities such as leukaemia, lymphoma, aplastic anaemia and myelodysplastic syndromes may present with features of HLH. Other malignancies, such as solid tumours, and autoimmune diseases, especially systemic onset juvenile rheumatoid arthritis and less frequently rheumatoid arthritis or systemic lupus erythematosus (SLE) have also been reported as being complicated by HLH. The term ‘macrophage activation syndrome’ (MAS) has been used, especially in HLH complicating autoimmune syndromes [92]. These cases of HLH presenting in adults or older children have been described as ‘secondary’ HLH. However, recent studies in haematological malignancies identified increased frequency of genetic defects such as those of perforin and SAP [26,93,94], implying a pre-existing genetic predisposition for the development of the disease in some cases. Although there are no accurate data about the incidence of secondary (acquired) HLH, secondary cases are more common overall than familial HLH.
Treatment
If left untreated, FHL is fatal with a median survival of 2 months. Patients will die of overwhelming infections, or uncontrolled systemic inflammation and multi-organ failure. The treatment involves initially the control of infectious triggers and of the immune dysregulation with chemotherapeutic regimens followed by definitive treatment with haematopoietic stem cell transplant (HSCT). Treatment should be started early once a clinical and laboratory diagnosis has been established, given the high mortality rate of FHL, and even if molecular diagnosis is not available.
Several chemotherapeutic regimens have been used in the past targeting activated macrophages/histiocytes and T cells. In 1994 the Histiocyte Society produced a consensus study protocol (HLH-94), which includes the combination of intravenous etoposide, oral or intravenous dexamethasone and, in patients with progressive neurological symptoms or abnormal cerebrospinal fluid (CSF), intrathecal methotrexate. Alternative regimens have used the T cell depleting agent anti-thymocyte globulin (ATG) [95]. A modified protocol (HLH-2004) with the addition of oral cyclosporin is being used currently (Table 3) [61,64]. Initial treatment lasts for 8 weeks followed by maintenance therapy with etoposide at lower doses, prednisolone and cyclosporin A in order to sustain remission until HSCT is performed. Supportive treatment includes broad-spectrum antibiotics, prophylactic co-trimoxazole, oral anti-mycotics, antiviral therapy if indicated and intravenous immunoglobulin.
Table 3.
Summary of the haemophagocytic lymphohistiocytosis (HLH)-2004 treatment protocol (the complete protocol is available on request at http://www.histiocytesociety.org).
| Initial therapy (weeks 1–8) |
| Etoposide intravenous 150 mg/m2twice weekly for 2 weeks and then weekly |
| Dexamethasone intravenous daily 10 mg/ m2for 2 weeks, 5 mg/ m2for 2 weeks, 2·5 mg/ m2for 2 weeks, 1·25 mg/ m2for 2 weeks, tapering for 1 week |
| Cyclosporin A oral daily, aiming at trough levels of 200 µg/l |
| Intrathecal methotrexate (up to 4 doses) weeks 3–6 if there is central nervous system involvement |
| Continuation therapy (week 9 until haematopoietic stem cell transplant) |
| Etoposide 150 mg/m2every second week alternating with dexamethasone |
| Dexamethasone intravenous 10 mg/ m2× 3 days every second week |
| Cyclosporin A daily oral aiming at trough levels of 200 µg/l |
| Reactivation therapy |
| Restart from week 2 of initial therapy |
If there is no response to the initial treatment, the prognosis is poor. Patients should be monitored closely for reactivation, especially of the CNS system. For CNS reactivation the use of dexamethasone and intrathecal therapy with methotrexate and prednisolone is recommended [12]. Cyclophosphamide may also be useful. Reactivations are common when the therapeutic intensity is reduced, after an infection or a vaccination. If reactivation of the disease occurs, the patient should be restarted at week 2 of the initial treatment. Satisfactory alternative treatment options are lacking, The use of drugs such as anti-TNF antibodies [96,97], anti-CD25 [98], Campath-1H [12] or even fludarabine [99] have been reported to be beneficial is selected cases, but the value of these therapies remains to be proved in larger series of patients. The therapeutic use of an anti-IFN-γ antibody resulted in recovery from HLH in the perforin-deficient and Rab27a-deficient mouse models infected with LCMV [100]. This suggests that neutralization of IFN-γ can potentially be used for symptom control in humans both for familial cases but also for secondary HLH. At present, early diagnosis, close monitoring and prompt treatment are the only ways to reduce mortality before definitive treatment is given.
The only curative treatment available to date is HSCT. The first case treated with a human leucocyte antigen (HLA)-matched HSCT was reported in 1986 [101]. The results of the HLH-94 protocol showed an overall 3-year probability of survival of 51% for the familial cases and 62% for all cases following HSCT [102]. The only independent association with improved survival is inactive disease after 2 months of HLH-94 therapy. Most deaths occur in the first year post-transplantation, and the likelihood of relapse is limited after the second year [103]. The overall survival is good when a matched donor is available, but following mismatched transplants survival is reduced [104].
Patients receiving HSCT are at risk of developing any of the usual complications of HSCT, but it seems that veno-occlusive disease and pneumonitis are the main causes of treatment-related mortality (TRM). A reduced intensity conditioning regimen (RIC) appears to be sufficient to cure the disease, even with mixed chimerism, while reducing TRM [104]. Although the risk of rejection may be higher, RIC has a favourable survival compared with conventional HSCT [105] and therefore it is the treatment of choice, at least for patients with pre-existing risk factors for HSCT.
Disease persistence or reoccurrence is a particular problem in HLH. Approximately 50% of patients develop mixed chimerism; sustained remission can be achieved with a stable chimerism of 10–20% of leucocytes [95], but long-term protection can be achieved with a donor chimerism limited to T cells [104].
Another important factor in determining outcome is disease activity at the time of transplantation. In patients who are transplanted with active disease, survival following HSCT is ∼50% [64,95,102]. It is therefore imperative to achieve optimal disease control at the time of HSCT in order to improve survival. Patients should be monitored closely for signs of reoccurrence before HSCT.
Given the cases with late-onset FHL, the possibility of the sibling carrying the disease should be considered when searching for an HSCT donor and all family members should be screened for FHL.
Conclusion
There has been a significant progress in the diagnosis and treatment since FHL was first recognized. Most of the genetic defects causing FHL have been identified. Further research to identify other as yet unknown genes resulting in the same clinical phenotype is still required. Epidemiological studies need to be performed to establish the true incidence of the disease and to characterize more clearly the genotype/phenotype correlation across the various ethnic groups.
With the establishment of consensus criteria for the diagnosis and treatment of the disease and increased awareness, the overall outcome for these patients has greatly improved. However, despite progress, disease mortality remains significantly high, making the need for improved treatment the major challenge for the future. The fact that HSCT is a curative treatment for FHL confirms that a genetic defect in haematopoietic cells is the primary cause of the disease, providing scope for gene therapy as an alternative treatment in the future.
Acknowledgments
This work was supported in part by the NIHR Biomedical Research Centres funding scheme.
Disclosure
None of the authors has any disclosures to make, HBG has the following statement to make: ‘HBG is supported by the Great Ormond Street Hospital Children's Charity’.
References
- 1.Janka G, Zur Stadt U. Familial and acquired hemophagocytic lymphohistiocytosis. Hematology (Am Soc Hematol Educ Program) 2005:82–8. doi: 10.1182/asheducation-2005.1.82. [DOI] [PubMed] [Google Scholar]
- 2.Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child. 1952;27:519–25. doi: 10.1136/adc.27.136.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arico M, Janka G, Fischer A, et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996;10:197–203. [PubMed] [Google Scholar]
- 4.Henter JI, Elinder G, Soder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand. 1991;80:428–35. doi: 10.1111/j.1651-2227.1991.tb11878.x. [DOI] [PubMed] [Google Scholar]
- 5.Ishii E, Ohga S, Tanimura M, et al. Clinical and epidemiologic studies of familial hemophagocytic lymphohistiocytosis in Japan. Japan LCH Study Group. Med Pediatr Oncol. 1998;30:276–83. doi: 10.1002/(sici)1096-911x(199805)30:5<276::aid-mpo3>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 6.Gurgey A, Gogus S, Ozyurek E, et al. Primary hemophagocytic lymphohistiocytosis in Turkish children. Pediatr Hematol Oncol. 2003;20:367–71. [PubMed] [Google Scholar]
- 7.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735–43. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 8.Menasche G, Feldmann J, Fischer A, de Saint Basile G. Primary hemophagocytic syndromes point to a direct link between lymphocyte cytotoxicity and homeostasis. Immunol Rev. 2005;203:165–79. doi: 10.1111/j.0105-2896.2005.00224.x. [DOI] [PubMed] [Google Scholar]
- 9.Badovinac VP, Tvinnereim AR, Harty JT. Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-gamma. Science (NY) 2000;290:1354–8. doi: 10.1126/science.290.5495.1354. [DOI] [PubMed] [Google Scholar]
- 10.Badovinac VP, Hamilton SE, Harty JT. Viral infection results in massive CD8+ T cell expansion and mortality in vaccinated perforin-deficient mice. Immunity. 2003;18:463–74. doi: 10.1016/s1074-7613(03)00079-7. [DOI] [PubMed] [Google Scholar]
- 11.Crozat K, Hoebe K, Ugolini S, et al. Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: a mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J Exp Med. 2007;204:853–63. doi: 10.1084/jem.20062447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2007;166:95–109. doi: 10.1007/s00431-006-0258-1. [DOI] [PubMed] [Google Scholar]
- 13.Osugi Y, Hara J, Tagawa S, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood. 1997;89:4100–3. [PubMed] [Google Scholar]
- 14.Schneider EM, Lorenz I, Muller-Rosenberger M, Steinbach G, Kron M, Janka-Schaub GE. Hemophagocytic lymphohistiocytosis is associated with deficiencies of cellular cytolysis but normal expression of transcripts relevant to killer-cell-induced apoptosis. Blood. 2002;100:2891–8. doi: 10.1182/blood-2001-12-0260. [DOI] [PubMed] [Google Scholar]
- 15.Takada H, Ohga S, Mizuno Y, et al. Oversecretion of IL-18 in haemophagocytic lymphohistiocytosis: a novel marker of disease activity. Br J Haematol. 1999;106:182–9. doi: 10.1046/j.1365-2141.1999.01504.x. [DOI] [PubMed] [Google Scholar]
- 16.Teruya-Feldstein J, Setsuda J, Yao X, et al. MIP-1alpha expression in tissues from patients with hemophagocytic syndrome. Lab Invest. 1999;79:1583–90. [PubMed] [Google Scholar]
- 17.Henter JI, Elinder G, Soder O, Hansson M, Andersson B, Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991;78:2918–22. [PubMed] [Google Scholar]
- 18.Henter JI, Carlson LA, Soder O, Nilsson-Ehle P, Elinder G. Lipoprotein alterations and plasma lipoprotein lipase reduction in familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand. 1991;80:675–81. doi: 10.1111/j.1651-2227.1991.tb11928.x. [DOI] [PubMed] [Google Scholar]
- 19.Stinchcombe JC, Griffiths GM. Secretory mechanisms in cell-mediated cytotoxicity. Annu Rev Cell Dev Biol. 2007;23:495–517. doi: 10.1146/annurev.cellbio.23.090506.123521. [DOI] [PubMed] [Google Scholar]
- 20.Ohadi M, Lalloz MR, Sham P, et al. Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3-22 by homozygosity mapping. Am J Hum Genet. 1999;64:165–71. doi: 10.1086/302187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science (NY) 1999;286:1957–9. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- 22.Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol. 2006;6:940–52. doi: 10.1038/nri1983. [DOI] [PubMed] [Google Scholar]
- 23.Risma KA, Frayer RW, Filipovich AH, Sumegi J. Aberrant maturation of mutant perforin underlies the clinical diversity of hemophagocytic lymphohistiocytosis. J Clin Invest. 2006;116:182–92. doi: 10.1172/JCI26217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Busiello R, Adriani M, Locatelli F, et al. Atypical features of familial hemophagocytic lymphohistiocytosis. Blood. 2004;103:4610–12. doi: 10.1182/blood-2003-10-3551. [DOI] [PubMed] [Google Scholar]
- 25.Clementi R, Emmi L, Maccario R, et al. Adult onset and atypical presentation of hemophagocytic lymphohistiocytosis in siblings carrying PRF1 mutations. Blood. 2002;100:2266–7. doi: 10.1182/blood-2002-04-1030. [DOI] [PubMed] [Google Scholar]
- 26.Clementi R, Locatelli F, Dupre L, et al. A proportion of patients with lymphoma may harbor mutations of the perforin gene. Blood. 2005;105:4424–8. doi: 10.1182/blood-2004-04-1477. [DOI] [PubMed] [Google Scholar]
- 27.Feldmann J, Le Deist F, Ouachee-Chardin M, et al. Functional consequences of perforin gene mutations in 22 patients with familial haemophagocytic lymphohistiocytosis. Br J Haematol. 2002;117:965–72. doi: 10.1046/j.1365-2141.2002.03534.x. [DOI] [PubMed] [Google Scholar]
- 28.Goransdotter Ericson K, Fadeel B, Nilsson-Ardnor S, et al. Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis. Am J Hum Genet. 2001;68:590–7. doi: 10.1086/318796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kogawa K, Lee SM, Villanueva J, Marmer D, Sumegi J, Filipovich AH. Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood. 2002;99:61–6. doi: 10.1182/blood.v99.1.61. [DOI] [PubMed] [Google Scholar]
- 30.Lee SM, Sumegi J, Villanueva J, et al. Patients of African ancestry with hemophagocytic lymphohistiocytosis share a common haplotype of PRF1 with a 50delT mutation. J Pediatr. 2006;149:134–7. doi: 10.1016/j.jpeds.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 31.Molleran Lee S, Villanueva J, Sumegi J, et al. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J Med Genet. 2004;41:137–44. doi: 10.1136/jmg.2003.011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suga N, Takada H, Nomura A, et al. Perforin defects of primary haemophagocytic lymphohistiocytosis in Japan. Br J Haematol. 2002;116:346–9. doi: 10.1046/j.1365-2141.2002.03266.x. [DOI] [PubMed] [Google Scholar]
- 33.Ueda I, Kurokawa Y, Koike K, et al. Late-onset cases of familial hemophagocytic lymphohistiocytosis with missense perforin gene mutations. Am J Hematol. 2007;82:427–32. doi: 10.1002/ajh.20878. [DOI] [PubMed] [Google Scholar]
- 34.Ueda I, Morimoto A, Inaba T, et al. Characteristic perforin gene mutations of haemophagocytic lymphohistiocytosis patients in Japan. Br J Haematol. 2003;121:503–10. doi: 10.1046/j.1365-2141.2003.04298.x. [DOI] [PubMed] [Google Scholar]
- 35.Zur Stadt U, Beutel K, Kolberg S, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. 2006;27:62–8. doi: 10.1002/humu.20274. [DOI] [PubMed] [Google Scholar]
- 36.Trambas C, Gallo F, Pende D, et al. A single amino acid change, A91V, leads to conformational changes that can impair processing to the active form of perforin. Blood. 2005;106:932–7. doi: 10.1182/blood-2004-09-3713. [DOI] [PubMed] [Google Scholar]
- 37.Feldmann J, Callebaut I, Raposo G, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3) Cell. 2003;115:461–73. doi: 10.1016/s0092-8674(03)00855-9. [DOI] [PubMed] [Google Scholar]
- 38.Santoro A, Cannella S, Bossi G, et al. Novel Munc13-4 mutations in children and young adult patients with haemophagocytic lymphohistiocytosis. J Med Genet. 2006;43:953–60. doi: 10.1136/jmg.2006.041863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishii E, Ueda I, Shirakawa R, et al. Genetic subtypes of familial hemophagocytic lymphohistiocytosis: correlations with clinical features and cytotoxic T lymphocyte/natural killer cell functions. Blood. 2005;105:3442–8. doi: 10.1182/blood-2004-08-3296. [DOI] [PubMed] [Google Scholar]
- 40.zur Stadt U, Schmidt S, Kasper B, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005;14:827–34. doi: 10.1093/hmg/ddi076. [DOI] [PubMed] [Google Scholar]
- 41.Arneson LN, Brickshawana A, Segovis CM, Schoon RA, Dick CJ, Leibson PJ. Cutting edge: syntaxin 11 regulates lymphocyte-mediated secretion and cytotoxicity. J Immunol. 2007;179:3397–401. doi: 10.4049/jimmunol.179.6.3397. [DOI] [PubMed] [Google Scholar]
- 42.Bryceson YT, Rudd E, Zheng C, et al. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood. 2007;110:1906–15. doi: 10.1182/blood-2007-02-074468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rudd E, Goransdotter Ericson K, Zheng C, et al. Spectrum and clinical implications of syntaxin 11 gene mutations in familial haemophagocytic lymphohistiocytosis: association with disease-free remissions and haematopoietic malignancies. J Med Genet. 2006;43:e14. doi: 10.1136/jmg.2005.035253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamamoto K, Ishii E, Horiuchi H, et al. Mutations of syntaxin 11 and SNAP23 genes as causes of familial hemophagocytic lymphohistiocytosis were not found in Japanese people. J Hum Genet. 2005;50:600–3. doi: 10.1007/s10038-005-0293-1. [DOI] [PubMed] [Google Scholar]
- 45.Cote M, Menager MM, Burgess A, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. 2009;119:3765–73. doi: 10.1172/JCI40732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.zur Stadt U, Rohr J, Seifert W, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85:482–92. doi: 10.1016/j.ajhg.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brochetta C, Vita F, Tiwari N, et al. Involvement of Munc18 isoforms in the regulation of granule exocytosis in neutrophils. Biochim Biophys Acta. 2008;1783:1781–91. doi: 10.1016/j.bbamcr.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 48.Martin-Verdeaux S, Pombo I, Iannascoli B, et al. Evidence of a role for Munc18-2 and microtubules in mast cell granule exocytosis. J Cell Sci. 2003;116:325–34. doi: 10.1242/jcs.00216. [DOI] [PubMed] [Google Scholar]
- 49.Ueda I, Ishii E, Morimoto A, Ohga S, Sako M, Imashuku S. Correlation between phenotypic heterogeneity and gene mutational characteristics in familial hemophagocytic lymphohistiocytosis (FHL) Pediatr Blood Cancer. 2006;46:482–8. doi: 10.1002/pbc.20511. [DOI] [PubMed] [Google Scholar]
- 50.Horne A, Ramme KG, Rudd E, et al. Characterization of PRF1, STX11 and UNC13D genotype–phenotype correlations in familial hemophagocytic lymphohistiocytosis. Br J Haematol. 2008;143:75–83. doi: 10.1111/j.1365-2141.2008.07315.x. [DOI] [PubMed] [Google Scholar]
- 51.Henter J. Hemophagocytic lymphohistiocytosis (HLH) – symptoms, signs and diagnosis of a rapidly fatal childhood disease. Histiocytosis Association of America. Available at: http://www.histiocytesociety.org/site/c.mqISL2PIJrH/b.4442719/k.C426/Educational_Materialnbspnbsp.htm.
- 52.Henter JI, Arico M, Elinder G, Imashuku S, Janka G. Familial hemophagocytic lymphohistiocytosis. Primary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 1998;12:417–33. doi: 10.1016/s0889-8588(05)70520-7. [DOI] [PubMed] [Google Scholar]
- 53.Allen M, De Fusco C, Legrand F, et al. Familial hemophagocytic lymphohistiocytosis: how late can the onset be? Haematologica. 2001;86:499–503. [PubMed] [Google Scholar]
- 54.Steinberg O, Yacobovich J, Dgany O, et al. Prolonged course of familial hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2006;28:831–3. doi: 10.1097/MPH.0b013e31802d3a96. [DOI] [PubMed] [Google Scholar]
- 55.Janka GE. Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr. 1983;140:221–30. doi: 10.1007/BF00443367. [DOI] [PubMed] [Google Scholar]
- 56.Henter JI, Ehrnst A, Andersson J, Elinder G. Familial hemophagocytic lymphohistiocytosis and viral infections. Acta Paediatr. 1993;82:369–72. doi: 10.1111/j.1651-2227.1993.tb12699.x. [DOI] [PubMed] [Google Scholar]
- 57.Sullivan KE, Delaat CA, Douglas SD, Filipovich AH. Defective natural killer cell function in patients with hemophagocytic lymphohistiocytosis and in first degree relatives. Pediatr Res. 1998;44:465–8. doi: 10.1203/00006450-199810000-00001. [DOI] [PubMed] [Google Scholar]
- 58.Arico M, Caselli D, Burgio GR. Familial hemophagocytic lymphohistiocytosis: clinical features. Pediatr Hematol Oncol. 1989;6:247–51. doi: 10.3109/08880018909034294. [DOI] [PubMed] [Google Scholar]
- 59.Komp DM, McNamara J, Buckley P. Elevated soluble interleukin-2 receptor in childhood hemophagocytic histiocytic syndromes. Blood. 1989;73:2128–32. [PubMed] [Google Scholar]
- 60.Gupta A, Tyrrell P, Valani R, Benseler S, Weitzman S, Abdelhaleem M. The role of the initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;51:402–4. doi: 10.1002/pbc.21564. [DOI] [PubMed] [Google Scholar]
- 61.Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18:29–33. [PubMed] [Google Scholar]
- 62.Janka GE. Familial hemophagocytic lymphohistiocytosis: diagnostic problems and differential diagnosis. Pediatr Hematol Oncol. 1989;6:219–25. doi: 10.3109/08880018909034290. [DOI] [PubMed] [Google Scholar]
- 63.Henter JI, Elinder G. Familial hemophagocytic lymphohistiocytosis. Clinical review based on the findings in seven children. Acta Paediatr Scand. 1991;80:269–77. doi: 10.1111/j.1651-2227.1991.tb11849.x. [DOI] [PubMed] [Google Scholar]
- 64.Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 65.Tabata Y, Villanueva J, Lee SM, et al. Rapid detection of intracellular SH2D1A protein in cytotoxic lymphocytes from patients with X-linked lymphoproliferative disease and their family members. Blood. 2005;105:3066–71. doi: 10.1182/blood-2004-09-3651. [DOI] [PubMed] [Google Scholar]
- 66.Betts MR, Brenchley JM, Price DA, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 67.Penack O, Gentilini C, Fischer L, et al. CD56dimCD16neg cells are responsible for natural cytotoxicity against tumor targets. Leukemia. 2005;19:835–40. doi: 10.1038/sj.leu.2403704. [DOI] [PubMed] [Google Scholar]
- 68.Marcenaro S, Gallo F, Martini S, et al. Analysis of natural killer-cell function in familial hemophagocytic lymphohistiocytosis (FHL): defective CD107a surface expression heralds Munc13-4 defect and discriminates between genetic subtypes of the disease. Blood. 2006;108:2316–23. doi: 10.1182/blood-2006-04-015693. [DOI] [PubMed] [Google Scholar]
- 69.zur Stadt U, Pruggmayer M, Jung H, et al. Prenatal diagnosis of perforin gene mutations in familial hemophagocytic lymphohistiocytosis (FHLH) Prenat Diagn. 2002;22:80–1. doi: 10.1002/pd.231. [DOI] [PubMed] [Google Scholar]
- 70.Coffey AJ, Brooksbank RA, Brandau O, et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet. 1998;20:129–35. doi: 10.1038/2424. [DOI] [PubMed] [Google Scholar]
- 71.Nagy N, Cerboni C, Mattsson K, et al. SH2D1A and SLAM protein expression in human lymphocytes and derived cell lines. Int J Cancer. 2000;88:439–47. [PubMed] [Google Scholar]
- 72.Ma CS, Nichols KE, Tangye SG. Regulation of cellular and humoral immune responses by the SLAM and SAP families of molecules. Annu Rev Immunol. 2007;25:337–79. doi: 10.1146/annurev.immunol.25.022106.141651. [DOI] [PubMed] [Google Scholar]
- 73.Dupre L, Andolfi G, Tangye SG, et al. SAP controls the cytolytic activity of CD8+ T cells against EBV-infected cells. Blood. 2005;105:4383–9. doi: 10.1182/blood-2004-08-3269. [DOI] [PubMed] [Google Scholar]
- 74.Rigaud S, Fondaneche MC, Lambert N, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444:110–14. doi: 10.1038/nature05257. [DOI] [PubMed] [Google Scholar]
- 75.Nichols KE, Ma CS, Cannons JL, Schwartzberg PL, Tangye SG. Molecular and cellular pathogenesis of X-linked lymphoproliferative disease. Immunol Rev. 2005;203:180–99. doi: 10.1111/j.0105-2896.2005.00230.x. [DOI] [PubMed] [Google Scholar]
- 76.Seemayer TA, Gross TG, Egeler RM, et al. X-linked lymphoproliferative disease: twenty-five years after the discovery. Pediatr Res. 1995;38:471–8. doi: 10.1203/00006450-199510000-00001. [DOI] [PubMed] [Google Scholar]
- 77.Arico M, Imashuku S, Clementi R, et al. Hemophagocytic lymphohistiocytosis due to germline mutations in SH2D1A, the X-linked lymphoproliferative disease gene. Blood. 2001;97:1131–3. doi: 10.1182/blood.v97.4.1131. [DOI] [PubMed] [Google Scholar]
- 78.Pastural E, Barrat FJ, Dufourcq-Lagelouse R, et al. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat Genet. 1997;16:289–92. doi: 10.1038/ng0797-289. [DOI] [PubMed] [Google Scholar]
- 79.Menasche G, Pastural E, Feldmann J, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25:173–6. doi: 10.1038/76024. [DOI] [PubMed] [Google Scholar]
- 80.Menasche G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1) J Clin Invest. 2003;112:450–6. doi: 10.1172/JCI18264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stinchcombe JC, Barral DC, Mules EH, et al. Rab27a is required for regulated secretion in cytotoxic T lymphocytes. J Cell Biol. 2001;152:825–34. doi: 10.1083/jcb.152.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Neeft M, Wieffer M, de Jong AS, et al. Munc13-4 is an effector of rab27a and controls secretion of lysosomes in hematopoietic cells. Mol Biol Cell. 2005;16:731–41. doi: 10.1091/mbc.E04-10-0923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nagle DL, Karim MA, Woolf EA, et al. Identification and mutation analysis of the complete gene for Chediak–Higashi syndrome. Nat Genet. 1996;14:307–11. doi: 10.1038/ng1196-307. [DOI] [PubMed] [Google Scholar]
- 84.Huizing M, Helip-Wooley A, Westbroek W, Gunay-Aygun M, Gahl WA. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet. 2008;9:359–86. doi: 10.1146/annurev.genom.9.081307.164303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wei ML. Hermansky–Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 2006;19:19–42. doi: 10.1111/j.1600-0749.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 86.Enders A, Zieger B, Schwarz K, et al. Lethal hemophagocytic lymphohistiocytosis in Hermansky–Pudlak syndrome type II. Blood. 2006;108:81–7. doi: 10.1182/blood-2005-11-4413. [DOI] [PubMed] [Google Scholar]
- 87.Arico M, Bettinelli A, Maccario R, Clementi R, Bossi G, Danesino C. Hemophagocytic lymphohistiocytosis in a patient with deletion of 22q11.2. Am J Med Genet. 1999;87:329–30. [PubMed] [Google Scholar]
- 88.Hisano M, Sugawara K, Tatsuzawa O, Kitagawa M, Murashima A, Yamaguchi K. Bacteria-associated haemophagocytic syndrome and septic pulmonary embolism caused by Burkholderia cepacia complex in a woman with chronic granulomatous disease. J Med Microbiol. 2007;56:702–5. doi: 10.1099/jmm.0.47071-0. [DOI] [PubMed] [Google Scholar]
- 89.Tapisiz A, Belet N, Ciftci E, Ince E, Dogru U. Hemophagocytic lymphohistiocytosis associated with visceral leishmaniasis. J Trop Pediatr. 2007;53:359–61. doi: 10.1093/tropej/fmm024. [DOI] [PubMed] [Google Scholar]
- 90.Gagnaire MH, Galambrun C, Stephan JL. Hemophagocytic syndrome: a misleading complication of visceral leishmaniasis in children – a series of 12 cases. Pediatrics. 2000;106:E58. doi: 10.1542/peds.106.4.e58. [DOI] [PubMed] [Google Scholar]
- 91.Rajagopala S, Dutta U, Chandra KS, Bhatia P, Varma N, Kochhar R. Visceral leishmaniasis associated hemophagocytic lymphohistiocytosis – case report and systematic review. J Infect. 2008;56:381–8. doi: 10.1016/j.jinf.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 92.Ravelli A, Magni-Manzoni S, Pistorio A, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr. 2005;146:598–604. doi: 10.1016/j.jpeds.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 93.Santoro A, Cannella S, Trizzino A, Lo Nigro L, Corsello G, Arico M. A single amino acid change A91V in perforin: a novel, frequent predisposing factor to childhood acute lymphoblastic leukemia? Haematologica. 2005;90:697–8. [PubMed] [Google Scholar]
- 94.Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, Trapani JA. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med. 2000;192:755–60. doi: 10.1084/jem.192.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ouachee-Chardin M, Elie C, de Saint Basile G, et al. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics. 2006;117:e743–50. doi: 10.1542/peds.2005-1789. [DOI] [PubMed] [Google Scholar]
- 96.Henzan T, Nagafuji K, Tsukamoto H, et al. Success with infliximab in treating refractory hemophagocytic lymphohistiocytosis. Am J Hematol. 2006;81:59–61. doi: 10.1002/ajh.20462. [DOI] [PubMed] [Google Scholar]
- 97.Prahalad S, Bove KE, Dickens D, Lovell DJ, Grom AA. Etanercept in the treatment of macrophage activation syndrome. J Rheumatol. 2001;28:2120–4. [PubMed] [Google Scholar]
- 98.Tomaske M, Amon O, Bosk A, Handgretinger R, Schneider EM, Niethammer D. Alpha-CD25 antibody treatment in a child with hemophagocytic lymphohistiocytosis. Med Pediatr Oncol. 2002;38:141–2. doi: 10.1002/mpo.1294. [DOI] [PubMed] [Google Scholar]
- 99.Schneider P, Greene V, Kanold J, Vannier JP. Fludarabine in the treatment of an active phase of a familial haemophagocytic lymphohistiocytosis. Arch Dis Child. 2001;84:373. doi: 10.1136/adc.84.4.373a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pachlopnik Schmid J, Ho CH, Chretien F, et al. Neutralization of IFNgamma defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO Mol Med. 2009;1:112–24. doi: 10.1002/emmm.200900009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fischer A, Cerf-Bensussan N, Blanche S, et al. Allogeneic bone marrow transplantation for erythrophagocytic lymphohistiocytosis. J Pediatr. 1986;108:267–70. doi: 10.1016/s0022-3476(86)81002-2. [DOI] [PubMed] [Google Scholar]
- 102.Henter JI, Samuelsson-Horne A, Arico M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367–73. doi: 10.1182/blood-2002-01-0172. [DOI] [PubMed] [Google Scholar]
- 103.Horne A, Janka G, Maarten Egeler R, et al. Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol. 2005;129:622–30. doi: 10.1111/j.1365-2141.2005.05501.x. [DOI] [PubMed] [Google Scholar]
- 104.Cooper N, Rao K, Gilmour K, et al. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lymphohistiocytosis. Blood. 2006;107:1233–6. doi: 10.1182/blood-2005-05-1819. [DOI] [PubMed] [Google Scholar]
- 105.Cooper N, Rao K, Goulden N, Webb D, Amrolia P, Veys P. The use of reduced-intensity stem cell transplantation in haemophagocytic lymphohistiocytosis and Langerhans cell histiocytosis. Bone Marrow Transplant. 2008;42(Suppl 2):S47–50. doi: 10.1038/bmt.2008.283. [DOI] [PubMed] [Google Scholar]