

Abstract
Graves' disease is a B cell-mediated and T cell-dependent autoimmune disease of the thyroid which is characterized by overproduction of thyroid hormones and thyroid enlargement by agonistic anti-thyrotrophin receptor (TSHR) autoantibody. In addition to antibody secretion, B cells have recently been recognized to function as antigen-presenting/immune-modulatory cells. The present study was designed to evaluate the efficacy of B cell depletion by anti-mouse (m) CD20 monoclonal antibody (mAb) on Graves' hyperthyroidism in a mouse model involving repeated injection of adenovirus expressing TSHR A-subunit (Ad-TSHR289). We observe that a single injection of 250 µg/mouse anti-mCD20 mAb eliminated B cells efficiently from the periphery and spleen and to a lesser extent from the peritoneum for more than 3 weeks. B cell depletion before immunization suppressed an increase in serum immunoglobulin (Ig)G levels, TSHR-specific splenocyte secretion of interferon (IFN)-γ, anti-TSHR antibody production and development of hyperthyroidism. B cell depletion 2 weeks after the first immunization, a time-point at which T cells were primed but antibody production was not observed, was still effective at inhibiting antibody production and disease development without inhibiting splenocyte secretion of IFN-γ. By contrast, B cell depletion in hyperthyroid mice was therapeutically ineffective. Together, these data demonstrate that B cells are critical not only as antibody-producing cells but also as antigen-presenting/immune-modulatory cells in the early phase of the induction of experimental Graves' hyperthyroidism and, although therapeutically less effective, B cell depletion is highly efficient for preventing disease development.
Keywords: autoimmunity, B cells, Graves' disease
Introduction
Organ-specific autoimmune diseases result from abnormal B and T cell recognition of self-autoantigen. Some of these diseases are mediated largely by humoral immune responses producing pathogenic autoantibodies, and others by cellular immune responses leading to destruction of target tissues by cytotoxic T cells. Graves' disease is representative of the former, characterized by stimulatory autoantibodies against the thyrotrophin receptor [thyroid stimulating hormone receptor (TSHR)] (thyroid stimulating antibody, TSAb), which cause overproduction of thyroid hormones and thyroid hyperplasia [1]. As antibody producing cells, B cells are crucial immune cells in the pathogenesis of Graves' disease. In addition, other important aspects of B cell function in immune reactions have been clarified recently, including antigen presentation, proinflammatory cytokine production, co-stimulatory molecule expression (CD80 and CD86), alterations in dendritic cell function, etc. [2]. Indeed, previous studies with mice genetically deficient for B cells [B cell knock-out (KO) mice] showed the requirement of B cells for development of autoimmune thyroiditis, type 1 diabetes and systemic lupus erythematosus (SLE) [3–5]. Impaired activation of TSHR-reactive T cells in B cell KO mice in a mouse Graves' model has also been demonstrated [6]. These data indicate the critical role of B cells not only for autoantibody production, but also for CD4+ T cell priming as professional antigen-presenting cells. B cells are therefore an ideal therapeutic target in terms of not only lowering activities of pathogenic antibodies, but also dampening pathogenic autoimmune responses per se in autoimmune diseases.
However, B cell KO mice have a serious problem, in that these mice have major qualitative and quantitative abnormalities in the immune system [7,8]. By contrast, B cell depletion may be a feasible approach to study the function of B cells in autoimmune diseases. Indeed monoclonal antibodies to B cell-specific cell surface molecules such as CD19, CD20, CD79 and to a B cell-surviving factor (B cell lymphocyte stimulator, BLyS) have been used successfully to deplete B cells in vivo and to treat numerous autoimmune and malignant haematopoietic diseases in humans and mice [2,9,10]. Transient depletion of B cells by these means can distinguish between the role of B cells during immune development and during immune responses.
CD20 is a B cell-specific molecule that is expressed on the cell surface during the transition of pre-B to immature B cells but is lost upon plasma cell differentiation [11]. In human autoimmune diseases, rituximab, a chimeric anti-human CD20 monoclonal antibody, has proved to be effective for treatment of autoimmune diseases, including rheumatoid arthritis, SLE, idiopathic thrombocytopenic purpura, haemolytic anaemia and pemphigus vulgaris [12]. In addition, preliminary clinical studies have shown the therapeutic efficacy of rituximab in a small fraction of Graves' patients with mild hyperthyroidism [13–16]. In mice, anti-mouse CD20 monoclonal antibodies (anti-mCD20 mAbs) which efficiently eliminate mouse B cells in vivo have been isolated recently [11,17], and used to treat mouse models of autoimmune thyroiditis, systemic sclerosis, collagen- or proteoglycan-induced arthritis, Sjögren's syndrome, SLE and type 1 diabetes [17–22]. Moreover, the soluble decoy receptor-Fc fusion proteins to block B cell surviving factors [BLyS and a proliferation-inducing ligand (APRIL)] reduced TSAb activities and thyroxine (T4) levels in a mouse model of Graves' disease [23].
In the present study, we evaluated the efficacy of anti-mCD20 mAb in a mouse model of Graves' disease we have established previously [23]. We found that this approach depleted B cells efficiently and that B cell depletion by this agent was effective for preventing Graves' hyperthyroidism. Our results indicate the requirement of antibody production and T cell activation by B cells in the early phase of disease initiation for the disease pathogenesis.
Materials and methods
Mice
Female BALB/c mice (6 weeks old) were purchased from Charles River Japan Laboratory Inc. (Tokyo, Japan) and were kept in a specific pathogen-free facility. Animal care and all experimental procedures were performed in accordance with the Guideline for Animal Experimentation of Nagasaki University with the approval of the Institutional Animal Care and Use Committee.
Experimental protocols
Construction, amplification, purification of non-replicative recombinant human adenovirus expressing the human TSHR-A subunit [adenovirus expressing (TSHR) A-subunit (Ad-TSHR289)] and determination of the viral particle concentration have been described previously [23].
Mice were injected intramuscularly in the quadriceps with 100 µl phosphate-buffered saline (PBS) containing 1010 particles of Ad-TSHR289 on three occasions at 3-week intervals (weeks 0, 3 and 6). Groups of mice were also treated by intraperitoneal (i.p.) injection of anti-mCD20 mAb (50 or 250 µg/mouse, single injection; 18B12, IgG2a) or control antibody (2B8, IgG2a) (gifts from R. Dunn and M. Kehry at Biogen Idec [17,18]) at the indicated time-points. Blood samples were obtained 2 weeks after the second immunization or 4 weeks after the third immunization.
T4 and anti-TSHR antibody measurements
Serum free T4 concentrations were measured with a radioimmunoassay (RIA) kit (DPC free T4 kit; Diagnostic Products, Los Angeles, CA, USA). The normal range was defined as the mean ± 3 standard deviations (s.d.) of control untreated mice.
Anti-TSHR antibodies in mouse sera were determined using two different methods, a biological TSAb assay and a flow cytometric assay with Chinese hamster ovary (CHO) cells stably expressing the full-length human TSHR, as described previously [24]. The former measures the stimulating antibodies responsible for hyperthyroidism, and the latter the titres of anti-TSHR antibodies recognizing the native TSHR expressed on the cell surface irrespective of their function.
Enzyme-linked immunosorbent assay (ELISA) for measuring serum IgG concentrations
ELISA wells were coated overnight with 100 µl goat anti-mouse Ig (diluted 1:1000; Southern Biotech, Birmingham, AL, USA) and were then incubated with mouse sera (diluted 1:2000). After incubation with horseradish peroxidase-conjugated anti-mouse IgG (diluted 1:3000; A3673; Sigma-Aldrich Corporation, St Louis, MO, USA), colour was developed using orthophenylene diamine and H2O2 as substrate, and optimal density (OD) was read at 492 nm.
Flow cytometry
Splenocytes were stained with fluorescein isothiocyanate (FITC) or phycoerythrin (PE)-conjugated anti-CD4 (H129·19), anti-CD44 (IM7), anti-CD62L (MEl-14), anti-B220 (RA3-6B2), anti-IgM (II/41) and anti-forkhead box P3 (FoxP3) (FJK-16s; FoxP3 staining kit) (PharMingen, San Diego, CA, USA or eBioscience, San Diego, CA, USA), and analysed on a FACSCanto II flow cytometry using fluorescence activated cell sorter (FACS) Diva software (BD Biosciences, San Diego, CA, USA).
Cytokine assays
Splenocytes were cultured (triplicate aliquots) at 5 × 105 cells/well in a 96-well round-bottomed culture plate in the presence or absence of 10 µg/ml TSHR289 protein, as described previously [25]. Four days later, the culture supernatants were collected. The concentrations of interferon (IFN)-γ were determined with Bio-Plex™ Suspension Array System (Bio-Rad, Tokyo, Japan). Cytokine production was expressed as pg/ml using a standard curve of recombinant mouse cytokines.
Statistical analysis
Levels of T4, antibodies and cytokines and incidences of hyperthyroidism were analysed by t-test or χ2 test, respectively. A P value of less than 0·05 was considered statistically significant.
Results
B cell depletion by anti-mCD20 mAb
To determine the efficacy of anti-mCD20 mAb for B cell depletion, BALB/c mice were treated with a single i.p. injection of 50 or 250 µg/mouse of either anti-mCD20 mAb or control mAb. Representative flow cytometric data on peripheral blood of naive, anti-mCD20 mAb-treated and control mAb-treated mice are shown in Fig. 1a. Anti-mCD20 mAb reduced B220+IgM+ B cell numbers in a dose-dependent manner, with 250 µg/mouse mAb resulting in the depletion of B cells to less than 5% of the baseline in the peripheral blood and spleen (Fig. 1b). The mAb was the least effective in the peritoneal cavity (Fig. 1b). This is thought to be due to inaccessibility of Fc receptor-bearing cells into the peritoneal cavity that mediate antibody-dependent cellular cytotoxicity [11,25]. The effect persisted for at least 3 weeks, with an approximately 80% recovery in 6 weeks (Fig. 1C). These data are essentially identical to those in the previous report that has studied the effect of anti-mCD20 mAb on different B cell subsets in BALB/c mice [22]. Despite effective B cell depletion in the peripheral blood and spleen, serum basal IgG levels remained unchanged (see below).
Fig. 1.
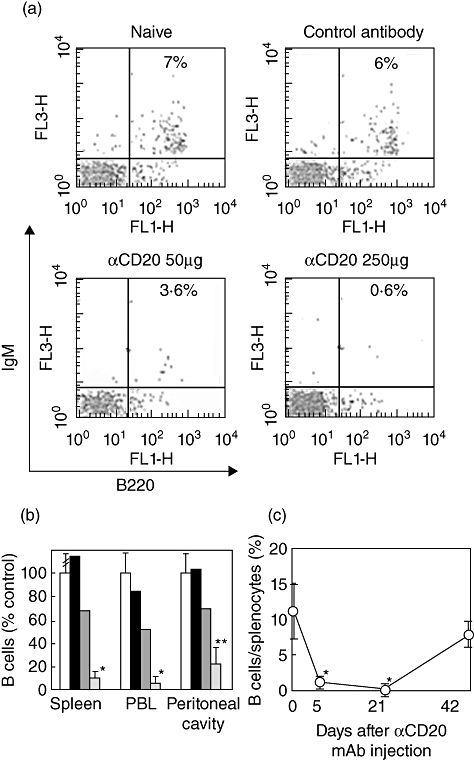
Flow cytometric analysis of B220+immunoglobulin (Ig)M+ B cells in mice. Mice were treated intraperitoneally with phosphate-buffered saline (PBS), 50 or 250 µg anti-mCD20 monoclonal antibody (mAb) or 250 µg control mAb, and B220 and IgM expression on splenocytes, the peripheral blood and the peritoneal cells was analysed at indicated time-points as described in the Materials and methods. (a) Representative flow cytometric data on peripheral blood from mice injected 5 days previously. (b) Dose-dependent effects of anti-mouse (m)CD20 mAb on B220+IgM+ B cell numbers in spleen, the peripheral blood (PB) and the peritoneal cavity 5 days after injection of anti-mCD20 mAb. Data are mean ± standard deviation (s.d.) (n = 4–6) or means of two mice. (c) Time–course of the effect of anti-mCD20 mAb on B cell numbers in spleen. Data are mean ± s.d. (n = 6–10). *P < 0·01 and **P < 0·05, compared to controls.
Regarding T cell subsets, the percentages of CD4+CD44-CD62L+ naive, CD4+CD44+CD62L+ activated, CD4+CD44+CD62L- memory and CD4+FoxP3+ regulatory T cells remained unaltered 2 weeks after anti-mCD20 mAb injection (data not shown).
Outcome of anti-mCD20 mAb treatment for Graves' hyperthyroidism and TSHR antibodies
The consequences of B cell depletion on Graves' hyperthyroidism were studied in a mouse model involving repeated injection of susceptible BALB/c mice with Ad-TSHR289 [23]. Antibody treatment (250 µg/mouse) was performed at three different time-points (experiments 1, 2 and 3 in Fig. 2) and sera were analysed at two time-points, 2 weeks after the second immunization (week 5) and 4 weeks after the third immunization (week 10).
Fig. 2.
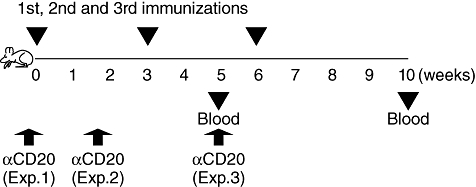
Experimental designs for B cell-depletion study. Mice were immunized thrice (weeks 0, 3 and 6) with adenovirus expressing thyroid stimulating hormone receptor (TSHR) A-subunit (Ad-TSHR289) as described in the Materials and methods. Anti-mouse (m)CD20 monoclonal antibody (mAb) was given 5 days before the first immunization (experiment 1), 10 days after the first immunization (experiment 2) or 2 weeks and 2 days after the second immunization (a time when development of Graves' hyperthyroidism was confirmed). Blood was taken 5 and 10 weeks after the first immunization.
In mice that received anti-mCD20 mAb 5 days before the first immunization (experiment 1 in Fig. 2), development of hyperthyroidism was suppressed completely at week 5 and reduced markedly at week 10 (Fig. 3a). Similarly, the titres of anti-TSHR antibodies were also inhibited almost completely at week 5 but began to increase at week 10 (Fig. 3b), presumably because of recovery of B cell numbers (see Fig. 1c). However, pathogenic TSAb activities were still low in the anti-mCD20 mAb-treated mice at this time-point (Fig. 3c), consistent with the lower incidence of hyperthyroidism (Fig. 3a). Thus, the ability of B cell depletion to suppress development of TSAb and Graves' hyperthyroidism is relatively long-lasting, even after circulating B cells recovered in the periphery. Thus, B cell depletion by anti-mCD20 mAb is extremely effective at preventing the development of Graves' hyperthyroidism.
Fig. 3.
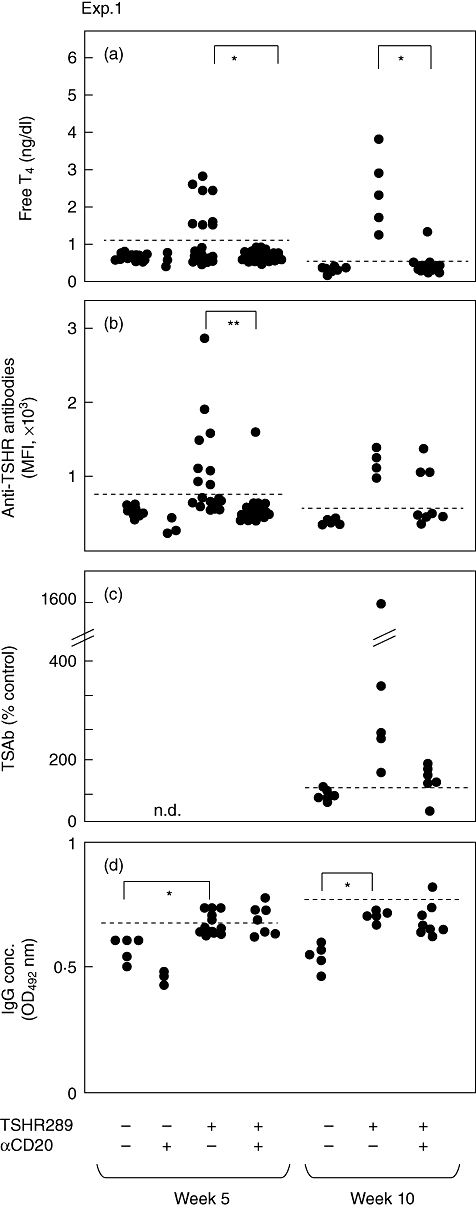
Thyroxine (T4)concentrations, anti-thyroid stimulating hormone receptor (TSHR) antibody titres and immunoglobulin (Ig)G levels in mice in experiment 1. Free T4 were determined by radioimmunoassay (RIA), anti-TSHR antibodies by flow cytometry and bioassay and serum IgG concentrations by enzyme-linked immunosorbent assay (ELISA) in mice from experiment 1. Data are shown for individual mice. The horizontal broken lines designate the normal upper limits of free T4, anti-TSHR antibodies and IgG. *P < 0·01; **P < 0·05.
We next tested the outcome of injecting anti-mCD20 mAb 10 days after the first immunization (experiment 2 in Fig. 2), a time-point at which we found previously that T cells were already primed but anti-TSHR antibodies or hyperthyroidism were not induced [26]. Albeit slightly less effective than pretreatment (Fig. 3), only 33% of immunized, anti-mCD20 mAb-treated mice became hyperthyroid compared with 73% in immunized, untreated mice (Fig. 4a). Again, the levels of anti-TSHR antibodies were significantly lower in mice that received anti-mCD20 mAb (Fig. 4b).
Fig. 4.
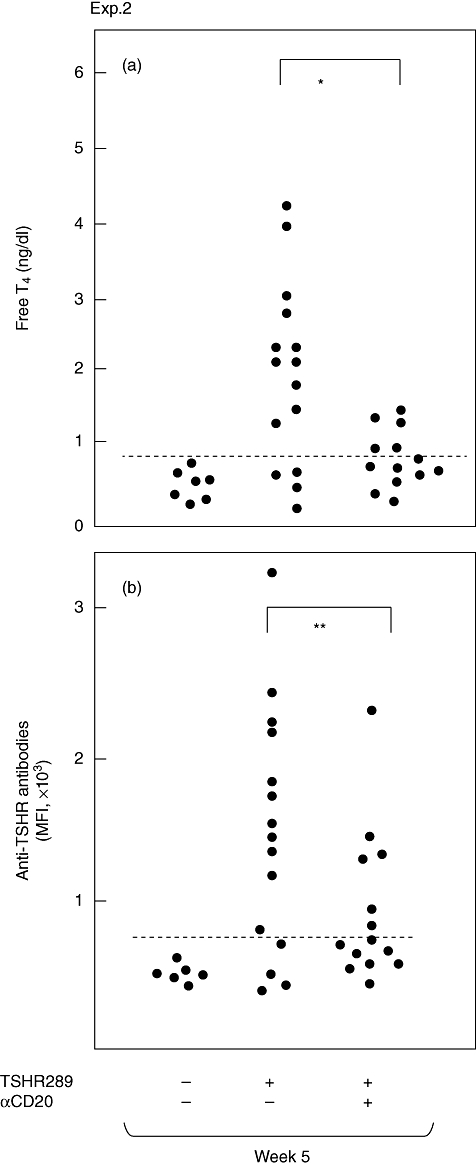
Thyroxine (T4) concentrations and anti-thyroid stimulating hormone receptor (TSHR) antibody titres in mice in experiment 2. Free T4 and anti-TSHR antibodies were determined as in Fig. 3. Data are shown for individual mice. The horizontal broken lines designate the normal upper limits of free T4 and anti-TSHR antibodies. *P < 0·01; **P < 0·05.
In the third approach, anti-mCD20 mAb was administered to hyperthyroid mice (experiment 3 in Fig. 2). This treatment proved to be ineffective. Thus, the incidences of hyperthyroidism were decreased from 90% in the immunized, untreated mice to 54% in the immunized, anti-mCD20 mAb-treated mice (Fig. 5a), which were statistically insignificantly different. Moreover, the differences in levels of anti-TSHR antibodies and TSAb activities were also insignificant between two groups (Fig. 3b,c).
Fig. 5.
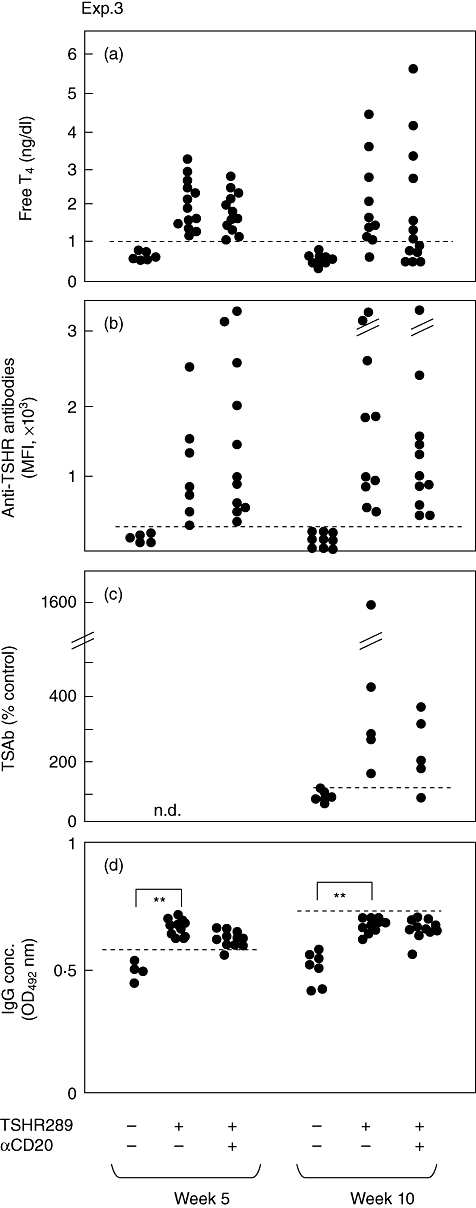
Thyroxine (T4) concentrations, anti-thyroid stimulating hormone receptor (TSHR) antibody titres and immunoglobulin (Ig)G levels in mice in experiment 3. Hyperthyroid mice selected at the fifth week were divided into two groups of untreated and αCD20-treated mice. The horizontal broken lines designate the normal upper limits of free T4, anti-TSHR antibodies and IgG. **P < 0·05.
Of interest, immunization with Ad-TSHR289 increased serum concentrations of IgG significantly (Figs 3d and 5d). However, anti-mCD20 mAb had no effect on the basal IgG levels (Fig. 3d).
Effect of B cell depletion on memory T cell responses
TSHR antigen-specific splenocyte secretion of IFN-γin vitro was used as a measure of T cell activation because we have found previously that this cytokine is indispensable for the pathogenesis of Graves' disease [27]. In the first experiment, splenocytes were prepared 2 weeks after a single injection of AdTSHR289 from mice which received anti-mCD20 mAb 5 days before immunization (experiment 1 in Fig. 2). Controls were splenocytes from immunized but not B cell-depleted mice, as well as splenocytes from unimmunized mice. In a T cell recall assay, splenocytes from Ad-TSHR289 immunized mice, but not from immunized and B cell-depleted mice, produced significantly increased amounts of IFN-γ in response to TSHR antigen (Fig. 6a). Thus, anti-mCD20 mAb suppressed antigen-specific IFN-γ synthesis by ∼50%. In the second experiment, T cell recall responses were studied in mice which received anti-mCD20 mAb 10 days after immunization with Ad-TSHR289 (experiment 2 in Fig. 3). Splenocytes were prepared 2 weeks after immunization from these B cell-depleted mice and from immunized but not B cell-depleted mice, as well as from unimmunized mice. In this case, splenocytes from both the immunized mice and the immunized and B cell-depleted mice produced comparably increased amounts of IFN-γ in response to TSHR antigen (Fig. 6b).
Fig. 6.
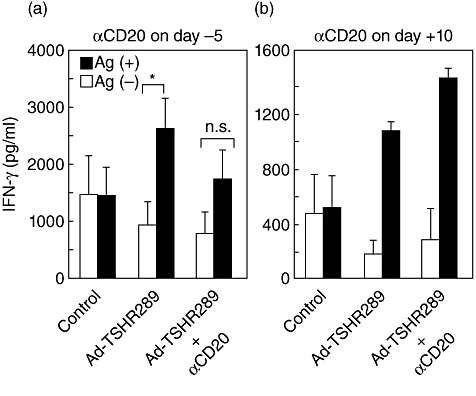
Antigen-specific splenocyte secretion of interferon (IFN)-γ in a T cell recall assay. (a) Mice injected with anti-mCD20 monoclonal antibody (mAb) 5 days previously were immunized with adenovirus expressing thyroid stimulating hormone receptor (TSHR) A-subunit (Ad-TSHR289). Two weeks later, splenocytes were prepared and subjected to a T cell recall assay as described in the Materials and methods. Data are means ± standard deviation (n = 3–5). *P < 0·05; n.s.: not significant. (b) Mice were injected with Ad-TSHR289 and 10 days later with anti-mouse (m)CD20 mAb. Four days later splenocytes were prepared. Data are means ± range (n = 2).
Overall, our findings indicate that B cells are important for disease initiation by stimulating T cell function and antibody production. However, B cell depletion prevents disease induction but is not efficacious once disease is manifested clinically.
Discussion
This study was designed to evaluate the prophylactic and therapeutic potentials of B cell depletion on Graves' hyperthyroidism in a mouse model. Although clinical trials of B cell depletion by rituximab have been performed previously in a small number of Graves' patients [13–16], we believed that studies on animal models would have an important role because of the substantial limitations of performing mechanistic studies on B cell contribution to Graves' disease in humans.
We first observed that anti-mCD20 mAb (18B12) efficiently depleted B cells in the periphery and spleen and to a lesser extent in the peritoneal cavity for a long time-period, in agreement with previous findings [17]. Baseline serum IgG levels were unaffected, presumably because the majority of antibodies are produced from CD20- plasma cells [11]. However, the outcomes of anti-CD20 mAb-mediated B cell depletion on T cell subsets in the previous studies are controversial. Thus, a slight increase in the percentages of naive CD4+ and CD8+ T cells (CD44lowCD62Lhigh) and a decrease in memory T cells (CD4+CD44highCD62Llow) were reported in one study [17] but not in another study [8]. Furthermore, expansion of regulatory T cells (Treg) was demonstrated recently in some studies [28,29] but not another study [30] in non-obese diabetic (NOD) mice. In this study we found no change in naive/activated/memory T cell subsets and also in Treg subsets.
In the Graves' mouse model we then showed the excellent prophylactic effect of anti-mCD20 mAb for blocking induction of anti-TSHR antibodies and preventing hyperthyroidism. This outcome could be expected because anti-mCD20 mAb eliminated antibody-producing B cells almost completely before immunization. However, B cell depletion before immunization also suppressed antigen-specific T cell activation significantly in a T cell recall assay. Previously, suppression of in vitro T cell proliferation and/or proinflammatory cytokine [IFN-γ and interleukin (IL)-17] secretion was reported [22,30], as well as in vivo proliferation of autoreactive T cells in response to endogenous autoantigens by B cell depletion [8]. Thus, elimination of both antigen-presentation and antibody production by B cells is possibly involved in this highly efficient prophylactic effect.
The effect of B cell depletion by anti-mCD20 mAb persisted even after the recovery of B cell numbers, as reported previously in diabetes [30]. B cell depletion may be able to ‘reset’ the immune system by breaking the self-perpetuating vicious cycle of autoreactive B cell generation and T cell activation. However, in other cases, continuous B cell depletion was necessary [19]. It is therefore critical to clarify the reason(s) of these differences for optimizing treatment strategies.
B cell depletion after the first immunization, when T cells were primed but anti-TSHR antibody production was not observed, was also effective at reducing hyperthyroidism, albeit to a lesser extent than when given before the first immunization. Because B cell depletion after immunization had no effect on already activated T cell function, the suppressive function of anti-mCD20 mAb at this time-point is probably attributed to elimination of activated, autoreactive B cells. Although Bouraziz et al. [8] have demonstrated elegantly that the presence of both dendritic cells and B cells are necessary for full CD4+ T cell activation, Yan et al. [31] have reported that B cells are the first subset of antigen-presenting cells for activating autoreactive T cells. Thus, it is likely that requirement of antigen-presenting function of B cells is limited at the early step of autoantigen presentation in induction of Graves' hyperthyroidism. By contrast, therapeutic effect was not observed when mAb was given to hyperthyroid mice. In this case, autoreactive B cells might already have differentiated into CD20- plasma cells, and/or the antigen-presenting ability of B cells may be no longer necessary once disease is manifested.
Preventive but not therapeutic effects of B cell depletion were reported in mouse models of systemic sclerosis, collagen-induced arthritis and Sjögren's syndrome [19–21]. The efficacy of B cell depletion on ongoing immune responses/inflammation was also reported when mAb were given prior to the onset of clinically manifested diseases in spontaneous mouse models of SLE and type 1 diabetes [17,30] and a proteoglycan-induced arthritis model [22]. Thus, in these autoimmune diseases, as in Graves' disease, B cells play a role in the early stages of autoimmunity during autoreactive T cell activation/expansion and autoantibody production. By contrast, therapeutic efficacy was observed in experimental autoimmune thyroiditis [18], suggesting the necessity of B cells to maintain the disease activity. These different outcomes may arise because of differential requirements for B cells in initiating disease versus maintaining disease in different disease models.
In contrast to a lack of therapeutic effect in the majority of mouse studies, some degree of therapeutic effect of rituximab was observed in human autoimmune diseases [2]. Thus, in human trials, rituximab therapy reduced levels of IgG autoantibodies to citrullinated protein, cytoplasmic neutrophil antigen, C1q and TSHR (TSAb), despite the lack of change in IgG levels [32–38]. It should be appreciated that most of the human studies that showed reduction in pathogenic antibodies and significant changes in some T cell subsets involved combination therapy of both rituximab and immunosuppressive drugs. However, autoantibody reduction does not always correlate with clinical efficacy [39,40], suggesting that the loss of other B cell functions contributes to suppression of autoimmune diseases. One reason for these differences between human and mouse studies may be that B cells augment T cell activation in response to continuous autoantigen challenge, and antibody-producing B cells/plasma cells are generated continuously in human diseases. For these reasons, it may be anticipated that B cell depletion therapy is more effective in humans than in mouse models.
In terms of antibody production, a drawback of anti-mCD20 mAb is its inability to deplete plasma cells, which do not express CD20. We considered that this problem could be overcome by the eventual demise of plasma cells, alone or in combination with B cell depletion. However, plasma cells have very long half-lives, measured in months or even years [11].
Finally, in this study we show that anti-mCD20 mAb depletes B cells efficiently and that, although therapeutically less effective, B cell depletion by this agent is highly efficient for preventing development of experimental Graves' hyperthyroidism. Our results indicate that B cells are critical not only as antibody-producing cells but also as antigen presenting/immune-modulatory cells in the early phase of the disease pathogenesis. Further studies are necessary to find efficient means to suppress the pathogenic autoantibody production therapeutically as novel therapeutic modalities for Graves' disease and also other autoantibody mediated autoimmune diseases.
Acknowledgments
We thank Drs R. Dunn and M. Kehry at Biogen Idec, San Diego, CA, for kind gifts of monoclonal anti-mCD20 (18B12) or control (2B8) antibodies, and Professors Sandra M. McLachlan and Basil Rapoport, at Autoimmune Disease Unit, Cedars-Sinai Medical Center and University of California Los Angeles, CA, for critical reading of the manuscript.
Disclosure
The authors have nothing to disclose.
References
- 1.Rapoport B, Chazenbalk GD, Jaume JC, McLachlan SM. The thyrotropin (TSH) receptor: interaction with TSH and autoantibodies. Endocr Rev. 1998;19:673–716. doi: 10.1210/edrv.19.6.0352. [DOI] [PubMed] [Google Scholar]
- 2.Yanaba K, Bouaziz JD, Matsushita T, Magro CM, William St Clair E, Tedder TF. B-lymphocyte contributions to human autoimmune disease. Immunol Rev. 2008;223:284–99. doi: 10.1111/j.1600-065X.2008.00646.x. [DOI] [PubMed] [Google Scholar]
- 3.Braley-Mullen H, Yu S. Early requirement for B cells for development of spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J Immunol. 2000;165:7262–69. doi: 10.4049/jimmunol.165.12.7262. [DOI] [PubMed] [Google Scholar]
- 4.Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. J Exp Med. 1994;180:1295–306. doi: 10.1084/jem.180.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serreze DV, Chapman HD, Varnum DS, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new ‘speed congenic’ stock of NOD.Ig mu null mice. J Exp Med. 1996;184:2049–53. doi: 10.1084/jem.184.5.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pichurin P, Aliesky H, Chen C-R, Nagayama Y, Rapoport B, McLachlan SM. Thyrotropin receptor-specific memory T cell responses require normal B cells in a murine model of Graves' disease. Clin Exp Immunol. 2003;134:396–402. doi: 10.1111/j.1365-2249.2003.02322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiu PP, Serreze DV, Danska JS. Development and function of diabetogenic T-cells in B-cell-deficient nonobese diabetic mice. Diabetes. 2001;50:763–70. doi: 10.2337/diabetes.50.4.763. [DOI] [PubMed] [Google Scholar]
- 8.Bouaziz J-D, Yanaba K, Venturi GM, et al. Therapeutic B cell depletion impairs adaptive and autoreactive CD4+ T cell activation in mice. Proc Natl Acad Sci USA. 2007;104:20878–83. doi: 10.1073/pnas.0709205105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Chen F, Putt M, et al. B cell depletion with Anti-CD79 mAbs ameliorates autoimmune disease in MRL/lpr mice. J Immunol. 2008;181:2961–72. doi: 10.4049/jimmunol.181.5.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zekavat G, Rostami SY, Badkerhanian A, et al. In vivo BLyS/BAFF neutralization ameliorates islet-directed autoimmunity in nonobese diabetic mice. J Immunol. 2008;181:8133–44. doi: 10.4049/jimmunol.181.11.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uchida J, Hamaguchi Y, Oliver JA, et al. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004;199:1659–69. doi: 10.1084/jem.20040119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanz I, Anolik JH, Looney RJ. B cell depletion therapy in autoimmune diseases. Front Biosci. 2007;12:2546–67. doi: 10.2741/2254. [DOI] [PubMed] [Google Scholar]
- 13.El Fassi D, Nielsen CH, Bonnema SJ, Hasselbalch HC, Hegedus L. B lymphocyte depletion with the monoclonal antibody rituximab in Graves' disease: a controlled pilot study. J Clin Endocrinol Metab. 2007;92:1769–72. doi: 10.1210/jc.2006-2388. [DOI] [PubMed] [Google Scholar]
- 14.Salvi M, Vannucchi G, Campi I, et al. Rituximab treatment in a patient with severe thyroid-associated ophthalmopathy: effects on orbital lymphocytic infiltrates. Clin Immunol. 2009;131:360–5. doi: 10.1016/j.clim.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 15.Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves' disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33–40. doi: 10.1530/eje.1.02325. [DOI] [PubMed] [Google Scholar]
- 16.Heemstra KA, Toes RE, Sepers J, et al. Rituximab in relapsing Graves' disease, a phase II study. Eur J Endocrinol. 2008;159:609–15. doi: 10.1530/EJE-08-0084. [DOI] [PubMed] [Google Scholar]
- 17.Ahuja A, Shupe J, Dunn R, Kashgarian M, Kehry MR, Mark J Shlomchik MJ. Depletion of B cells in murine lupus: efficacy and resistance. J Immunol. 2007;179:3351–61. doi: 10.4049/jimmunol.179.5.3351. [DOI] [PubMed] [Google Scholar]
- 18.Yu S, Dunn R, Kehry MR, Braley-Mullen H. B cell depletion inhibits spontaneous autoimmune thyroiditis in NOD.H2-h4 mice. J Immunol. 2008;180:7706–13. doi: 10.4049/jimmunol.180.11.7706. [DOI] [PubMed] [Google Scholar]
- 19.Yanaba K, Hamaguchi Y, Venturi GM, Steeber DA, William St Clair E, Tedder TF. B cell depletion delays collagen-induced arthritis in mice: arthritis induction requires synergy between humoral and cell-mediated immunity. J Immunol. 2007;179:1369–80. doi: 10.4049/jimmunol.179.2.1369. [DOI] [PubMed] [Google Scholar]
- 20.Hasegawa M, Hamaguch Y, Yanaba K, et al. B-lymphocyte depletion reduces skin fibrosis and autoimmunity in the tight-skin mouse model for systemic sclerosis. Am J Pathol. 2006;169:954–66. doi: 10.2353/ajpath.2006.060205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayakawa I, Tedder TF, Zhuang Y. B-lymphocyte depletion ameliorates Sjögren's syndrome in Id3 knockout mice. Immunology. 2007;122:73–9. doi: 10.1111/j.1365-2567.2007.02614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiu Y, Wong CP, Bouaziz JD, et al. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in FcγR effector functions. J Immunol. 2008;180:2863–75. doi: 10.4049/jimmunol.180.5.2863. [DOI] [PubMed] [Google Scholar]
- 23.Chen C-R, Pichurin P, Nagayama Y, Latrofa F, Rapoport B, McLachlan SM. The thyrotropin receptor autoantigen in Graves' disease is the culprit as well as the victim. J Clin Invest. 2003;111:1897–904. doi: 10.1172/JCI17069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saitoh O, Abiru N, Nakahara M, Nagayama Y. CD8+CD122+ T cells, newly identified regulatory T cells, negatively regulate Graves' disease in a murine model. Endocrinology. 2007;148:6040–6. doi: 10.1210/en.2007-0300. [DOI] [PubMed] [Google Scholar]
- 25.Hamaguchi Y, Uchida J, Cain DW, et al. The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. J Immunol. 2005;174:4389–99. doi: 10.4049/jimmunol.174.7.4389. [DOI] [PubMed] [Google Scholar]
- 26.Nagayama Y, Mizuguchi H, Hayakawa T, Niwa M, McLachlan SM, Rapoport B. Prevention of autoantibody-mediated Graves'-like hyperthyroidism in mice by IL-4, a Th2 cytokine. J Immunol. 2003;170:3522–7. doi: 10.4049/jimmunol.170.7.3522. [DOI] [PubMed] [Google Scholar]
- 27.Nagayama Y, Saitoh O, McLachlan SM, Rapoport B, Kano H, Kumazawa Y. TSH receptor-adenovirus-induced Graves' disease is attenuated in both interferon-γ and interleukin-4 knockout mice; implications for the Th1/Th2 paradigm. Clin Exp Immunol. 2004;138:417–22. doi: 10.1111/j.1365-2249.2004.02641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu CY, Rodriguez-Pinto D, Du W, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007;117:3857–67. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fiorina P, Vergani A, Dara S, et al. Targeting CD22 reprograms B-cells and reversed autoimmune diabetes. Diabetes. 2008;57:3013–24. doi: 10.2337/db08-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilbert JA, Kalled SL, Moorhead J, et al. Treatment of autoimmune hyperthyroidism in a murine model of Graves' disease with tumor necrosis factor-family ligand inhibitors suggests a key role for B cell activating factor in disease pathology. Endocrinology. 2006;147:4561–8. doi: 10.1210/en.2006-0507. [DOI] [PubMed] [Google Scholar]
- 31.Yan J, Harvey BP, Gee RJ, Shlomchik MJ, Mamula MJ. B cells drive early T cell autoimmunity in vivo prior to dendritic cell-mediated autoantigen presentation. J Immunol. 2006;177:4481–7. doi: 10.4049/jimmunol.177.7.4481. [DOI] [PubMed] [Google Scholar]
- 32.Vallerskog T, Gunnarsson I, Widhe M, et al. Treatment with rituximab affects both the cellular and the humoral arm of the immune system in patients with SLE. Clin Immunol. 2006;122:62–74. doi: 10.1016/j.clim.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 33.Stasi R, Stipa E, Del Poeta G, Amadori S, Newland AC, Provan D. Long-term observation of patients with anti-neutrophil cytoplasmic antibody-associated vasculitis treated with rituximab. Rheumatology (Oxf) 2006;45:1432–6. doi: 10.1093/rheumatology/kel098. [DOI] [PubMed] [Google Scholar]
- 34.Stasi R, Poeta GD, Stipa E, et al. Response to B-cell-depleting therapy with rituximab reverts the abnormalities of T-cell subsets in patients with idiopathic thrombocytopenic purpura. Blood. 2007;110:2924–30. doi: 10.1182/blood-2007-02-068999. [DOI] [PubMed] [Google Scholar]
- 35.El Fassi D, Banga JP, Bilbert JA, Padoa C, Hegedus L, Nielsen CH. Treatment of Graves' disease with rituximab specifically reduces the production of thyroid stimulating autoantibodies. Clin Immunol. 2009;130:252–8. doi: 10.1016/j.clim.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 36.Ferraro AJ, Drayson MT, Savage COS, MacLennan ICM. Levels of autoantibodies, unlike antibodies to all extrinsic antigen groups, fall following B cell depletion with Rituximab. Eur J Immunol. 2008;38:292–8. doi: 10.1002/eji.200737557. [DOI] [PubMed] [Google Scholar]
- 37.Teng YK, Levarht EW, Hashemi M, et al. Immunohistochemical analysis as a means to predict responsiveness to rituximab treatment. Arthritis Rheum. 2007;56:3909–18. doi: 10.1002/art.22967. [DOI] [PubMed] [Google Scholar]
- 38.Thurlings RM, Vos K, Wijbrandts CA, Zwinderman AH, Gerlag DM, Tak PP. Synovial tissue response to rituximab: mechanism of action and identification of biomarkers of response. Ann Rheum Dis. 2008;67:917–25. doi: 10.1136/ard.2007.080960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose-escalation trial of rituximab. Arthritis Rheum. 2004;50:2580–89. doi: 10.1002/art.20430. [DOI] [PubMed] [Google Scholar]
- 40.Joly P, Mouquet H, Roujeau JC, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357:545–52. doi: 10.1056/NEJMoa067752. [DOI] [PubMed] [Google Scholar]