

Abstract
Kawasaki disease (KD) is an acute vasculitis affecting mainly infants and children. Human B cells express Toll-like receptor (TLR)-9, whose natural ligands are unmethylated cytosine–guanine dinucleotide (CpG) motifs characteristic of bacterial DNA. The aim of this study was to clarify the pathogenesis of KD analysing the activation status of peripheral blood mononuclear cells (PBMC), focusing on B lymphocyte activation and functions. Ten patients and 10 age-matched healthy donors were recruited from the Bambino Gesù Hospital of Rome, Italy and enrolled into this study. We determined phenotype profile and immunoglobulin (Ig) production of PBMC from KD patients and age-matched controls. We found that the frequency of CD19+ B lymphocytes and CD19+/CD86+ activated B lymphocytes from KD patients during the acute phase before therapy was increased significantly. Moreover, B lymphocytes of acute-phase KD patients were more prone to CpG oligodeoxynucleotide (ODN) activation compared with the age-matched controls, as assessed by a significant increase of the number of IgA-secreting cells (SC). In the same patients we found a marked increase of IgM, IgG, interleukin (IL)-6 and tumour necrosis factor (TNF)-α production compared with the control group. In addition, in two convalescent KD patients, conventional treatment with intravenous immunoglobulin (IVIG) restored the normal frequency of CD19+ B cells, the number of IgA-, IgM- and IgG-SC and the production of IL-6 and TNF-α. Our findings indicate that the percentages of peripheral B lymphocytes of acute-phase KD patients are increased and are prone to bacterial activation in terms of increased numbers of IgA-SC and increased production of IL-6 and TNF-α inflammatory cytokines. Thus, our data support the hypothesis of an infectious triggering in KD.
Keywords: B lymphocytes, humoral immune response, Ig, Kawasaki disease, TLR-9
Introduction
Kawasaki disease (KD) is an acute multi-system vasculitis affecting mainly infants and children [1]. Coronary artery aneurysms develop in 20–30% of untreated children with KD, leading to ischaemic heart disease, myocardial infarction and even death [2].
The exact cause of KD is unknown, and is the subject of considerable debate. However, clinical and epidemiological features strongly suggest an undefined infectious trigger in genetically predisposed individuals [3]. Although KD has been reported all over the world, the disease is over-expressed among Asian populations, especially Japanese. The Japanese incidence (135–200/100 000, 5 years of age) is 10–15 times greater than among Caucasians (9–17/100 000, 5 years of age) [4].
To date, there is much controversy as to whether a superantigen or conventional antigen plays a role in KD [5,6]. Immunological mechanisms play a key role in KD onset, progression and complications [7,8]. The acute phase of KD is characterized by marked immune activation [9–11] associated with the generation of cytotoxic anti-endothelial cell antibodies [12] and increased cytokine production [13]. These alterations could contribute to the endothelial injury observed in KD.
Toll-like receptors (TLRs) are key molecules of the innate immune system that recognize molecular patterns on microorganisms and alert the host rapidly to the presence of potentially dangerous organisms [14]. Members of the TLR family are expressed differently on human immune cells, and their stimulation triggers different immune responses depending on the specific expression pattern of these receptors. In humans, expression of TLR-9 is restricted to B cells and plasmacytoid dendritic cells (DC). TLR-9 recognizes the cytosine–guanine dinucleotide (CpG) motif in unmethylated bacterial DNA and both in vivo and in vitro CpG oligodeoxynucleotides (ODN) can be used to stimulate TLR-9 [14].
It has been reported that in KD the TLR-4 pathway is activated significantly during the acute phase, thus causing dysregulation of the immune response [15].
Although therapeutic strategies with intravenous immunoglobulin (IVIG) to control inflammation have reduced morbidity and mortality associated with KD, the lack of a known aetiological agent and incomplete understanding of the molecular mechanisms mediating either the pathological changes of KD or the mechanism of action of IVIG have hampered the development of targeted and more effective treatment options. Moreover, until now neither diagnostic test is available nor is prevention feasible.
The immunological studies on KD involving the activation status of peripheral blood mononuclear cells (PBMC) remain controversial. In particular, few reports have investigated the activation of peripheral blood B cells in KD.
Therefore, the aim of this study was to clarify the pathogenesis and pathophysiology of KD analysing the activation status of PBMC, focusing on B cell activation and functions.
Materials and methods
Patients
The investigation conforms with the principles outlined in the Declaration of Helsinki. Informed consent from parents was obtained. This study was approved by the Institutional Review Board of ‘Bambino Gesù’ Hospital of Rome, Italy. Ten paediatric KD patients (Orpha 2331) aged between 6 and 56 months, comprising seven males and three females, and 10 age-matched healthy donors, were recruited from the Bambino Gesù Hospital of Rome (Italy) and enrolled into this study. Diagnosis of KD was based on the classic clinical criteria [16].
Nine patients were diagnosed as complete KD: they presented fever ≥5 days associated with ≥4 diagnostic criteria (polymorphous exanthema, bilateral non-exudative conjunctival injection, changes in lips and oral cavity, changes in the extremities, including erythema or indurative oedema and cervical lymphadenopathy, often unilateral and large (≥1·5 cm), or fever ≥5 days associated with <4 diagnostic criteria and coronary artery abnormalities, or fever on the fourth day with ≥4 diagnostic criteria and coronary artery abnormalities.
Only one patient presented an incomplete KD: she presented the typical fever without a sufficient number of diagnostic criteria, with coronary artery injuries [16].
At diagnosis, nine patients presented high inflammatory index [white blood cell (WBC), C-reactive protein (CRP), erythrocyte sedimentation rate (ESR)] while two patients presented hyperechogenicity areas of coronary arteries.
To assess disease severity we used the scoring model established by Kobayashi et al. [17] for predicting IVIG resistance: high serum levels of aspartate aminotransferase (AST), CRP, high percentage of neutrophils, young age at disease onset, low serum levels of sodium, low platelet counts and early administration of IVIG in the course of illness are independent risk factors. The points scored for each variable were as follows: sodium ≤133 mmol/l, 2 points; days of illness at initial treatment ≤4, 2 points; AST ≥100 IU/l, 2 points; % neutrophils ≥80%, 2 points; CRP ≥10 mg/dl, 1 point; age ≤12 months, 1 point; and platelet count ≤300 × 103/mm3, 1 point. This scoring system reflects clinical data as the consequences of increased inflammatory cascades. The low-risk patients' (score 0–4) clinical and coronary outcomes were reported to be better than those of the high-risk patients (score 5 or more). All the patients studied belong to the low-risk category.
All patients had blood sampled before the start of therapy with IVIG and aspirin. Two KD patients had pre- and post-IVIG blood samples collected.
Reagent
Unmethylated CpG ODN 2006 (sequence: 5′-TCG TC G TTT TGT CGT TTT GTC GTT-3′) was purchased from Operon Biotechnologies (Cologne, Germany) and used at 2·5 µg/ml.
Cell isolation and culture
Human PBMC were isolated by Ficoll-Hypaque (Flow Laboratories, Irvine, UK) gradient separation of peripheral blood of KD patients and of control subjects. PBMC were suspended at 1 × 106/ml in RPMI-1640 (Lonza, Verviers, Belgium) supplemented with 10% heat-inactivated fetal bovine serum (HyClone, Southlogan, UT, USA), L-glutamine and 1% penicillin–streptomycin (Euroclone, Milan, Italy) stimulated with CpG ODN 2006. The cultures were incubated at 37°C in a humidified 5% CO2 incubator. After 7 days, cells were assayed for IgM, IgG- and IgA-secreting cells (SC) by enzyme-linked immunospot (ELISPOT) assay. IgM-, IgG- and IgA-SC were expressed per number of recovered cells at the end of the culture period. Viable cell counts were performed on the harvested samples by trypan blue staining.
Flow cytometry
Analysis of cell surface markers was performed using the following mouse monoclonal antibodies (mAbs): fluorescein isothiocyanate (FITC)-conjugate anti-CD19 and anti-CD25, phycoerythrin (PE)-conjugated anti-CD3, anti-CD56 and anti-CD86 (all from BD Biosciences San Jose, CA, USA). Negative controls included directly labelled isotype-matched irrelevant mAbs. Cells were analysed with a fluorescence activated cell sorter (FACScan) flow cytometer and CellQuest software (Becton Dickinson, Franklin Lakes, NJ, USA).
ELISPOT assay for Ig-SC
Ig-SC were enumerated by a modification of the ELISPOT test [18]. To enumerate Ig-SC, 96-well flat-bottomed microtitre plates (Costar, New York, NY, USA) were coated overnight at 4°C with 0·1 ml/well of optimal dilution of goat anti-human IgM, IgG and IgA (Southern Biotechnology, Birmingham, AL, USA) in carbonate–bicarbonate buffer, pH 9·7. Plates were washed twice with phosphate-buffered saline (PBS)-0·05% Tween 20 (Sigma Chemical Co., St. Louis, MO, USA) and incubated for 1 h at 37°C with PBS containing 3% gelatin (Sigma Chemical Co.) as blocking agent. The plates were rinsed and the cells were then added and incubated at 37°C in 5% CO2 incubator for 3 h. The wells were subsequently washed and then incubated overnight at 4°C with 100 µl/well of an optimal dilution of alkaline phosphatase (AP)-conjugated goat anti-human IgM, IgG and IgA (Southern Biotechnology). After extensive washings with PBS-Tween, 100 µl of 1 mg/ml AP substrate 5-bromo-4-chloro-3-indolylphosphate (BCIP; Sigma Chemical Co.) in 1 m 2-amino-2 methyl-1-propanol buffer (AMP; Sigma Chemical Co.) containing 5 mm MgCl2, 0·01% Triton X-405 (Sigma Chemical Co.) and 0·01% NaN3 was added to each well. The plates were incubated for 1 h at room temperature. The supernatants were then discarded, the plates were allowed to dry and the spots were enumerated under a stereomicroscope with 40-fold magnification.
ELISPOT assay for IL-17-SC
The ELISPOT test, used to enumerate IL-17-SC, was based essentially on the method described by Versteegen et al. [19]. In brief, 96-well plates (Millipore, County Cork, Ireland) were coated with 0·1 ml/well mouse anti-human IL-17 (R&D Systems, Minneapolis, MN, USA) 10 µg/ml in carbonate–bicarbonate buffer pH 9·7, overnight at 4°C. Plates were washed with PBS-0·05% Tween 20 (Sigma Chemical Co.) and then blocked with 3% gelatine (Sigma Chemical Co.) in PBS. PBMC were plated at 2 × 105 per well in triplicate and stimulated with anti-CD2 (0·25 µg/ml), anti-CD3 (0·25 µg/ml) and anti-CD28 (0·25 µg/ml) using a T cell activation/expansion kit (Miltenyi Biotec, Bergisch Gladbach, Germany). After incubation for 18–24 h at 37°C, the cells were removed and incubated with an optimal dilution of biotinylated anti-human IL-17 detection antibody (R&D Systems) for 2 h at room temperature. After washing, wells were developed for 20 min with an optimal dilution of AP–streptavidin (Vector Laboratories, Peterborough, UK), then washed and 100 µl of 1 mg/ml AP substrate BCIP (Sigma Chemical Co.) in 1 m AMP (Sigma Chemical Co.) containing 5 mm MgCl2, 0·01% Triton X-405 (Sigma Chemical Co.) and 0·01% NaN3 was added to each well. The plates were incubated for 1 h at room temperature. The supernatants were then discarded and the wells rinsed with deionized water. The plates were allowed to dry and the spots were counted using an automated ELISPOT reader (AELVis 4·0; Hannover, Germany). The number of IL-17-SC was calculated as the number of spots/number of total PBMC per well, and was adjusted as the number of IL-17-SC/106 PBMC.
Cytokine assay
Cell culture supernatants were recovered 7 days after CpG ODN stimulation. IL-6 and TNF-α levels were measured by enzyme-linked immunosorbent assay (ELISA) (BD Opteia; ELISA sets, San Diego, CA, USA), according to the manufacturer's instructions.
Statistical analysis
Statistical analysis was performed using unpaired t-test with Welch's correction when appropriate. P-values <0·05 were considered significant.
Results
Distribution of T, B and natural killer (NK) cells during the acute phase (pre-IVIG) and during the convalescent phase (after IVIG) of KD
We first analysed the frequency of T, B and NK cells and the frequency of T, B and NK cells expressing the activation markers CD25 and CD86 in the peripheral blood of KD patients in acute phase before treatment, in comparison with age-matched healthy control subjects (Fig. 1a).
Fig. 1.
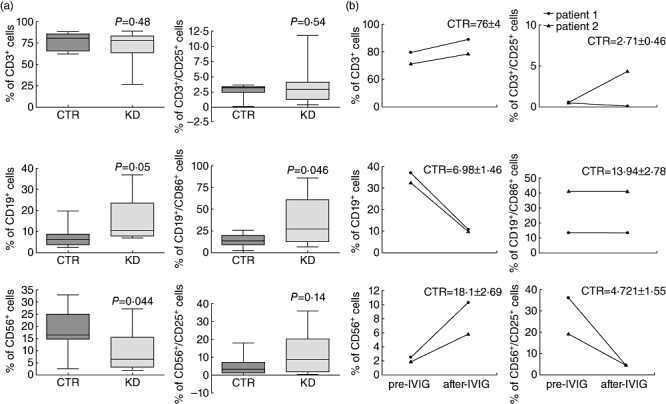
Changes in T, B and natural killer (NK) cell subsets in patients with Kawasaki disease (KD) in acute phase versus healthy controls. (a) Percentage of CD3+, CD19+, CD56+ cells (left) and proportions of CD3+/CD25+, CD19+/CD86+ and CD56+/CD25+ cells (right) in the peripheral blood of age-matched control subjects (CTR, n = 10) and acute-phase KD patients before treatment (n = 10). Analysis of cell surface markers was performed by flow cytometry. Comparisons by unpaired t-test revealed significant difference in the percentages of CD19+, CD19+/CD86+ and CD56+ cells between the CTR and KD groups. P-values <0·05 were considered significant. (b) Percentage of CD3+, CD19+, CD56+ cells (left) and proportions of CD3+/CD25+, CD19+/CD86+ and CD56+/CD25+ cells (right) in the peripheral blood of two patients before [pre-intravenous immunoglobulin (IVIG)] and after (post-IVIG) therapy. Mean ± standard error of the mean from 10 age-matched CTR subjects are reported.
We found that the percentage as well as the absolute number of CD3+ T cells during the acute phase of KD did not change compared with healthy control subjects [2·186 × ± 0·36 × 103/µl versus 2·590 ± 0·59 × 103/µl; mean ± standard deviation (SD)]. No significant changes were observed in the percentage of CD25+ cells among CD3+ T cells in KD patients compared with healthy controls. Among the T cell population the proportion of CD4+ and CD4+/CD25+ T cells did not change in acute-phase KD patients before treatment with respect to age-matched control subjects (data not shown).
We then analysed the proportion of CD19+ B cells and we found that the percentage of CD19+ B cells was increased significantly in KD patients during the acute phase in comparison with age-matched control subjects (P = 0·05). We found that the absolute number of CD19+ B lymphocytes was increased significantly in KD patients during the acute phase compared with control subjects (1·013 ± 0·16 × 103/µl versus 0·730 ± 0·30 × 103/µl; P = 0·02). The percentage of CD19+ B cells expressing the CD86 activation marker was increased significantly in KD patients during the acute phase in comparison with age-matched control subjects (P = 0·046). We did not find any correlation between the increased frequency of resting and activated B lymphocytes with respect to illness severity.
The percentage of CD56+ NK cells was reduced significantly in KD patients during the acute phase compared with age-matched control subjects (P = 0·044), as well as their absolute number (0·225 ± 0·12 × 103/µl versus 0·705 ± 0·32 × 103/µl; P = 0·01). The percentage of CD56+ NK cells expressing the activation marker CD25 increased, although not significantly, in KD patients during the acute phase in comparison with age-matched controls.
We next compared the frequency of T, B and NK cells and the frequency of T, B and NK cells expressing the activation markers CD25 and CD86 in the peripheral blood of two patients collected before (pre-IVIG) and after (post-IVIG) therapy (Fig. 1b).
We found that in both patients the percentage of CD3+ T cells in the acute phase did not change after therapy, whereas in one of two patients we found that the percentage of CD3+/CD25+ T lymphocytes increased in the convalescent phase after therapy. The increased percentage of CD19+ B cells, observed in both patients during the acute phase (pre-IVIG), decreased after IVIG therapy to a level similar to that of the control group. However, the percentage of CD19+ B cells expressing the activation marker CD86, increased in one of two patients, did not reverse after IVIG therapy. Finally, the percentage of CD56+ NK cells in both patients reversed partially after IVIG therapy, while the percentage of CD56+/CD25+ decreased following IVIG therapy to a level similar to that of age-matched control subjects.
CpG-induced immunoglobulin production during the acute phase (pre-IVIG) and during the convalescent phase (post-IVIG) of KD
Human B cells express TLR-9, whose natural ligands are unmethylated CpG motifs. CpG motifs are prevalent in bacterial genomic DNA. Therefore, to evaluate the responsiveness of peripheral B cells obtained from KD patients in both the acute and convalescent phases, we stimulated the PBMC of these patients with CpG ODN to evaluate IgM, IgA and IgG production (Fig. 2a).
Fig. 2.
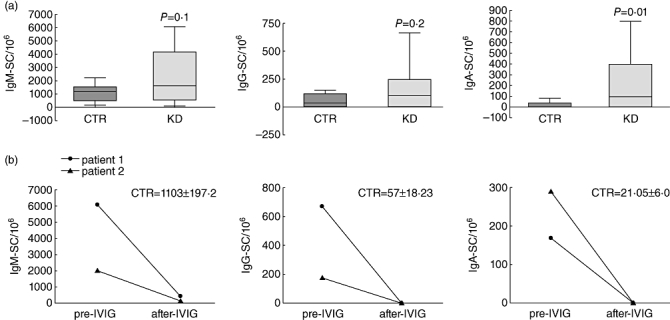
Changes in cytosine–guanine dinucleotide (CpG)-induced immunoglobulin production in patients with Kawasaki disease (KD) in acute phase versus healthy controls. (a) Peripheral blood mononuclear cells (PBMC) from 10 acute-phase KD patients and 10 age-matched controls were stimulated with CpG oligodeoxynucleotide (ODN) and after 7 days immunoglobulin (Ig)M-, IgG- and IgA-secreting cells (SC) were enumerated by enzyme-linked immunospot (ELISPOT) assay. Comparisons by unpaired t-test revealed significant differences in the number of IgA-SC between the control (CTR) and KD groups. P-values <0·05 were considered significant. (b) Number of IgM-, IgG- and IgA-SC in two KD patients before (pre-IVIG) and after (post-IVIG) therapy. Mean ± standard error of the mean from 10 age-matched CTR subjects are reported.
We found that activation of B lymphocytes by CpG ODN induced a significant increase in the number of IgA-SC in acute-phase KD patients compared with age-matched controls. In addition, the numbers of IgM- and IgG-SC were greatly increased in acute-phase KD patients compared with age-matched control subjects. We did not find any correlation between increased numbers of IgA-SC with respect to illness severity.
Notably, we found that the numbers of IgM-, IgG- and IgA-SC were restored in convalescent patients following IVIG therapy (Fig. 2b).
Cytokine production during the acute phase (pre-IVIG) and during the convalescent phase (post-IVIG) of KD
Because CpG ODN stimulates the secretion of cytokines by B lymphocytes [14], we analysed the pattern of production of these mediators in acute-phase KD patients and convalescent phases as well as in age-matched controls. To this aim, IL-6 and TNF-α accumulation was measured in culture supernatants of CpG ODN-stimulated PBMC (Fig. 3a).
Fig. 3.
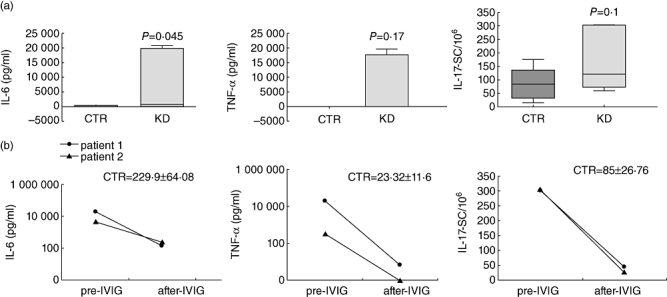
Changes in interleukin (IL)-6, tumour necrosis factor (TNF)-α and IL-17 production in patients with Kawasaki disease (KD) in acute phase versus healthy controls. (a) Peripheral blood mononuclear cells (PBMC) from 10 acute-phase KD patients and 10 age-matched controls were stimulated with cytosine–guanine dinucleotide oligodeoxynucleotide (CpG ODN), and after 7 days IL-6 and TNF-α were measured by enzyme-linked immunosorbent assay (ELISA). IL-17-SC were enumerated by enzyme-linked immunospot (ELISPOT) assay in PBMC cultures stimulated for 24 h with anti-CD2, -CD3 and -CD28 monoclonal antibodies (n = 10). Comparisons by unpaired t-test revealed significant differences in IL-6 secretion between control (CTR) and KD groups. P-values <0·05 were considered significant. (b) IL-6, TNF-α and IL-17 production in two KD patients before (pre-IVIG) and after (post-IVIG) therapy. Mean ± standard error of the mean from 10 age-matched CTR subjects are reported.
We found that CpG ODN induced a significant increase of IL-6 and a marked increase of TNF-α production from B cells after 7 days of culture in acute-phase KD patients in comparison with age-matched control subjects.
We next analysed IL-6 and TNF-α accumulation in culture supernatants of two patients in the acute and convalescent phases (post-IVIG). We found that the levels of IL-6 and TNF-α reversed in the convalescent phase following IVIG therapy (Fig. 3b).
We then analysed the production of IL-17, a proinflammatory cytokine produced by T lymphocytes, to evaluate its involvement in KD pathogenesis. To this aim we stimulated PBMC with anti-CD2, -CD3 and -CD28 mAbs. Compared with age-matched control subjects, the frequency of IL-17-SC was elevated markedly in acute-phase KD patients (Fig. 3a) and decreased in the convalescent phase after IVIG (Fig. 3b).
Discussion
In the present study we analysed the immunological status of PBMC from KD patients focusing on B lymphocyte activation and function. We provide evidence that the percentage of CD19+ as well as CD19+/CD86+ B lymphocytes of acute-phase KD patients before IVIG was increased significantly. Moreover, we found that B lymphocytes from acute-phase KD patients were more prone to be activated by the CpG ODN polyclonal bacterial stimulus in comparison with age-matched controls, as assessed by the significant increase in IgA, IL-6 and TNF-α production. In addition, we found that in two convalescent KD patients, after IVIG therapy, either the number of B cells and IgA, IgM and IgG-SC or the production of IL-6 and TNF-α decreased to a level similar to that of age-matched controls.
Despite 30 years of research, the aetiology of KD is still unknown and there is much controversy as to whether a superantigen or conventional antigen plays a role in KD [5,6].
Immunological mechanisms play a critical role in KD, and until now immunological studies on PBMC activation during acute KD appear to be still controversial.
We found that the percentage of CD3+ T lymphocytes as well as the percentage of CD3+/CD25+ lymphocytes did not change in acute-phase KD patients in comparison with age-matched controls. However, in our study we did not set up specific experiments to assess the functional activity of CD3+ T lymphocytes from acute-phase KD patients; thus, we cannot rule out that CD3+ T lymphocytes are functionally activated.
At present, the status of peripheral T cell activation remains controversial [8–10]. Ikeda et al. showed that the proportion of CD3+/CD25+ cells was significantly lower in KD patients with respect to control subjects, while the proportion of CD3+/CD69+ cells was significantly higher in KD patients with respect to control subjects [8]. Similarly, Ehara et al. showed a marked increase in the proportion of CD69+/CD8+ T cells [20]. Conversely, various reports have shown that CD4 and CD8 T cell activation did not change, as determined by the expression of the CD69 early activation antigen [9,10,21]. These discrepancies could be attributed to the differences in the timing of blood sampling by illness onset. Moreover, the immune systems of very young children are not fully mature, and children of this age group receive vaccinations against various viruses which can complicate further the results of the study.
As innate immunity plays a central role in host defence against viral and bacterial infection, we analysed the NK cell phenotype in peripheral blood from KD patients. We found that the percentage of CD56+ NK cells decreased significantly in acute-phase KD patients and reverted partially after IVIG therapy. The percentage of CD56+ NK cells expressing the CD25 activation marker increased in KD patients before IVIG and decreased after IVIG to a level similar to the control group. Our data are in agreement with the results of Furukawa et al. [21] and Shingadia et al. [22] showing a significant reduction in the percentage of circulating CD16+ NK cells during the acute phase of KD. This intriguing result could be due to an increased turnover or enhanced migration out of peripheral blood into tissues.
Many conflicting data regarding PBMC numbers in acute KD have been reported [8,11,22–24]. However, we hypothesize that the age difference of the patients and persistence of concomitant infection at the time of KD could, at least partially, explain these discrepancies.
Early reports studying the humoral response in peripheral blood of KD patients during the acute phase showed a polyclonal B cell activation and increased production of serum immunoglobulin (IgA, IgM, IgG) [25,26].
Human B cells express TLR-9, whose natural ligands are unmethylated CpG motifs prevalent in bacterial genomic DNA [27]. We found that CpG ODN stimulation of acute KD patient B cells induced a significant increase in the numbers of IgA-SC. The numbers of IgM- and IgG-SC were also increased in KD patients during the acute phase. Furthermore, we found that in two patients the numbers of IgA- IgM- and IgG-SC reversed in the convalescent phase after IVIG.
After cell stimulation with CpG ODN, TLR-9 signalling is triggered by recruitment of the MyD88 adaptor molecule [28]. Wang et al. [29] reported that in monocyte/macrophage of acute-phase KD patients MyD88 mRNA levels were elevated significantly. Therefore, we hypothesize that increased numbers of Ig-SC observed in acute-phase KD may be due to an up-regulation of MyD88 gene expression in B lymphocytes, accounting for the increased responsiveness to TLR-9 engagement. Alternatively, it can be hypothesized that the pre-engagement of TLR-9 in KD patients by a putative infectious agent, causing a proteolitic cleavage prerequisite for TLR-9 signalling, [28], may render KD B cells more prone to CpG ODN activation with respect to B cells obtained from healthy subjects.
Takeshita et al. [30] demonstrated increased levels of serum anti-lipid A IgA and IgM, thus suggesting that a conventional bacterial antigen derived from Gram-negative bacteria may contribute to KD onset.
Rowley et al. [31] reported that IgA plasma cells infiltrate the bronchial-associated vascular tissue in the acute phase of fatal KD, thus suggesting that the KD aetiological agent enters through the upper respiratory tract, resulting in an IgA immune response, with subsequent systemic spread [32]. They also demonstrated that the IgA produced in acute KD vascular tissue is oligoclonal, consistent with an antigen-driven immune response [33].
Parallel with increased Ig production, TLR-9 stimulation induced a higher level of IL-6 and TNF-α, and in agreement with previous data [34] we found that IL-17 was increased markedly in KD patients. More importantly, we found that IL-17 levels reversed in the convalescent phase after IVIG therapy, suggesting that IL-17 may be involved in the inflammatory state typical of acute-phase KD.
Our findings indicate that B lymphocytes of acute-phase KD patients are increased and are more prone to bacterial activation in terms of a significantly increased number of IgA-SC and increased production of IL-6 and TNF-α inflammatory cytokines. Thus, our data suggest that TLR-9 engagement by conventional or ubiquitous bacteria may activate the humoral mucosal immune response, contributing to the pathogenesis of KD.
Acknowledgments
This work was supported by a grant from Ministero della Sanità to M.V. and W.M.
Disclosure
None.
References
- 1.Kawasaki T, Kosaki F, Okawa S, Shigematsu I, Yanagawa H. A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics. 1974;54:271–6. [PubMed] [Google Scholar]
- 2.Landing BH, Larson EJ. Pathological features of Kawasaki disease (mucocutaneous lymph node syndrome) Am J Cardiovasc Pathol. 1987;1:218–29. [PubMed] [Google Scholar]
- 3.Rowley AH, Shulman ST. New developments in the search for the etiologic agent of Kawasaki disease. Curr Opin Pediatr. 2007;19:71–4. doi: 10.1097/MOP.0b013e328012720f. [DOI] [PubMed] [Google Scholar]
- 4.Burns JC, Glode MP. Kawasaki syndrome. Lancet. 2004;364:533–44. doi: 10.1016/S0140-6736(04)16814-1. [DOI] [PubMed] [Google Scholar]
- 5.Rowley AH, Baker SC, Orenstein JM, Shulman ST. Searching for the cause of Kawasaki disease – cytoplasmic inclusion bodies provide new insight. Nat Rev Microbiol. 2008;6:394–401. doi: 10.1038/nrmicro1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsubara K, Fukaya T. The role of superantigens of group A Streptococcus and Staphylococcus aureus in Kawasaki disease. Curr Opin Infect Dis. 2007;20:298–303. doi: 10.1097/QCO.0b013e3280964d8c. [DOI] [PubMed] [Google Scholar]
- 7.Yeung RSM. Pathogenesis and treatment of Kawasaki disease. Curr Opin Rheumatol. 2005;17:617–23. doi: 10.1097/01.bor.0000174184.15901.ee. [DOI] [PubMed] [Google Scholar]
- 8.Ikeda K, Yamaguchi K, Tanaka T, et al. Unique activation status of peripheral blood mononuclear cells at acute phase of Kawasaki disease. Clin Exp Immunol. 2010;160:246–55. doi: 10.1111/j.1365-2249.2009.04073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsubara T, Ichiyama T, Furukawa S. Immunological profile of peripheral blood lymphocytes and monocytes/macrophages in Kawasaki disease. Clin Exp Immunol. 2005;141:381–7. doi: 10.1111/j.1365-2249.2005.02821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brogan PA, Shah V, Clarke LA, Dillon MJ, Klein N. T cell activation profiles in Kawasaki syndrome. Clin Exp Immunol. 2008;151:267–74. doi: 10.1111/j.1365-2249.2007.03567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee HH, Park IH, Shin JS, Kim DS. Immunoglobulin V(H) chain gene analysis of peripheral blood IgM-producing B cells in patients with Kawasaki disease. Yonsei Med J. 2009;50:493–504. doi: 10.3349/ymj.2009.50.4.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grunebaum E, Blank M, Cohen S, et al. The role of anti-endothelial cell antibodies in Kawasaki disease –in vitro and in vivo studies. Clin Exp Immunol. 2002;130:233–40. doi: 10.1046/j.1365-2249.2002.02000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin CY, Lin CC, Hwang B, Chiang B. Serial changes of serum interleukin-6, interleukin-8, and tumor necrosis factor alpha among patients with Kawasaki disease. J Pediatr. 1992;121:924–6. doi: 10.1016/s0022-3476(05)80343-9. [DOI] [PubMed] [Google Scholar]
- 14.Krieg AM, Vollmer J. Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol Rev. 2007;220:251–69. doi: 10.1111/j.1600-065X.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 15.Wang GB, Li CR, Zu Y. Change of MyD88-independent signal transduction of Toll-like receptor 4 in immunological pathogenesis of Kawasaki disease. Zhonghua Er Ke Za Zhi. 2007;45:818–23. [PubMed] [Google Scholar]
- 16.Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management in Kawasaki disease: a statement for health professionals from the committee on rheumatic fever, endocarditis and Kawasaki disease, Council on Cardiovascular Disease in the Young. American Heart Association. Pediatrics. 2004;114:1708–33. doi: 10.1542/peds.2004-2182. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi T, Inoue Y, Takeuchi K, et al. Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease. Circulation. 2006;113:2606–12. doi: 10.1161/CIRCULATIONAHA.105.592865. [DOI] [PubMed] [Google Scholar]
- 18.Sedgwick JD, Holt PG. ELISA-plaque assay for the detection of single antibody-secreting cells. In: Kemeny DM, Challacombe SJ, editors. ELISA and other solid phase immunoassays. Chichester: Wiley; 1988. pp. 241–63. [Google Scholar]
- 19.Versteegen JM, Logtenberg T, Ballieux RE. Enumeration of IFN-gamma-producing human lymphocytes by spot-ELISA. A method to detect lymphokine-producing lymphocytes at the single-cell level. J Immunol Methods. 1988;111:25–9. doi: 10.1016/0022-1759(88)90055-5. [DOI] [PubMed] [Google Scholar]
- 20.Ehara H, Kiyohara K, Izumisawa Y, Ito T. Early activation does not translate into effector differentiation of peripheral CD8T cells during the acute phase of Kawasaki disease. Cell Immunol. 2010;265:57–64. doi: 10.1016/j.cellimm.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 21.Furukawa S, Matsubara T, Tsuji K, et al. Comparison of Kawasaki disease and infectious mononucleosis in terms of natural killer cell and CD8+ T cell subsets. Infect Dis. 1991;163:416–17. doi: 10.1093/infdis/163.2.416. [DOI] [PubMed] [Google Scholar]
- 22.Shingadia D, O'Gorman M, Rowley AH, Shulman ST. Surface and cytoplasmic immunoglobulin expression in circulating B-lymphocytes in acute Kawasaki disease. Pediatr Res. 2001;50:538–43. doi: 10.1203/00006450-200110000-00019. [DOI] [PubMed] [Google Scholar]
- 23.Jason J, Gregg L, Han A, et al. Imunoregulatory changes in Kawasaki disease. Clin Immunol Immunopathol. 1997;84:296–306. doi: 10.1006/clin.1997.4376. [DOI] [PubMed] [Google Scholar]
- 24.Furukawa S, Matsubara T, Yabuta K. Mononuclear cell subsets and coronary artery lesions in Kawasaki disease. Arch Dis Child. 1992;67:706–8. doi: 10.1136/adc.67.6.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leung DY, Chu E, Wood N, Grady S, Meade R, Geha R. Immunoregulatory T cell abnormalities in mucocutaneous lymph node syndrome. J Immunol. 1983;130:2002–4. [PubMed] [Google Scholar]
- 26.Leung DY, Siegel R, Grady S, et al. Immunoregulatory abnormalities in mucocutaneous lymph node syndrome. Clin Immunol Immunopathol. 1982;23:100–12. doi: 10.1016/0090-1229(82)90075-7. [DOI] [PubMed] [Google Scholar]
- 27.Hornung V, Rothenfusser S, Britsch S, et al. Quantitative expression of Toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–7. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 28.Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat Immunol. 2008;9:1407–14. doi: 10.1038/ni.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang GB, Li CR, Zu Y, Yang WG. Changes and significance for regulatory factors for signal pathways of Toll-like receptors in immunological pathogenesis of Kawasaki disease. Zhonghua Er Ke Za Zhi. 2008;46:49–54. [PubMed] [Google Scholar]
- 30.Takeshita S, Kawase H, Shimizu T, Yoshida M, Sekine I. Increased production of serum IgA-class antibody to lipid A in Kawasaki disease. Pediatr Int. 2002;44:5–11. doi: 10.1046/j.1442-200x.2002.01506.x. [DOI] [PubMed] [Google Scholar]
- 31.Rowley AH, Eckerley CA, Jäck HM, Schulman ST, Baker SC. IgA plasma cells in vascular tissue of patients with Kawasaki syndrome. J Immunol. 1997;159:5946–55. [PubMed] [Google Scholar]
- 32.Rowley AH, Shulman ST, Mask CA, et al. IgA plasma cell infiltration of proximal respiratory tract, pancreas, kidney, and coronary artery in acute Kawasaki disease. J Infect Dis. 2000;182:1183–91. doi: 10.1086/315832. [DOI] [PubMed] [Google Scholar]
- 33.Rowley AH, Shulman ST, Spike BT, Mask CA, Baker SC. Oligoclonal IgA response in the vascular wall in acute Kawasaki disease. J Immunol. 2001;166:1334–43. doi: 10.4049/jimmunol.166.2.1334. [DOI] [PubMed] [Google Scholar]
- 34.Sohn MH, Noh SY, Chang W, Shin KM, Kim DS. Circulating interleukin 17 is increased in the acute stage of Kawasaki disease. Scand J Rheumatol. 2003;32:364–6. doi: 10.1080/03009740410005034. [DOI] [PubMed] [Google Scholar]