

Abstract
Vaccination with autologous cancer cells aims to enhance adaptive immune responses to tumour-associated antigens. The incorporation of Fms-like tyrosine kinase 3-ligand (FLT3L) treatment to the vaccination scheme has been shown previously to increase the immunogenicity of cancer vaccines, thereby enhancing their therapeutic potential. While evidence has been provided that FLT3L confers its effect through the increase of absolute dendritic cell (DC) numbers, it is currently unknown which DC populations are responsive to FLT3L and which effect FLT3L treatment has on DC functions. Here we show that the beneficial effects of FLT3L treatment resulted predominantly from a marked increase of two specific DC populations, the CD8 DCs and the recently identified merocytic DC (mcDC). These two DC populations (cross)-present cell-associated antigens to T cells in a natural killer (NK)-independent fashion. FLT3L treatment augmented the absolute numbers of these DCs, but did not change their activation status nor their capacity to prime antigen-specific T cells. While both DC populations effectively primed CD8+ T cell responses to cell-associated antigens, only mcDC were capable to prime CD4+ T cells to cell-associated antigens. Consequentially, the transfer of tumour vaccine-pulsed mcDC, but not of CD8 DCs, protected mice from subsequent tumour challenge in a vaccination model and resulted in eradication of established tumours in a therapeutic approach. These results show that the beneficial effect of FLT3L is associated with the induction of mcDC and suggests that selective targeting to mcDC or instilling mcDC ‘characteristics’ into conventional DC populations could significantly enhance the efficacy of tumour vaccines.
Keywords: cytotoxic T cell response, dendritic cell, tumour, vaccine
Introduction
Autologous tumour cell vaccines are intended to drive specific activation of the adaptive immune system for therapy of existing malignancies. The resulting in vivo destruction of tumour cells leads to an additional release of tumour antigens that further amplifies tumour-specific T cell responses [1–3]. This secondary antigenic boost has been suggested to help to enhance and sustain anti-tumour T cell responses and prevent recurrences and metastases.
Dendritic cells (DC) are the only antigen-presenting cells that can adequately prime naive T cells. The (cross)-presentation of tumour antigens by DC upon uptake of dying tumour cells/tumour cell debris has also been shown to be critical for the induction of endogenous anti-tumour T cell responses [4,5].
DCs are phenotypically and functionally heterogeneous. At least six DC subsets have been described in mice and humans: plasmacytoid DCs (pDCs), three blood-derived subsets (CD4+ DCs, CD8α+ DCs and CD4-CD8- DCs [6,7]) and two tissue-derived subsets (Langerhans' cells and dermal/interstitial DCs) – all of which appear to be distinct sublineages and not precursor-product-related [8–10]. However, this classification has been proved to be a simplified subdivision, as we and others have recently identified novel DC subsets that are either present in common lymphoid tissues or associated with specific organs [11–15].
Even though most DC subsets can capture proteins and cell-associated antigens and can activate CD4+ and CD8+ T cells when pulsed with cognate peptides, only few DC populations have the actual capacity to process and present tumour-derived antigens to T cells [16,17]. Cross-presentation of cell-associated antigens to CD8+ T cells in particular is believed to be limited to just one or two DC populations [17,18].
Moreover, besides the fact that only few DC subsets can present both CD4+ and CD8+ T cell epitopes from cell-associated antigens, both human and mouse studies have shown that detection and subsequent clearance of apoptotic cells leads to a tolerogenic state in DC [19–22]. Phagocytosis of apoptotic material has, in fact, been shown to prevent DC maturation and inhibit proinflammatory cytokine production. In addition, the uptake of apoptotic cells by various lineages of phagocytes has been shown to induce specific immunoregulatory factors, including interleukin (IL)-10, transforming growth factor (TGF)-β and prostaglandin E2, that dampen adaptive immune responses [19–22]. While this process is beneficial for maintaining tissue homeostasis and preventing autoimmunity, it is clearly an impediment in the induction of anti-tumour responses.
We have recently identified a novel naturally occurring DC population [CD11c+CD11b-CD8α-PDCA-1- merocytic DC (mcDC)] that, in contrast with other DC subsets, produces proinflammatory type I IFN after uptake of dying cells and potently (cross)-primes both CD4+ and CD8+ T cells to cell-associated antigens [12,23,24]. T cells primed by mcDC display a greater capacity for primary expansion, cytokine production and memory formation on a per cell basis than those primed by other DC subsets. Because mcDCs are not susceptible to tolerance induction by apoptotic cells, we hypothesize that the selective expansion of mcDCs would be therapeutically more beneficial than the expansion of all DC populations.
The incorporation of the cytokine Fms-like tyrosine kinase 3-ligand (FLT3L) with various treatment strategies has been shown recently to increase the immunogenic and thereby therapeutic potential of cancer vaccines [25–29]. FLT3L by itself promotes tumour regression in some solid tumour models, presumably through the activation of natural killer (NK) cells [30–32]. However, poorly immunogenic tumours are seldom rejected by this means alone. The primary mechanism of FLT3L is attributed currently to its support of the survival, proliferation and differentiation of haematopoietic progenitors into DCs [33–36]. Although there is consensus that the increase in DC numbers is one of the main mechanisms for the enhanced anti-tumour responses upon FLT3L treatment, many details on the relative contribution of distinct DC populations or the possible effect of FLT3L on their functions are still unclear.
Here we show that FLT3L confers its immunostimulatory effect to prime CD4+ and CD8+ T cells to tumour-associated antigens through the preferential expansion of specific DC subsets rather than through changing the capacity of DC subtypes.
Materials and methods
Mice, cell lines and peptides
C57Bl/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Mice expressing chicken ovalbumin (ActmOVA) were a kind gift from M. Jenkins [37] and were bred onto the B6.C-H2bm1/ByJ (B6.Kbm1) background. OT-1 (OVA-specific transgenic CD8 T cells) were bred onto the CD45·1 (B6.SJL.Ptpcra) background and OT-2 (OVA-specific transgenic CD4 T cells) were bred onto the CD90·1 (B6.PL-Thy1a/CyJ) background in our facility. Mice were maintained under specific pathogen-free conditions in accordance with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International.
PK136 (NK1·1 depleting antibody), EL-4mOVA and the parental line EL-4 were cultured in Iscove's modified Dulbecco's medium (IMDM) (Invitrogen Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (FCS), 50 µM 2-mercaptoethanol (ME), 2 mM l-glutamine, 20 U/ml penicillin and 20 µg/ml streptomycin. OVA257–264 (SIINFEKL), tyrosine-related protein-2 tyrosinase-related protein (TRP)-2180–188 (SVDYDFFDWL), OVA323–339 (ISQAVHAAHAEINEAGR) and lymphocytic choriomeningitis virus–glycoprotein (LCMV GP)61–80 (GLKGPDIYKGVYQFKSVEFD) were obtained from A&A Laboratories (San Diego, CA, USA).
DC and T cell isolation
DC were isolated from spleens of naive mice or mice treated for 9 days with 10 µg human recombinant (hr)FLT3L as described previously [34]. hrFLT3L was a kind gift from Amgen (Thousand Oaks, CA, USA). DC were analysed for the expression of CD4, CD8α, CD11b, CD11c, CD40, CD54, CD80, CD86, Kb, Db and I-A/E by flow cytometric analysis (antibodies/isotype controls; eBioscience/Biolegend, San Diego, CA, USA; DCs were subsorted by flow cytometry based on their expression of CD11c, CD11b, CD8α or PDCA-1 by flow cytometry to purity of >95% and viability >95% (7-AAD staining).
OT-1 and OT-2 T cells were isolated using CD8 or CD4 microbeads (Miltenyi Biotec, Auburn, CA, USA) and labelled with 5,6-carboxy-succinimidyl-fluorescein-ester (CFSE) (Molecular Probes, Eugene, OR, USA) as described previously [38]. Purity of sorted cells was >98% and viability was >97% as determined by CD4/CD8/Vα2/Vβ5 expression and 7-AAD staining.
Cytokine induction in DC
Purified DCs (1 × 105) were cultured with irradiated splenocytes in a 1:3 ratio in 96-well U-bottomed plates. After 3, 6 and 16 h supernatant was analysed for type I IFN by reporter assay [39] and IL-10, tumour necrosis factor (TNF)-α and TGF-β by quantitative polymerase chain reaction (PCR) using SybrGreen and the following primers: ml32 forward 5-GAAACTGGCGGAAACCCA-3, ml32 reverse 5-GGATCTGGCCCTTGAACCTT-3, TNF-α forward 5-GTACTGGCATGTGTATGTCA-3, TNF-α reverse 5-TGGTTGAGGGAATCATT-3, IL-10 forward 5-GGTTGCCAAGCCTTATCGGA-3, IL-10 reverse 5-ACCTGCTCCACTGCCTTGCT-3, TGF-β forward 5-GACCGCAACAACGCCATCTA-3, TGF-β reverse 5-GGCGTATCAGTGGGGGTCAG-3. The fold increase of specific RNA (mRNA after apoptotic cells exposure/mRNA before apoptotic cells) was determined after normalization to L32 for each sample.
In vitro priming by DCs
Purified DCs (1 × 105) were cultured with irradiated purified ActmOVA-Kbm1 T cells in a 1:3 ratio in 96-well U-bottomed plates. After 24 h, 1 × 105 CFSE-labelled OT-1 or OT-2 T cells were added to the wells. This experimental set-up allows us to study exclusively cross-priming by the DC subsets because the mutated peptide binding groove of Kbm1 cannot bind the OVA257–264 peptide [40] and the lack of MHC class II on the T cells prevents direct activation of the OT-2 T cells [41]. As positive control, DCs were pulsed with OVA peptides for 10 min and washed thoroughly.
OT-1 and OT-2 T cell proliferation and survival were determined after 70 h by analysis of CFSE dilution together with staining for Vα2, CD4/CD8 and 7-AAD. Expansion of OT-1/OT-2 T cells was determined by dividing the number of live T cells at the end of the culture by the number of cells added at the start of culture [12]. In parallel studies, 0·3 µM [3H]-thymidine was added after 60 h of culture, and incorporation was determined 12 h later. Cytokine production in the supernatant was determined by standard sandwich enzyme-linked immunosorbent assay (ELISA) for IL-2, IL-4, TNF-α and IFN-γ (Biolegend, San Diego, CA, USA).
In vivo priming by DCs
For in vivo priming, B6 mice received intravenous (i.v.) 4 × 105 purified DC that were incubated with irradiated ActmOVA-Kbm1 T cells, as described above. Apoptotic cells were removed from the DC populations using the apoptotic cell removal kit (Miltenyi Biotec, Auburn, CA, USA). CD8+ T cell responses were analysed in spleens 7 days after DC transfer using intracellular cytokine staining to IFN-γ and TNF-α upon incubation with OVA257–264 (5 µg/ml) or control peptide TRP-2180–188 (5 µg/ml) for 5 h in the presence of brefeldin A. Surface staining for CD8 and CD44 and intracellular cytokine staining for IFN-γ was performed using a Cytofix/Cytoperm kit (BD Pharmingen, La Jolla, CA, USA), according to the manufacturer's instructions [12,41]. For memory CD8+ T cell assessment, an in vivo cytotoxicity assay was performed 28 days after DC treatment. Briefly, mice received CFSEhigh-labelled splenocyte pulsed with OVA257–264 (target cells) mixed with an equal number of CFSEmedium-labelled control cells. Twenty-four h later the ratio of CFSElow/CFSEhigh cells was determined by flow cytometry [42].
OVA-specific CD4+ T helper type 1 (Th1) and Th2 cells were enumerated by enzyme-linked immunospot assay (ELISPOT) 10 days after DC transfer after a 48-h in vitro stimulation with OVA323–339 (10 µg/ml), control peptide GP61–80 (10 µg/ml) or concanavalin A (ConA) (2 µg/ml; positive control), as described previously [43].
Tumour model studies
Challenge model
Mice received i.v. 5 × 105 purified DC that were incubated with irradiated ActmOVA-Kbm1 T cells. Seven days later, mice were challenged by subcutaneous (s.c.) injection of 2 × 106 EL-4-mOVA cells in the left flank and 2 × 106 EL-4 cells in the right flank. Tumour growth was measured every second day with vernier calipers. Tumour size was calculated as the product of bisecting tumour diameters.
Therapeutic model
In the therapeutic approach, mice were inoculated with 2 × 106 live EL-4-mOVA cells on the left flank and 2 × 106 EL-4 as control on the right flank. As soon as palpable tumours had formed, mice received 1 × 106 purified DC that had been exposed to irradiated ActmOVA cells, and tumour growth was monitored daily with a vernier caliper. In parallel studies mice received only EL-4-mOVA cells in the left flank to determine long-term survival, reoccurrence of tumours and possible loss of OVA-tumour antigen.
Statistics
Unless stated otherwise, the data are expressed as means [standard error of the mean (s.e.m.)]. Survival responses were analysed by Kaplan–Meyer using a log-rank test. All other data were evaluated using an analysis of variance followed by a Dunnett test. A probability value of P < 0·05 was considered statistically significant.
Results
Immunostimulatory effect of FLT3L to cell-associated antigens is independent of NK T cells
We first established the immunostimulatory capacity of FLT3L in our model. To this end, mice pretreated with PBS or FLT3L were immunized s.c. with irradiated EL-4mOVA cells and OVA257–264 specific CD8+ T cell responses in spleens were determined 7 days later by intracellular cytokine staining upon stimulation with OVA257–264 or with control peptide. As expected, FLT3L-treated mice showed a greater induction of OVA257–264-specific IFNγ-producing CD8+ T cells compared to PBS-treated mice (Fig. 1a and b). FLT3L-treated, but not PBS-treated, mice were protected from EL-4-mOVA challenge 35 days after the initial immunization (Fig. 1c). This protection was CD8+ T cell-dependent, as antibody-mediated depletion of CD8+ T cells before tumour challenge resulted in tumour growth comparable to that observed in naive mice (data not shown).
Fig. 1.
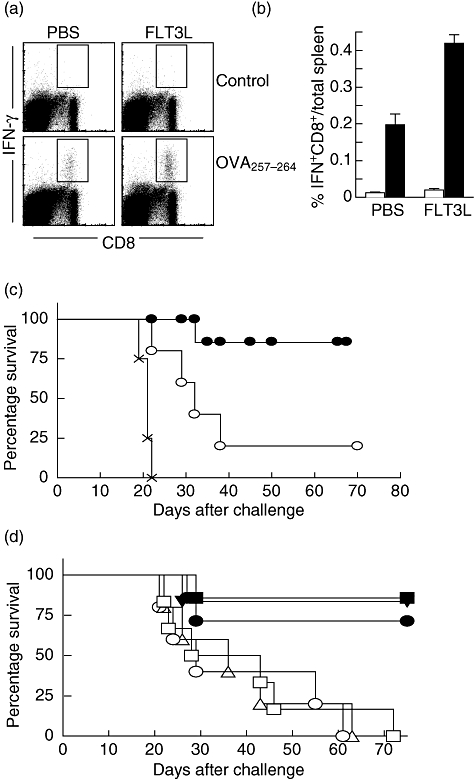
Increased induction of tumour-specific CD8+ T cell responses and tumour rejection upon Fms-like tyrosine kinase 3-ligand (FLT3L) treatment. Phosphate-buffered saline (PBS)- and FLT3L-treated mice were immunized subcutaneously with 10 × 106 irradiated EL-4 cells expressing chicken ovalbumin (EL4-mOVA). (a,b) Frequency of OVA257–264-specific interferon (IFN)-γ-producing CD8+ T cells 7 days after immunization, as assessed by intracellular cytokine staining upon a brief incubation with cognate peptide (OVA257–264; black bar) or control peptide (white bar) for 5 h. Data in (a) show representative dot-plots of one mouse/group (representative of two independent experiments; four mice per group). Data in (b) are shown as mean ± standard error of the mean (n = 4). (c) PBS- (open circles) and FLT3L (closed circles)-treated mice were immunized with irradiated EL-4-mOVA cells and 35 days later challenged with live EL-4mOVA. Naive untreated mice (x) served as control (five to seven mice per group). (d) PBS (open circles)- and FLT3L (closed symbols)-treated mice were immunized with irradiated EL-4-mOVA cells and 35 days later challenged with live EL-4mOVA. Natural killer T cells were depleted 2 days before immunization (squares) or 2 days before tumour challenge (circles) by intraperitoneal administration of 300 µg PK136 antibody (five to seven mice per group).
As FLT3L has been shown to increase NK cell numbers and their activation status [44,45], we determined if NK cells played a role in the increased CD8+ T cell priming in FLT3L-treated mice. Temporary elimination of NK T cells by antibody depletion prior to immunization did not affect the magnitude of the antigen-specific T cell response or survival upon tumour challenge in PBS- and FLT3L-treated mice. Moreover, NK T cell depletion after immunization (but before tumour challenge) did not affect the FLT3L-mediated protection from tumour outgrowth, demonstrating that both the protection to tumour growth and increased OVA257–264-specific CD8+ T cell response in FLT3L-treated mice was NK T cell-independent (Fig. 1d, and data not shown).
FLT3L treatment alters the composition of the DC population but not DC functions
As FLT3L treatment has been shown to expand DCs in the spleen and secondary lymphoid organs [34], we next analysed the effect of FLT3L treatment on frequency of total DC, the frequency of different DC subsets (CD11b DCs, CD11c+CD11b+PDCA-1-CD8α-; CD8 DCs, CD11c+CD11b-PDCA-1-CD8α+; pDC, CD11c+CD11b-PDCA-1+CD8α-; mcDC, CD11c+CD11b-PDCA-1-CD8α- (Fig. 2a) and their functional capacity. Importantly, not only the absolute number of DC but also the distribution of different DC populations within the CD11+ population changed dramatically upon FLT3L treatment (Fig. 2b). While total CD11b DCs expanded ∼ twofold (2·2 ± 0·3) upon FLT3L treatment, CD8 DCs, mcDC and pDC expanded ∼ ninefold (9·6 ± 2·3-, 9·2 ± 1·6- and 8·3 ± 1·1-fold, respectively).
Fig. 2.
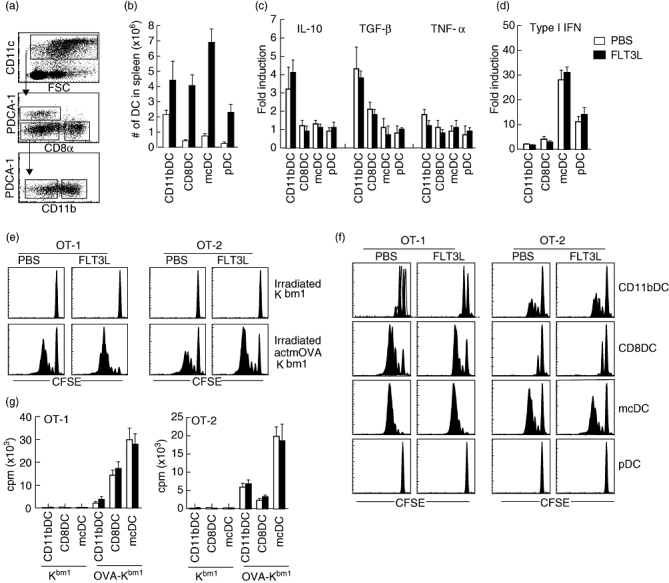
Increased absolute dendritic cell (DC) numbers by Fms-like tyrosine kinase 3-ligand (FLT3L) treatment that prime T cells to cell-associated antigen in vitro. (a) Flow cytometric analysis of DC populations in spleen. (b) Total numbers of indicated DC populations in spleens 24 h after the last injection with phosphate-buffered saline (PBS) (white bar) and FLT3L (black bar) (four mice per group). (c) Cytokine induction in DC from PBS- and FLT3L-treated mice as determined by quantitative polymerase chain reaction (PCR) upon co-culture with apoptotic cells. Data are expressed as fold increase (mRNA apoptotic cells exposed DC/mRNA naive DC) after normalization of each sample to L32. (d) Type I interferon (IFN) production by DC from PBS- and FLT3L-treated mice as determined by reporter assay. (e,f,g) Capacity of DC from PBS- and FLT3L-treated mice to prime T cells to cell-associated antigens. Total DC were isolated from PBS- and FLT3L-treated mice. After overnight culture with irradiated ActmOVA-Kbm1 T cells total DC were recovered (e) or DC were subsorted based on their expression of CD11c, CD11b, CD8, B220 and plasmacytoid DC antigen-1 (PDCA-1) (f). DC were cultured with 5,6-carboxy-succinimidyl-fluorescein-ester (CFSE)-labelled transgenic T cells specific for OVA257–264 (OT-1 CD8+ T cells) or OVA323–339 (OT-2 CD4+ T cells). Proliferation of OT-1 and OT-2 T cells as determined by CFSE dilution upon 72 h culture. Antibody staining for Vα2, CD90·1/CD45·1, and 7-aminoactinomycin D (7-AAD) was used to identify live T cells. Representative plots are shown for seven independent experiments. (g) Proliferation of OT-1 and OT-2 T cells as determined by [3H]-thymidine incorporation upon 72 h of culture. Data are expressed as counts per minute mean ± standard error of the mean (triplicate wells).
Interestingly, FLT3L treatment did not affect the functional profile of the DC supsets. The expression levels of major histocompatibility complex (MHC) I/II or co-stimulatory molecules [CD40, CD54, CD80, CD86, CD274 programmed cell death ligand 1 (PD-L1), CD273 (PD-L2)] were comparable with the corresponding DC populations from PBS-treated mice (data not shown). In addition, the cytokine induction by DCs upon interaction with apoptotic cells was also unaltered (Fig. 2c). Specifically, CD11b DCs displayed increased mRNA levels in both FLT3L- and PBS-treated mice of the anti-inflammatory TGF-β and IL-10 upon interaction with apoptotic cells, while CD8 DCs showed a modest increase in TGF-β only. Importantly, mcDC, and to a lesser degree pDC, produced the proinflammatory type I IFN upon uptake of apoptotic cells (Fig. 2d). Together these data show that FLT3L treatment induces the proliferation but not the functional profile of specific DC subsets.
Comparable T cell priming capacity of DC subsets from PBS- and FLT3L-treated mice
To study T cell priming to cell-associated antigens in vitro we used a culturing system where DC were cultured with irradiated ActmOVA cells that lacked MHC-I/II before CFSE-labelled OVA-specific OT-1 (CD8+) and OT-2 (CD4+) T cells were added [12].
Bulk DC from FLT3L-treated mice induced more proliferation in both OT-1 and OT-2 T cells than bulk DC from PBS-treated mice (Fig. 2e), showing that the increased T cell activation in vivo (Fig. 1a and b) could be recapitulated in vitro. The increased activation of both CD4+ and CD8+ T cells primed by FLT3L-DC was also measurable by elevated levels of the cytokines IFN-γ and IL-2 (CD4+ T cells) and IFN-γ and TNF-α (CD8+ T cells) (data not shown). To determine whether the increased T cell activation in FLT3L-DC resulted from the altered composition of the DC population or rather from altered functionality of one or more specific DC populations, we repeated the experiment with purified DC populations. CD11b DCs induced poor OT-1 T cell proliferation and intermediate OT-2 T cell proliferation (Fig. 2f and g). In contrast, CD8 DCs from both treatment groups induced good proliferation of CD8+ OT-1 T cells, but poor proliferation in OT-2 cells. mcDC potently induced both OT-1 and OT-2 responses, while pDC failed to induce significant T cell responses (Fig. 2f and g). Cytokine analysis of the primed OT-1 and OT-2 T cells showed similar results (data not shown). Importantly, we could not detect significant differences between DC populations that were isolated from PBS- and FLT3L-treated mice. This finding again shows that DC functions were not altered upon FLT3L treatment and indicates that the increased T cell priming observed upon FLT3L treatment results from changes in the composition of the DC population.
mcDC potently induce endogenous CD8+ and CD4+ T cell responses
To determine the effect of FLT3L treatment in the capacity of DC to prime endogenous CD8+ T cell responses in vivo, DC subpopulations (purified from PBS- and FLT3L-treated mice) were incubated with irradiated ActmOVA-Kbm1T cells, repurified and transferred i.v. into naive mice. Seven days later the frequency of endogenous OVA257–264-specific CD8+ T cells was determined by intracellular IFN-γ staining. pDCs failed to induce OVA257–264-specific CD8+ T cell responses and CD11b DC-treated mice showed poor induction of OVA257–264-specific responses, and FLT3L treatment did not change this phenotype (Fig. 3a and b). In contrast, priming by CD8 DCs was robust, and mcDC showed superior priming of endogenous OVA257–264-specific CD8+ T cells. OVA257–264-specific CD8+ T cells induced by CD8 DCs and mcDC produced both IFN-γ and TNF-α, but IL-2 or IL-4 production was undetectable (data not shown). Importantly, mcDC transfer induced CD8+ T cell memory. When mice were challenged with OVA257–264-pulsed target cells 28 days after DC transfer, mcDC-treated mice showed robust killing of target cells. This antigen-specific killing was superior to the killing observed in CD8 DC-transferred mice (Fig. 3c).
Fig. 3.
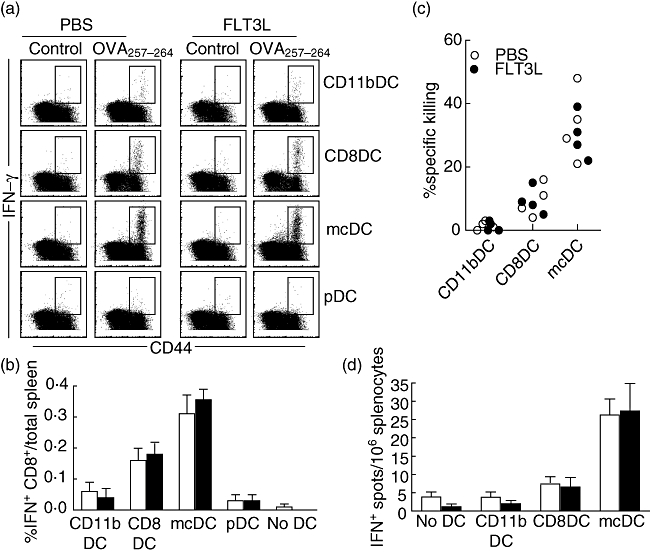
No functional effect of Fms-like tyrosine kinase 3-ligand (FLT3L) on endogenous T cells primed by distinct dendritic cell (DC) populations in vivo. DC were isolated from phosphate-buffered saline (PBS)- and FLT3L-treated mice. After overnight culture with irradiated ActmOVA-Kbm1 T cells DC were subsorted based on their expression of CD11c, CD11b, CD8, B220 and plasmacytoid DC antigen-1 (PDCA-1). Four ×105 cells of indicated DC populations were transferred intravenously (i.v.) into naive mice. (a) Frequency of ovalbumin (OVA)257–264-specific interferon (IFN)-γ-producing CD8+ T cells in the spleen 7 days after DC transfer. (b) Absolute number of IFN-γ-producing CD8+ T cells 7 days after indicated DC transfer. Data are expressed as mean ± standard error of the mean (four mice per group). (c) In vivo killing of OVA257–264-expressing target cells 28 days after DC immunization. Data of individual mice are shown. (d) Frequency of OVA323–339-specific CD4+ T cells in the spleen 10 days after DC transfer as determined by IFN-γ enzyme-linked immunospot assay. Data are shown as mean ± standard error of the mean (n = 4).
We next determined the induction of OVA323–339-specific CD4+ T cell responses by the different DC subsets. CD11b DCs, pDC and CD8 DCs showed poor priming of OVA323–339-specific CD4+ T cell responses as determined by ELISPOT for IFN-γ 10 days after DC transfer (Fig. 3d). Importantly, mcDC transfer resulted in a significantly stronger priming of IFN-γ-producing OVA323–339-specific CD4+ T cells (P < 0·05). We could not detect the cytokines IL-4 and IL-5 by ELISPOT upon mcDC transfer, indicating that mcDCs induce CD4+ T cell responses of a Th1 phenotype. Comparable to the in vitro data, DC populations from PBS- and FLT3L-treated mice had the same capacity to activate endogenous CD4+ and CD8+ T cell responses, showing that the DC functions also remain unaltered in vivo by FLT3L treatment.
mcDC display the greatest capacity to induce protective anti-tumour responses
To determine the capacity of the different DC populations to induce protective anti-tumour responses, mice received DC populations from FTL3L-treated mice that had been cultured with irradiated ActmOVA-Kbm1 T cells in vitro. Seven days after the transfer of 0·5 × 106 DC, mice were challenged on the left flank with EL-4-mOVA cells and on the right flank with EL-4 parental cells. In naive mice, EL-4 and EL-4-mOVA tumours grew with comparable kinetics (data not shown). Pretreatment of the mice with CD11b DCs did not affect tumour growth of either EL-4 or EL-4-mOVA (Fig. 4a). Pretreatment of the mice with CD8 DCs delayed tumour growth of the EL-4-mOVA but not the parental EL-4 tumour. Strikingly, mcDC pretreatment protected the mice completely from EL-4-mOVA tumour challenge but not EL-4-tumour challenge (Fig. 4a), highlighting their potency to induce protective tumour-specific immunity. Similar outcomes were seen when mcDC were isolated from PBS-treated mice (Fig. 4b), which was expected given their similar capacity to prime endogenous T cell responses to cell-associated antigens in vivo. Moreover, the protection to EL-4-mOVA but not EL-4 parental tumour challenge demonstrated the specificity of the DC treatments.
Fig. 4.
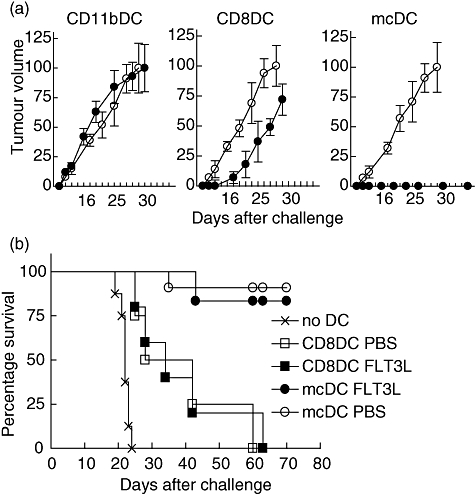
Merocytic dendritic cells (mcDCs) confer the most potent protective anti-tumour T cell response in vivo. DC from phosphate-buffered saline (PBS) and Fms-like tyrosine kinase 3-ligand (FLT3L) mice were isolated, cultured with irradiated ActmOVA-Kbm1 cells and subsorted based on their expression of CD11c, CD11b, CD8, B220 and plasmacytoid DC antigen-1 (PDCA-1). Naive mice received 5 × 105 DC. (a) Specificity of anti-tumour responses. Seven days after the DC transfer mice were inoculated subcutaneously (s.c.) with 2 × 106 EL-4-mOVA cells on the left flank (closed circle) and 2 × 106 EL-4 parental cells on the right flank (open circle). Tumour growth of both tumours was monitored (five mice per group). (b) FLT3L does not alter DC function. Seven days after the DC transfer mice were inoculated s.c. with 2 × 106 EL-4-mOVA cells on the left flank and survival was monitored (five to nine mice per group).
Differences in capacity for therapeutic intervention
We next determined the therapeutic potential of tumour cell vaccine presentation by the different DC populations in tumour-bearing mice. Mice received EL-4-mOVA cells on one flank and the parental EL-4 on the other flank. As soon as palpable tumours had formed, mice were treated with purified DC that had been exposed to irradiated ActmOVA-Kbm1 cells in vitro.
Treatment with CD11b DCs did not affect tumour growth, and both EL-4 tumour and EL-4-mOVA tumour growth was comparable with the tumour growth in untreated mice (Fig. 5a). CD8 DC treatment resulted in a significant but temporary inhibition of the EL-4-mOVA tumour, but not the EL-4 tumour. Importantly, treatment with mcDC resulted in specific rejection of the EL-4-mOVA tumour (Fig. 5a). The observed tumour rejection was complete, as parallel studies using mice that received EL-4-mOVA tumours (but not EL-4 tumours) did not show tumour re-occurrences or metastases for >70 days after mcDC treatment (Fig. 5b and data not shown).
Fig. 5.
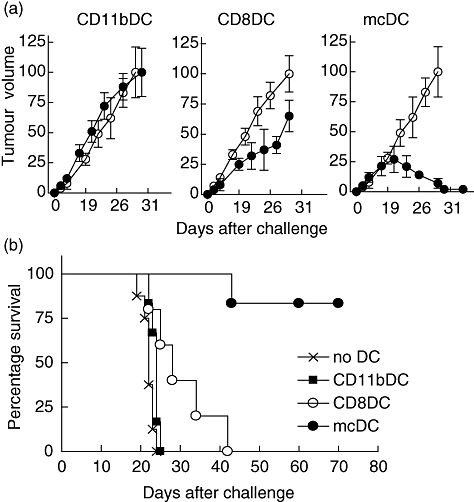
Merocytic dendritic cells (mcDCs) are superior for therapeutic intervention. Naive mice were inoculated subcutaneously with 2 × 106 EL-4-mOVA cells on the left flank, and 2 × 106 EL-4-parental cells on the right flank. As soon as palpable tumour had formed, mice received subsorted DC from Fms-like tyrosine kinase 3-ligand (FLT3L)-treated mice that had been exposed to irradiated ActmOVA-Kbm1 cells in vitro. (a) Tumour growth of both tumours upon treatment with indicated DC populations (five mice per group). (b) EL-4-mOVA tumour-bearing mice were treated with indicated DC from FLT3L-treated mice and long-term survival was monitored (five to nine mice per group).
Discussion
In this study we show that the beneficial effects of FLT3L administration before treatment with autologous tumour vaccine result predominantly from the increase of CD8 DCs and mcDC, two specific DC populations that have the capacity to (cross)-present cell-associated antigens to T cells in an NK-independent fashion. Interestingly, FLT3L treatment solely augmented the numbers of these DC populations, but did not change the activation status of DCs upon interaction with tumour cell vaccines or their capacity to prime antigen-specific CD4+ and CD8+ T cells. This was also evidenced by the fact that T cell priming was equally efficient by DCs derived from PBS- and FLT3L-treated mice.
FLT3L is essential for DC development. Its receptor, FLT3, a type-III receptor tyrosine kinase, is expressed continuously from progenitor cells to steady-state DC. The development from precursor into specific DC subpopulation may be both stochastic or defined by cytokines and other extrinsic factors [15,36]. Previously it has been shown that FLT3L of mice treatment results in massive expansion of the pDC and CD8 DC populations [33,34]. Here we show that the recently described mcDC expand to a similar degree.
pDC are known for their capacity to produce type I IFN upon infection of the host and are generally considered poor presenters of cell-associated antigens. Recent studies showed that human pDC have the capacity to prime T cells to cell-associated antigens, especially in the context of infection or Toll-like receptor (TLR) ligation. pDC have been implicated in the development of autoimmune diseases where type I IFN production is thought to amplify the immune responses to self. Conversely, pDC have also been shown to suppress ongoing immune responses through their production of immune suppressive molecules such as IL-10 or indoleamine-2,3 dioxygenase (IDO), or signalling via the PD-L1–PD-1 or inducible co-stimulator–inducible co-stimulator ligand (ICOS–ICOSL) pathways (reviewed in [46]).
In our studies, pDC showed some capacity for uptake of apoptotic materials and subsequent type I IFN production. However, pDC failed to prime T cells in vitro and in vivo. In addition, OT-1 and OT-2 T cells cultured with pDC did not express activation markers such as CD69/CD44 (data not shown), suggesting that in this setting the lack of T cell responses did not result from induction of anergy or tolerance but rather from a lack of activation. Although pDC did not seem to have a direct effect on anti-tumour vaccine priming, it is extremely possible that they amplified indirectly the immune response through their production of type I IFNs that have been shown to induce DC maturation, enhance antigen processing and presentation and enhance T cell recruitment, proliferation and accumulation by inhibition of apoptosis [47–51].
CD8 DCs are considered the classic cross-presenting DC and, for a long time, have been assumed to be the only mouse DC population with the ability to cross-present cell-associated antigens to CD8+ T cells. CD8 DCs display more efficient phagocytic uptake of dead cells and loading of antigenic peptides into MHC class I than many other DC populations. In addition, CD8 DCs are able to produce high levels of bioactive IL-12p70 that helps in their induction of Th1/Tc1 responses. However, their capacity to present antigens in MHC class II to CD4+ T cells under conditions of limiting antigen is relatively poor (reviewed in [52]).
Our studies show that FLT3L treatment greatly expanded the recently described mcDC population, that potently primes both CD4+ and CD8+ T cell to cell-associated antigens [12,23]. Importantly, T cells primed to cell-associated antigens by mcDC displayed greater primary expansion and development into memory cells than those primed by other DC populations.
The superior T cell priming capacity of mcDC can be contributed to several mechanisms. mcDC store phagoytosed materials in non-acid organelles and use this as an antigen depot which allows for prolonged antigen presentation [24]. Increasing the length of antigenic stimulation has been shown to positively affect T cell expansion, acquisition of effector functions and memory development [53–56]. Secondly, the type I IFN production by mcDC upon uptake of apoptotic material is likely to provide an adjuvant effect in both an autocrine and paracrine fashion (manuscript in preparation). Moreover, our previous observations indicated that mice deficient in type I IFN sensing failed to induce protective CD8+ T cell responses when treated with autologous tumour vaccines [12,23]. Besides the production of type I IFN, the mcDCs capacity to prime strong CD4+ T cell responses to cell-associated antigens is also instrumental in the induction of anti-tumour CD8+ T cell responses. We and others have shown that CD4+ T cell help during priming of CD8+ T cells is required for optimal CD8+ T cell activation, primary expansion, acquisition of effector function and the development of memory [42,57,58]. Supportively, increasing CD4+ T cell help through transfer of (transgenic) CD4+ T cells or preimmunization of mice enhances the induction of CD8+ T cell responses [59,60]. In addition, ample studies indicate that CD4+ T cell help plays a supporting role in the maintenance, reactivation and expansion of existing memory cells [61–63].
FLT3L was shown recently to increase a DC population that had the ability to cross-present cell-associated antigens to CD8+ T cells without the need to express CD8α[64]. These cells converted into CD8 DCs without dividing upon transfer in vivo or manipulation in vitro, suggesting that these cells could be immediate precursors of CD8 DCs (preCD8 DCs). Due to their increased lifespan compared to CD8 DCs, the preCD 8DCs displayed an increased capacity to prime CD8+ T cells [64]. In contrast to preCD8 DCs, mcDCs do not convert into CD8 DCs upon transfer in vivo and have a similar lifespan as CD8 DCs [24]. Moreover, their type I IFN production upon uptake of apoptotic material and generation of antigen depots in non-acidic organelles are characteristic features of mcDC that are essential for their T cell priming capacity [24]. Based on these functional data, mcDC seem to represent a distinct DC population, but further elucidation of their developmental pathways and lineage commitment may demonstrate a close relationship to other DC populations with cross-priming capacities.
Given the therapeutic potential of the mcDC, it will be of extreme interest to identify the human equivalent of this population. Recent publications discussing the capacity of human pDC and CD141+ DC to present cell-associated antigens in the presence and absence of infection [18,65–69] indicate that novel human DC subpopulations or new functions within existing populations remain to be discovered.
Collectively, our data suggest that FLT3L expands DC populations with capacity to (cross)-present cell-associated antigens while having a limited effect on DC populations that are associated with the induction of tolerance (such as CD11b DCs). The expansion of CD8 DCs will be beneficial in the induction of CD8+ T cell responses, whereas mcDC will increase both CD8+ and CD4+ T cell responses. Selective targeting to especially mcDC or instilling mcDC ‘traits’ into conventional DC populations could enhance tumour vaccine efficacy significantly.
Acknowledgments
We would like to thank Amgen for the rhFLT3L and Dr K. Prilliman for critical reading of the manuscript. This work is supported by NIH/NIAID grant AI079545 and NIH/NCI grant CA138617 to EMJ.
Disclosure
None.
References
- 1.Aymeric L, Apetoh L, Ghiringhelli F, et al. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res. 2010;70:855–8. doi: 10.1158/0008-5472.CAN-09-3566. [DOI] [PubMed] [Google Scholar]
- 2.Palucka K, Ueno H, Zurawski G, Fay J, Banchereau J. Building on dendritic cell subsets to improve cancer vaccines. Curr Opin Immunol. 2010;22:258–63. doi: 10.1016/j.coi.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–26. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 4.Petersen TR, Dickgreber N, Hermans IF. Tumor antigen presentation by dendritic cells. Crit Rev Immunol. 2010;30:345–86. doi: 10.1615/critrevimmunol.v30.i4.30. [DOI] [PubMed] [Google Scholar]
- 5.Palucka AK, Ueno H, Fay J, Banchereau J. Dendritic cells: a critical player in cancer therapy? J Immunother. 2008;31:793–805. doi: 10.1097/CJI.0b013e31818403bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vremec D, Pooley J, Hochrein H, Wu L, Shortman K. CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J Immunol. 2000;164:2978–86. doi: 10.4049/jimmunol.164.6.2978. [DOI] [PubMed] [Google Scholar]
- 7.Vremec D, Zorbas M, Scollay R, et al. The surface phenotype of dendritic cells purified from mouse thymus and spleen: investigation of the CD8 expression by a subpopulation of dendritic cells. J Exp Med. 1992;176:47–58. doi: 10.1084/jem.176.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Keeffe M, Hochrein H, Vremec D, et al. Mouse plasmacytoid cells: long-lived cells, heterogeneous in surface phenotype and function, that differentiate into CD8(+) dendritic cells only after microbial stimulus. J Exp Med. 2002;196:1307–19. doi: 10.1084/jem.20021031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamath AT, Henri S, Battye F, Tough DF, Shortman K. Developmental kinetics and lifespan of dendritic cells in mouse lymphoid organs. Blood. 2002;100:1734–41. [PubMed] [Google Scholar]
- 10.Grouard G, Rissoan MC, Filgueira L, Durand I, Banchereau J, Liu YJ. The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J Exp Med. 1997;185:1101–11. doi: 10.1084/jem.185.6.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belz GT, Bedoui S, Kupresanin F, Carbone FR, Heath WR. Minimal activation of memory CD8(+) T cell by tissue-derived dendritic cells favors the stimulation of naive CD8(+) T cells. Nat Immunol. 2007;8:1060–6. doi: 10.1038/ni1505. [DOI] [PubMed] [Google Scholar]
- 12.Janssen E, Tabeta K, Barnes MJ, et al. Efficient T cell activation via a Toll-interleukin 1 receptor-independent pathway. Immunity. 2006;24:787–99. doi: 10.1016/j.immuni.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 13.Caminschi I, Ahmet F, Heger K, et al. Putative IKDCs are functionally and developmentally similar to natural killer cells, but not to dendritic cells. J Exp Med. 2007;204:2579–90. doi: 10.1084/jem.20071351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu CH, Miyake Y, Kaise H, Kitamura H, Ohara O, Tanaka M. Novel subset of CD8{alpha}+ dendritic cells localized in the marginal zone is responsible for tolerance to cell-associated antigens. J Immunol. 2009;182:4127–36. doi: 10.4049/jimmunol.0803364. [DOI] [PubMed] [Google Scholar]
- 15.Liu K, Nussenzweig MC. Origin and development of dendritic cells. Immunol Rev. 2010;234:45–54. doi: 10.1111/j.0105-2896.2009.00879.x. [DOI] [PubMed] [Google Scholar]
- 16.Allan RS, Waithman J, Bedoui S, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25:153–62. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 17.Belz GT, Shortman K, Bevan MJ, Heath WR. CD8alpha+ dendritic cells selectively present MHC class I-restricted noncytolytic viral and intracellular bacterial antigens in vivo. J Immunol. 2005;175:196–200. doi: 10.4049/jimmunol.175.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoeffel G, Ripoche AC, Matheoud D, et al. Antigen crosspresentation by human plasmacytoid dendritic cells. Immunity. 2007;27:481–92. doi: 10.1016/j.immuni.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 19.Williams CA, Harry RA, McLeod JD. Apoptotic cells induce dendritic cell-mediated suppression via interferon-gamma-induced IDO. Immunology. 2008;124:89–101. doi: 10.1111/j.1365-2567.2007.02743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng YF, Elkon KB. Peripheral CD8 T-cell responses to apoptotic cell proteins and peptides. Crit Rev Immunol. 2007;27:357–65. doi: 10.1615/critrevimmunol.v27.i4.50. [DOI] [PubMed] [Google Scholar]
- 21.Ferguson TA, Kazama H. Signals from dying cells: tolerance induction by the dendritic cell. Immunol Res. 2005;32:99–108. doi: 10.1385/IR:32:1-3:099. [DOI] [PubMed] [Google Scholar]
- 22.Saas P, Bonnefoy F, Kury-Paulin S, Kleinclauss F, Perruche S. Mediators involved in the immunomodulatory effects of apoptotic cells. Transplantation. 2007;84:S31–4. doi: 10.1097/01.tp.0000269113.59857.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katz JD, Ondr JK, Opoka RJ, Garcia Z, Janssen EM. Cutting edge: merocytic dendritic cells break T cell tolerance to {beta} cell antigens in nonobese diabetic mouse diabetes. J Immunol. 2010;185:1999–2003. doi: 10.4049/jimmunol.1001398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reboulet R, Hennies CM, Garcia Z, Nierkens S, Janssen EM. Prolonged antigen storage endows merocytic dendritic cells with enhanced capacity to prime anti-tumor responses in tumor-bearing mice. J Immunol. 2010;185:3337–47. doi: 10.4049/jimmunol.1001619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curran MA, Allison JP. Tumor vaccines expressing flt3 ligand synergize with ctla-4 blockade to reject preimplanted tumors. Cancer Res. 2009;69:7747–55. doi: 10.1158/0008-5472.CAN-08-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramakrishna E, Woller N, Mundt B, et al. Antitumoral immune response by recruitment and expansion of dendritic cells in tumors infected with telomerase-dependent oncolytic viruses. Cancer Res. 2009;69:1448–58. doi: 10.1158/0008-5472.CAN-08-1160. [DOI] [PubMed] [Google Scholar]
- 27.King GD, Kroeger KM, Bresee CJ, et al. Flt3L in combination with HSV1-TK-mediated gene therapy reverses brain tumor-induced behavioral deficits. Mol Ther. 2008;16:682–90. doi: 10.1038/mt.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chakravarty PK, Guha C, Alfieri A, et al. Flt3L therapy following localized tumor irradiation generates long-term protective immune response in metastatic lung cancer: its implication in designing a vaccination strategy. Oncology. 2006;70:245–54. doi: 10.1159/000096288. [DOI] [PubMed] [Google Scholar]
- 29.Kutzler MA, Weiner DB. Developing DNA vaccines that call to dendritic cells. J Clin Invest. 2004;114:1241–4. doi: 10.1172/JCI23467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Braun SE, Chen K, Blazar BR, et al. Flt3 ligand antitumor activity in a murine breast cancer model: a comparison with granulocyte-macrophage colony-stimulating factor and a potential mechanism of action. Hum Gene Ther. 1999;10:2141–51. doi: 10.1089/10430349950017130. [DOI] [PubMed] [Google Scholar]
- 31.Lynch DH, Andreasen A, Maraskovsky E, Whitmore J, Miller RE, Schuh JC. Flt3 ligand induces tumor regression and antitumor immune responses in vivo. Nat Med. 1997;3:625–31. doi: 10.1038/nm0697-625. [DOI] [PubMed] [Google Scholar]
- 32.Silver DF, Hempling RE, Piver MS, Repasky EA. Flt-3 ligand inhibits growth of human ovarian tumors engrafted in severe combined immunodeficient mice. Gynecol Oncol. 2000;77:377–82. doi: 10.1006/gyno.2000.5782. [DOI] [PubMed] [Google Scholar]
- 33.Brasel K, McKenna HJ, Morrissey PJ, et al. Hematologic effects of flt3 ligand in vivo in mice. Blood. 1996;88:2004–12. [PubMed] [Google Scholar]
- 34.Maraskovsky E, Brasel K, Teepe M, et al. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J Exp Med. 1996;184:1953–62. doi: 10.1084/jem.184.5.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKenna HJ, Stocking KL, Miller RE, et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. 2000;95:3489–97. [PubMed] [Google Scholar]
- 36.Schmid MA, Kingston D, Boddupalli S, Manz MG. Instructive cytokine signals in dendritic cell lineage commitment. Immunol Rev. 2010;234:32–44. doi: 10.1111/j.0105-2896.2009.00877.x. [DOI] [PubMed] [Google Scholar]
- 37.Ehst BD, Ingulli E, Jenkins MK. Development of a novel transgenic mouse for the study of interactions between CD4 and CD8 T cells during graft rejection. Am J Transplant. 2003;3:1355–62. doi: 10.1046/j.1600-6135.2003.00246.x. [DOI] [PubMed] [Google Scholar]
- 38.Lyons AB. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J Immunol Methods. 2000;243:147–54. doi: 10.1016/s0022-1759(00)00231-3. [DOI] [PubMed] [Google Scholar]
- 39.Jiang Z, Georgel P, Du X, et al. CD14 is required for MyD88-independent LPS signaling. Nat Immunol. 2005;6:565–70. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 40.Nikolic-Zugic J, Carbone FR. The effect of mutations in the MHC class I peptide binding groove on the cytotoxic T lymphocyte recognition of the Kb-restricted ovalbumin determinant. Eur J Immunol. 1990;20:2431–7. doi: 10.1002/eji.1830201111. [DOI] [PubMed] [Google Scholar]
- 41.Benedict CA, Loewendorf A, Garcia Z, Blazar BR, Janssen EM. Dendritic cell programming by cytomegalovirus stunts naive T cell responses via the PD-L1/PD-1 pathway. J Immunol. 2008;180:4836–47. doi: 10.4049/jimmunol.180.7.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–6. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 43.McBride S, Hoebe K, Georgel P, Janssen E. Cell-associated double-stranded RNA enhances antitumor activity through the production of type I IFN. J Immunol. 2006;177:6122–8. doi: 10.4049/jimmunol.177.9.6122. [DOI] [PubMed] [Google Scholar]
- 44.Colucci F, Di Santo JP. The receptor tyrosine kinase c-kit provides a critical signal for survival, expansion, and maturation of mouse natural killer cells. Blood. 2000;95:984–91. [PubMed] [Google Scholar]
- 45.Williams NS, Klem J, Puzanov IJ, Sivakumar PV, Bennett M, Kumar V. Differentiation of NK1.1+, Ly49+ NK cells from flt3+ multipotent marrow progenitor cells. J Immunol. 1999;163:2648–56. [PubMed] [Google Scholar]
- 46.Swiecki M, Colonna M. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol Rev. 2010;234:142–62. doi: 10.1111/j.0105-2896.2009.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–8. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 48.Hoebe K, Janssen EM, Kim SO, et al. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat Immunol. 2003;4:1223–9. doi: 10.1038/ni1010. [DOI] [PubMed] [Google Scholar]
- 49.Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med. 1999;189:521–30. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ogasawara K, Hida S, Weng Y, et al. Requirement of the IFN-alpha/beta-induced CXCR3 chemokine signalling for CD8+ T cell activation. Genes Cells. 2002;7:309–20. doi: 10.1046/j.1365-2443.2002.00515.x. [DOI] [PubMed] [Google Scholar]
- 51.Le Bon A, Tough DF. Type I interferon as a stimulus for cross-priming. Cytokine Growth Factor Rev. 2008;19:33–40. doi: 10.1016/j.cytogfr.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 52.Shortman K, Heath WR. The CD8+ dendritic cell subset. Immunol Rev. 2010;234:18–31. doi: 10.1111/j.0105-2896.2009.00870.x. [DOI] [PubMed] [Google Scholar]
- 53.Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol. 2001;2:415–22. doi: 10.1038/87720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spierings DC, Lemmens EE, Grewal K, Schoenberger SP, Green DR. Duration of CTL activation regulates IL-2 production required for autonomous clonal expansion. Eur J Immunol. 2006;36:1707–17. doi: 10.1002/eji.200635929. [DOI] [PubMed] [Google Scholar]
- 55.van Stipdonk MJ, Hardenberg G, Bijker MS, et al. Dynamic programming of CD8+ T lymphocyte responses. Nat Immunol. 2003;4:361–5. doi: 10.1038/ni912. [DOI] [PubMed] [Google Scholar]
- 56.van Stipdonk MJ, Lemmens EE, Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol. 2001;2:423–9. doi: 10.1038/87730. [DOI] [PubMed] [Google Scholar]
- 57.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science (NY) 2003;300:339–42. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science (NY) 2003;300:337–9. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 59.Hamilton-Williams EE, Lang A, Benke D, Davey GM, Wiesmuller KH, Kurts C. Cutting edge: TLR ligands are not sufficient to break cross-tolerance to self-antigens. J Immunol. 2005;174:1159–63. doi: 10.4049/jimmunol.174.3.1159. [DOI] [PubMed] [Google Scholar]
- 60.Krawczyk CM, Shen H, Pearce EJ. Memory CD4 T cells enhance primary CD8 T-cell responses. Infect Immun. 2007;75:3556–60. doi: 10.1128/IAI.00086-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ryu SJ, Jung KM, Yoo HS, et al. Cognate CD4 help is essential for the reactivation and expansion of CD8 memory T cells directed against the hematopoietic cell-specific dominant minor histocompatibility antigen, H60. Blood. 2009;113:4273–80. doi: 10.1182/blood-2008-09-181263. [DOI] [PubMed] [Google Scholar]
- 62.Novy P, Quigley M, Huang X, Yang Y. CD4 T cells are required for CD8 T cell survival during both primary and memory recall responses. J Immunol. 2007;179:8243–51. doi: 10.4049/jimmunol.179.12.8243. [DOI] [PubMed] [Google Scholar]
- 63.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. 2004;5:927–33. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bedoui S, Prato S, Mintern J, et al. Characterization of an immediate splenic precursor of CD8+ dendritic cells capable of inducing antiviral T cell responses. J Immunol. 2009;182:4200–7. doi: 10.4049/jimmunol.0802286. [DOI] [PubMed] [Google Scholar]
- 65.Bachem A, Guttler S, Hartung E, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med. 2010;207:1273–81. doi: 10.1084/jem.20100348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crozat K, Guiton R, Contreras V, et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med. 2010;207:1283–92. doi: 10.1084/jem.20100223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jongbloed SL, Kassianos AJ, McDonald KJ, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. 2010;207:1247–60. doi: 10.1084/jem.20092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Di Pucchio T, Chatterjee B, Smed-Sorensen A, et al. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat Immunol. 2008;9:551–7. doi: 10.1038/ni.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mouries J, Moron G, Schlecht G, Escriou N, Dadaglio G, Leclerc C. Plasmacytoid dendritic cells efficiently cross-prime naive T cells in vivo after TLR activation. Blood. 2008;112:3713–22. doi: 10.1182/blood-2008-03-146290. [DOI] [PMC free article] [PubMed] [Google Scholar]