

Abstract
Significant effort continues to be exerted toward the improvement of transfection mediated by nonviral vectors. These endeavors are often focused on the design of particulate carriers with properties that encourage efficient accumulation at the membrane surface, particle uptake, and endosomal escape. Despite its demonstrated importance in successful nonviral transfection, relatively little investigation has been done to understand the pressures driving internalized vectors into favorable nondegradative endocytic pathways. Improvements in transfection efficiency have been noted for complexes delivered with a substrate-mediated approach, but the reasons behind such enhancements remain unclear. The phenotypic changes exhibited by cells interacting with nano- and micro-featured substrates offer hints that may explain these effects. This review describes nanoscale particulate and substrate parameters that influence both the uptake of nonviral gene carriers and the endocytic phenotype of interacting cells, and explores the molecular links that may mediate these interactions. Substrate-mediated control of endocytosis represents an exciting new design parameter that will guide the creation of efficient transgene carriers.
Keywords: Endocytosis, Transfection, Nanotopography, Microtopography, Cell-substrate interactions, Nonviral gene delivery, Nanoparticles, Substrate-mediated
1. Introduction
Gene transfer techniques and therapies have enjoyed steady interest due to their current and potential application against a diversity of human illnesses [1] including diabetes, hemophilia, peanut anaphylaxis, and cystic fibrosis. In an effort to increase transfection efficiency and specificity, researchers and physicians often turn to carrier systems to deliver engineered genetic material to target cells and tissues. Generally, such carriers fall into one of two broad categories – viral or nonviral vectors (though the distinction may be blurred for virus-like particles). Viruses are naturally professional gene therapists, and have been reengineered to carry a myriad of therapeutic gene sequences. Despite their high efficiency, there are a number of drawbacks associated with viral vectors: they are typically more immunogenic than their nonviral counterparts, limited in DNA packaging capacity, and susceptible to shutdown of transgene expression due to immune response. These potential issues motivate the ongoing search for suitable alternatives, frequently in the form of particulate polymer- and lipid-DNA complexes, which are less toxic, able to carry larger genes, and amenable to formulation optimization such as prolonged circulation, targeted delivery, and storage stability.
In spite of their wide application, the mechanisms whereby DNA complexes are able to traverse the cellular, lysosomal, and nuclear membrane barriers to then induce transgene expression are only recently being elucidated, often with contradictory results for seemingly similar carriers. Efficient cellular internalization of the carrier-DNA complex is crucial to nonviral gene transfer. Increasing consideration has been given to nanoscale particulate parameters including size, shape, ligand decoration, and surface charge. These parameters have been shown to dictate the extent and pathway of endocytic uptake, and the subsequent ability of the DNA to arrive intact within the nucleus, able to induce transgene expression.
While clearly important for successful nonviral transfection, particulate parameters do not fully account for the differences in transfection efficiency in vitro and in vivo; particle- and cell-substrate interactions have also been demonstrated to influence the uptake and expression of particulate nonviral vectors. When delivered from a surface, the ability of nonviral particles to induce gene expression depends not only on their local concentration, but also on the tightness of their adsorption, the presence of extracellular matrix (ECM) proteins, and substrate surface chemistry. Substrates with micro- and nano-patterned topographies could also directly influence the endocytic behavior and transfectability of interacting cells by inducing changes in proliferation, spreading, morphology, cytoskeletal arrangement, differentiation, and protein expression. Though potential molecular links have been described by molecular biologists, little is known about the functional interactions between DNA complexes, cell substrates, focal adhesions, and the cytoskeletal and endocytic machineries; this presents an exciting opportunity for the design of particles and substrates that are able to probe and exploit beneficial aspects of the endocytic process.
While nanoscale particulate parameters gain prominence, the contribution of nanoscale substrate-mediated effects on DNA complexes and cells that interact with them is generally ignored during the optimization of nonviral gene carriers. In this review we highlight the body of evidence supporting the importance of nanoscale particulate parameters for gene delivery, and also prospect routes whereby nanoscale substrate parameters may influence the uptake, processing, and expression of these particles. A full understanding of the interaction of cells with nonviral gene carriers depends not only on an awareness of particulate parameters and their implications on endocytosis and transgene expression, but also on a clear knowledge of the substrative context that the particles are presented.
2. Barriers to gene transfer
DNA complexes must overcome a series of barriers to gain access to the membrane surface, cytoplasmic compartment, and nucleus of a target cell, and to translate transgenes into protein (Figure 1). As particles encounter each of these barriers, they are subject to a certain probability of success or failure in overcoming each. The cumulative probability of success for the entire journey is reflected in the transfection efficiency for a given system. Certain portions of the trip may be more limiting than others for a given carrier system. If an easily-crossed barrier is not subject to saturation and is upstream of a bottleneck barrier, increasing the efficiency of crossing the upstream barrier will help to increase the number of particles that appear downstream of the limiting barrier.
Figure 1. Barriers to nonviral gene delivery.

(1) Transgenic DNA can be lost due to incomplete complexation with cationic polymer/lipid. (2) Complexes may be cleared from the circulation before they are able to bind to the cell surface. (3) Some of the complexes bound to the cell surface will not be internalized. (4) Following endocytosis, a portion of DNA may be degraded within the acidic late endosomes and lysosomes. (5) DNA successfully escaping the endosomal compartment may be further degraded by cytoplasmic DNAse. (6) A portion of the DNA reaching the nucleus may be unable to induce transcription. (7) Some of the exported mRNA may be incapable of translation into useful transgenic protein.
This probabilistic way of thinking about nonviral gene delivery is supported by a study finding that noncoding DNA can enhance the efficiency of transfection by polyethylenimine- (PEI) DNA complexes [2]. A constant quantity of reporter plasmid was diluted with noncoding “junk” DNA, followed by delivery of either one population of particles containing both coding and noncoding DNA, or co-delivery of two different particle populations – one containing coding DNA and one without. It is important to note that because the total amount of coding DNA remains the same in each case, the number of particles including coding DNA is increased in the case of particles formed with both coding and noncoding DNA. The mean expression per cell was not affected, but the number of expressing cells was increased when a larger number of coding particles were delivered. The authors suggest that fusion with the nucleus is a very inefficient process, so increasing the number of coding particles can overcome this bottleneck by increasing the occurrence of this rare event.
The formation of DNA complexes usually proceeds by condensation of anionic DNA with cationic lipid (lipoplex) or polymer (polyplex); these interactions must be strong enough to keep particles stable during exposure to charged serum components. The next barriers facing systemically delivered particles are extracellular, and include: serum proteases and nucleases, variations in pH, opsonization, and clearance during passage through the kidneys and liver [3]. Upon arrival near the cell, complexes must associate with the cell surface, either through electrostatic interactions, physical concentration at the cell substrate via adsorption, or by ligand-receptor binding. Originally, it was thought that lipoplexes could then enter target cells by direct fusion with the cell membrane. It is now well-accepted that both nonspecifically- and ligand-bound complexes enter cells principally via endocytic processes [4] (considered in detail in the following section).
After escape from the endocytic compartment, the complexes must gain entry to the nucleus, and unpack their DNA cargo. Upon arrival at this step, it becomes a liability for carriers to bind their cargo too tightly; overbinding prohibits access by the translational machinery (striking an appropriate balance between protection and release has been reviewed by Grigsby et al. [5]). Indeed, it is worth mentioning that translational inefficiencies may generally be one of the most rate-limiting obstacles in nonviral gene delivery. In one excellent exploration of this barrier, the efficiency of each step in transfection mediated by adenovirus and lipofectamine (LF) was compared [6–7]. LF was shown to require a dose three thousand times higher than that delivered by adenovirus to support the same level of expression. Though LF encouraged higher levels of DNA uptake on a per-carrier basis, adenovirus was eight thousand times more efficient at completing transcription and translation of the transgenes delivered to the nucleus. PEI polyplexes have been observed to unpack more efficiently compared to DOTAP lipoplexes following direct injection into the nucleus, lowering the translational barrier for this polyplex upon arrival in the nucleus [8].
3. Endocytic pathways involved in transfection
A basic discussion of relevant endocytic pathways is required to describe the uptake of nonviral vectors. All cells perform some form of endocytosis to maintain the homeostasis of intracellular species. Endocytosis is broadly divided here into clathrin-and caveolae-mediated endoctysosis, and fluid-phase macropinocytosis (Figure 2). These three pathways are not inclusive of all the forms of endocytic uptake of nonviral carriers [9], but are the most studied in this context, and are the focus of this review.
Figure 2. Endocytic pathways traversed by nonviral carriers.

Cationic particles bind to anionic heparan sulfate proteoglycans (HSPGs) and may be internalized via macropinocytosis (A), a form of fluid-phase endocytosis. Macropinosomes are fluid-filled vesicles formed by actin-driven membrane ruffling; these vesicles may fuse with degradative late endosomes, or may be trafficked directly to the nucleus. Nonviral vectors can also be internalized by clathrin-mediated endocytosis (B), which progresses by receptor clustering, formation of the clathrin coat, and actin-driven internalization, forming early endosomes. Some early endosomes are recycled to the cell surface, while others are uncoated, acidified, and progress to late endosomes and lysosomes on their way to the nucleus. Caveolae-mediated endocytosis (C) proceeds by oligomerization of caveolin, actin-dependent internalization of caveolae to form cavicles, and merger with the degradative lysosomal compartment, or non-degradative trafficking to the nucleus via caveosomes. Each pathway relies on microtubules for rapid transport of endocytic vesicles.
Clathrin-mediated endocytosis is the most well-understood endocytic pathway [10], and is involved in nutrient uptake and signal transduction through internalization of ligand-bound receptors [3]. Low density lipoprotein (LDL), transferrin (Tf), and epidermal growth factor (EGF) are prototypic species transported via clathrin-mediated endocytosis. Upon receptor-ligand binding, the receptors cluster in clathrin-coated pits, mediated by the adapter protein AP-2 [11]. Dynamin, a GTPase, then frees the coated pit into the cell interior, fusing with and forming early endosomes [4, 10]. Depending on the cargo molecule, early endosomes are either uncoated and trafficked to acidic lysosomes via microtubule transport, or shuttled back to the cell surface via recycling endosomes. Exposure to the acidic and degradative lysosomal compartment reduces the transfection efficiency of nonviral vectors. Therefore, enhanced escape from the acidic endosomes by the proton sponge effect [12], or by chemical and physical endosomolytic agents, have been pursued to help surmount this barrier [13]. Clathrin-mediated endocytosis is synonymous with “receptor-mediated endocytosis” in the literature, but this terminology has become antiquated with the discovery that other forms of endocytosis also proceed by ligand-receptor binding.
Caveolae-mediated endocytosis is associated with the uptake of glycosphingolipids, and is involved with transcytosis of serum proteins across endothelium [11]. Caveolae are caveolin-coated, cholesterol- and sphingolipid-enriched flask-shaped invaginations in the cell membrane. These structures are relatively static compared to clathrin-coated pits, and generally proceed via slower uptake kinetics compared to clathrin-mediated endocytosis [14]. Upon dynamin- and actin-mediated uptake, cavicles are shuttled toward the nucleus via microtubules. Importantly for nonviral gene delivery, certain forms of caveolae-mediated endocytosis are thought to avoid the degradative lysosomal compartment [15]. Caveolae-mediated endocytosis has been shown to be initiated by viruses binding to and clustering integrins, resulting in their uptake [16] in a manner similar to that observed in the ligand-receptor binding of clathrin-mediated endocytosis.
Macropinocytosis, a form of fluid-phase endocytosis, is the uptake of fluid and solutes by actin-driven ruffling of the plasma membrane. Macropinocytosis can be induced by platelet derived growth factor (PDGF) activating Rho-family GTPases, which trigger actin assembly and internalization of surrounding extracellular fluid [11]. Long-range transport of macropinosomes along microtubules is inhibited by nocodazole. The rate of macropinocytosis of a solute is typically proportional to its concentration in solution (non-saturable kinetics). Like caveolae-mediated endocytosis, particles endocytosed by macropinocytosis may bypass the lysosomal compartment, making it an attractive pathway for efficient nonviral gene delivery.
4. Dependence of nonviral transfection on endocytosis
A number of studies have unambiguously implicated macropinocytosis and clathrin- and caveolae-mediated endocytosis as necessary processes for the uptake and subsequent expression of both poly- and lipoplexes. The contribution of each of these pathways also varies by cell type and cargo identity. The degradative processes following particle uptake vary by endocytic pathway and have been shown to be important barriers to nonviral gene delivery.
Clathrin-mediated endocytosis has been demonstrated to support transfection by lipoplexes through studies employing electron microscopy and co-localization with labeled transferrin [17]. Cells subject to inhibition of clathrin-mediated endocytosis by potassium or cholesterol depletion and those expressing dominant negative mutant Eps15 do not internalize lipoplexes, and subsequently support lower levels of transgene expression (wild-type Eps15 allows docking of AP-2 to the plasma membrane, which goes on to assemble clathrin-coated pits) [18]. Endocytosed lipoplexes eventually co-localize with acidic lysosomes stained with LysoTracker, providing evidence that particles taken up by this pathways are subject to low pH [19]. The use of nocodazole to interfere with microtubule function has also been shown to increase the nuclear accumulation and transgene expression of lipoplexes [20]. The authors suggest that because nocodazole uncouples endosomes from their trafficking to the lysosomal compartment [21], lipoplexes are able to skirt degradative processing following endocytosis, thereby increasing their transfection efficiency.
The ability of a nonviral vector to escape from the endosomal compartment determines that carrier’s transfection ability. Polyplexes and lipoplexes are believed to escape endosomes using different mechanisms. Carriers with a strong ability to buffer the influx of protons during endosome acidification increase the accumulation of H+ and Cl− ions and osmotic pressure within the vesicles, eventually leading to bursting and vector escape [10]. Inhibition of the activity of proton pumps decreases the transfection efficiency of PEI polyplexes [22], and endosomes have been observed to accumulate greater amounts of Cl− and swelling after delivery of highly-buffering polyplexes [12]. In contrast to this “proton sponge effect”, lipoplexes containing lipids that encourage formation of nonlamellar phases may escape endosomes through direct fusion and release into the cytosol [23]. Nonviral carriers must have the ability to escape the endocytic vesicles encountered along the endocytic pathway(s) they traverse.
A study relying on direct microscopic visualization has elucidated the relationship between endocytosis and transfection efficiency of PEI polyplexes, through observation of the uptake of polyplexes stained with YOYO-1 (a fluorescent green DNA intercalator) by fibroblasts labeled with FM4–64 (a lipophilic stain which fluoresces red upon binding the outer leaf of cell membranes) [24]. Labeled particles were observed to co-localize strongly with the membrane marker for endocytosis for the duration of the transfection process. Since endocytic uptake of the cell membrane and the macropinocytic uptake of extracellular fluid proceed by the same intracellular pathways, the polyplexes in this study were believed to be taken up by fluid-phase endocytosis. Given the co-localization persisted through maturation of the early endosomes into lysosomes, a large fraction of the complexes were sequestered in the endocytic compartment, with only a small population escaping to the nucleus to induce transgene expression.
Douglas et al. noted cell-line dependent differences in the endocytic processes of 293T, COS7, and CHO cells as the cause for varying levels of transfection using identical preparations of alginate-chitosan-DNA nanoparticles [25]. By measuring the variable inhibition of particle uptake by the clathrin-mediated inhibitor chlorpromazine and caveolin-mediated inhibitor genistein, the authors were able to determine that both routes were used for COS7 and 293T cells, whereas CHO cells endocytosed particles by a clathrin-independent mechanism. Furthermore, the induction of macropinocytosis by phorbol myristate acetate (PMA) did not result in an increase in complex internalization for any cell type. For COS7 and 293T cells, transfection was only supported by particles taken up by clathrin-mediated endocytosis, whereas CHO cells did not produce significant transgene product, possibly because they lack the mannose receptor indicated in the clathrin-mediated uptake of chitosan. In this case, clathrin-mediated endocytosis was thought to be superior because the particles were visualized escaping the acidic lysosomes, presumably via the proton sponge effect, whereas particles trafficked to non-degradative caveosomes lacked an escape mechanism and were therefore sequestered. These results highlight the dependence of trafficking on particle and cell type.
Caveolae- and clathrin-mediated endocytosis have been demonstrated to be required for uptake and expression of polyplexes and lipoplexes, respectively, in a single study [14]. The internalization of DOTAP lipoplexes was inhibited by chlorpromazine and potassium depletion (clathrin-mediated) but was not affected by filipin and genistein (caveolae-mediated), and uptake of PEI polyplexes was down-regulated by all four inhibitors. Transfection by DOTAP particles was also abolished by inhibitors of clathrin-mediated endocytosis, whereas transfection by PEI particles was only inhibited by removal of caveolae-mediated uptake. Rejman et al. suggest that PEI polyplexes (which are unable to fuse directly with endocytic vesicles due to a lack of lipid content) were only able to avoid degradation if trafficked via non-degradative caveolae-mediated endocytosis. On the other hand, the lipoplexes taken up by clathrin rapidly escaped the degradative pathway before acidification by direct fusion with the vesicle membranes.
Macropinocytosis is a major route of entry for positively charged complexes, particularly for those coated with arginine [26]. Membrane-bound negatively-charged heparan sulfates act as receptors for positively charged particles; liposomes modified with octaarginine co-localized with neutral dextran, a tracer of fluid-phase endocytosis [27]. The uptake of these particles was inhibited by amiloride, which interferes with a Na+/H+ exchanger required for macropinocytosis. The lipoplexes internalized by macropinocytosis did not co-localize with acidic lysosomes, lending support to macropinocytosis as an attractive non-degradative pathway for gene delivery.
5. Relationship between particulate surface nanotechnology and uptake and transfection efficiency
As has been described, nonviral vectors can be transported to the cytoplasmic compartment by a diversity of endocytic mechanisms. Each of these pathways may support a different level of transfection mediated by a given lipo- or polyplex delivery system. An emerging paradigm for the design of effective gene carriers is the modification of particulate parameters to encourage entry via a preferable endocytic pathway. These parameters include size, shape, charge, chemistry, and ligand modification. These factors are often difficult to vary independently, so the contribution of each is difficult to generalize. Furthermore, endocytic processes vary by cell type, so the behavior of particles in one culture system may not be predictive of another. For example, HepG2 cells lack endogenous caveolin and are therefore unable to internalize particles by caveolae-mediated endocytosis [28]. Some of the key findings of this section are summarized in Figure 3.
Figure 3. Particulate parameter modifications and the resulting effects on endocytic uptake, trafficking, and transgene expression.

DOTAP lipoplexes are taken up by clathrin-mediated endocytosis, while expression-competent PEI polyplexes are endocytosed by a caveolae-dependant process. Large lipoplexes are generally taken up more efficiently, and large PLGA particles tend to depend more heavily on caveolae-mediated processing. Positively-charged particles usually bind to and are taken up by cells more efficiently than those with negative zeta potentials. Low aspect ratio PEG cylinders show a significantly lower extent of uptake than high aspect ratio equi-volume counterparts. Particles with a high density of octaargine functionalization induce macropinocytosis and are taken up more efficiently than those with sparse modification. Each of these differences has been supported by peer-reviewed publications, but may differ by cell type and culture conditions.
5.1 Particle chemistry
Altering the chemistry of a particle can modulate its hydrophilicity, the tendency for it to fuse with endocytic vesicles, and its susceptibility to serum inhibition, all with implications for uptake and transfection efficiency. For instance, a comparison of six phosphatidylcholine lipoplexes with varying hydrophobic chain lengths revealed that those with short chains mediated much higher levels of transfection in endothelial cells, both in the presence and absence of serum [29]. X-ray diffraction showed the particles supported formation of an inverted cubic phase, which is believed to resemble the membrane structure produced during fusion between lipid bilayers [30]. Masotti et al. have shown identical particle sizes or charge ratios of DMRIE/Chol, Cellfectin, Lipofectamine, Lipofectamine 2000, Lipofectin, and Fugene lipoplexes induce transfection levels that vary over many orders of magnitude in rat glioma cells [31].
PEGylation of cyclodextrin polyplexes endowed the particles with enhanced stability in the presence of salt, but decreased their uptake and transfection efficiency in BHK-21 cells [32]. PEGylated PEI polyplexes were taken up to a similar extent as unmodified particles, but were unable to transfect cells as efficiently; the addition of PEG may have interfered with the proton sponge effect. Using EM and fluorescence microscopy, the authors observed that PEGylated particles remained separate and stable during their journey toward the nucleus and were unable to unpack their DNA cargo, whereas unmodified particles aggregated into larger masses, which released their DNA into the cytoplasm and the nucleus.
The molecular weight and degree of branching in PEI has been investigated in PEI/liposome/DNA complexes (polylipoplexes). Branched and linear PEI induced similar levels of transfection, but PEI with lower molecular weight performed better than the larger PEI. Improvements in transfection efficiency with low over high molecular weight PEI have also been observed for pure PEI polyplexes [33]. PLGA nanoparticles made more hydrophilic with an increasing fraction of PVA emulsifier at their surface had a similar size and surface charge as more hydrophobic particles, but were taken up significantly less by smooth muscle cells [34].
5.2 Particle size
Nanoparticles are necessarily described by characteristic size parameters. It is important to note that DNA complex size can be a moving target, and is not completely defined by a single number. That is, particles often aggregate with time and, like polymers, require reporting of a polydispersity parameter for a full description of their size characteristics. Caution is warranted in comparison of particle sizes measured with different techniques and at different hydration states. For example, a particle’s hydrodynamic diameter measured in solution with dynamic light scattering may differ significantly from the same particle measured after dehydration and visualization by TEM. The endocytic machinery and cell membrane have well-defined geometries and flexibility that may restrict entry of incompatibly large or small particles. Modifications and procedures to create or stabilize a target nanoparticle size can direct gene carriers to endocytic routes that are supportive of high expression levels.
A linear relationship between the size of DC/Chol/DOPE lipoplexes, uptake, and transfection efficiency was observed over a range of 300–2000 nm; this relationship held true regardless of whether the particle size was changed by altering the cation to DNA charge ratio, or serum concentration [35]. Interestingly, particles formed in the presence of increasing serum concentration bound to cell membranes to the same extent, but larger particles were endocytosed much more efficiently. The size of DOTAP/DOPE lipoplexes can also be increased by pre-incubation with free PEG in solution prior to the onset of transfection [36]. PEG dehydrates and destabililizes lipid bilayers, leading to increased aggregation, fusion, and generation of micron-size particles from 500 – 800 nm nanoparticles. This increase in size leads to an increase in cellular association and uptake of the particles in multiple cell types. Transfection efficiency also improved for micron-size particles, but the increase in uptake did not account for the difference observed, suggesting the larger particles may have been preferentially trafficked into a more favorable endocytic route.
Extrusion of various multilamellar vesicles through a 100 nm filter results in lower transfection efficiency, but the identity of the cationic lipids used had a stronger effect on transfection in Neuro2A cells [37]. Similarly, Li et al. observed an increase in uptake and transfection efficiency of DOTAP and Lipotap complexes when their size was increased with subsequent layer-by-layer self-assembly of positively charged gold nanoparticles with uncomplexed DNA on the surface of the lipoplexes [38]. Altered endocytic trafficking should be added to the authors’ list of possible mechanisms whereby the larger particles increased transfection efficiency, which included elevated sedimentation, DNA payload, and charge-shielding. Larger, low molecular weight PEI polyplexes (590 nm) have also been observed to transfect NIH-3T3, HEK293, COS-7, CHO, HeLa, and Jurkat cells more efficiently than small high molecular weight polyplexes (156 nm), though it is unclear what contribution size made relative to weaker DNA condensation for the larger particles [33].
Prabha et al. separated a bimodal preparation of PLGA nanoparticles into fractions with hydrodynamic diameters of 150 and 300 nm [39]. The smaller nanoparticles produced 27× higher transfection efficiency in COS-7 cells, despite similar levels of particle uptake by mass and slower DNA release by the smaller particles. However, it was revealed with calculation that the smaller particles were taken up 20× more efficiently by number. Therefore, it is difficult to determine if the increase in transfection efficiency was a result of differential trafficking, or simply due to an increase in particle count. Considering that nonviral carriers are typically delivered on a per-mass basis, increase in particle count with decreasing diameter may influence transfection efficiency in many studies without notice. Other polymeric systems produce similar size-dependencies on uptake; only sub-micron polystyrene particles are efficiently taken up by Caco-2, HepG2, and Hepa 1–6 cells [40]. Very small (< 25 nm) particles may traffic through a unique non-clathrin- and non-caveolae pathway that could be interesting for nonviral gene delivery [41].
Particle size clearly affects the extent to which particles are taken up and are able to transfect target cells. Though many of the results presented here have suggested larger polymer and lipid particles (still in the submicron rage) are taken up more efficiently than smaller ones, some studies have claimed particles smaller than 100 nm offer good transfection efficiency, particularly when they must first pass through a capillary network (in vivo) [30]. Beyond controlling the extent of uptake, particle size has also been demonstrated to control the endocytic uptake pathway of nanoparticles [42]. In a comparison of the uptake of fluorescent latex nanospheres by B16 cells, uptake was only observed for particles 50–500 nm in diameter, but not for 1 um particles. Inhibitors of clathrin-mediated endocytosis were less effective at blocking uptake of large particles, whereas inhibitors of uptake by caveolae were only effective against the uptake of 500 nm nanospheres. Small particles (50 – 100 nm) were taken up within 30 minutes and appeared in the lysosomal compartment, whereas larger particles were taken up over a span of hours and did not colocalize with the late endosomes. These results suggest that large particles may be preferentially trafficked through a slow, non-degradative, caveolae-mediated route, and may explain why larger lipoplexes often produce higher transfection efficiencies. It would be interesting to extend these results to other chemistries and surface charges to determine if the size cutoffs for each pathway are intrinsic to some geometry of the endocytic machinery.
5.3 Surface charge
Along with size, surface charge (zeta potential) is a ubiquitous particulate parameter that is important for the understanding of uptake mechanisms and transfection efficiency. Particles that may be of an appropriate size to traverse a desired endocytic pathway may not be able to access that pathway if cellular binding is diminished by a significantly negative zeta potential. Also, the stability of a particulate gene delivery system can often be predicted by its zeta potential.
It is generally believed that positively-charged nanoparticles perform better for in vitro transfection of cells through their enhanced binding to negatively-charged proteoglycans on cell surfaces [27, 43]. Indeed, grafting polymerization of MMA onto carboxymethyl chitosan or chitosan hydrochloride can generate 150 nm nanoparticles with widely varying surface charge, leading to charge-dependant differences in endocytic uptake [44]. Particles with more-positive zeta potentials encouraged the highest rates of uptake in L02 and SMMC-7721 cells.
The dependence of uptake efficiency on surface charge is extendable to particles carrying genetic payloads. Optimization of a nonviral carrier often involves an empirical modification of the charge ratio (cationic polymer or lipid to anionic DNA) aimed to balance competing effects on cellular binding and uptake, DNA protection and release, and complex size and stability. Despite this wide parameter space for optimization, carriers with positive surface charges are often the most effective. This is likely due to increased binding to anionic cell surfaces, as well as more complete DNA complexation at high (+/−) charge ratios. Almofti et al. demonstrated that increasing the DNA content of DC-Chol-DOPE lipoplexes results in a decrease in zeta potential [45]. Particle size and liposome-liposome fusion were maximal at neutral charge ratios where the particles were unable to repel each other electrostatically, however transfection in A431 cells was greatest at a slightly positive charge ratio and was abolished by endocytic inhibitors. EPC-Chol lipoplexes modified with octaarginine demonstrate a similar dependence on surface charge; increasing densities of octaarginine produce increasingly cationic particles, leading to an increase in macropinocytosis, uptake, and transgene expression [26].
Though a positive zeta potential may be desirable to increase cellular uptake in vitro, positively charged particles may interact with negatively charged serum proteins in vivo, leading to charge neutralization, opsonization, increased particle size, and clearance [46]. With this in mind, cationic particles can be pre-neutralized with plasma-compatible proteins to increase transfection efficiency [47–48]. Unlike their cationic precursors, BSA-neutralized particles do not accumulate extra protein or increase in size upon exposure to plasma, and are endocytosed more effectively than PEG-shielded liposomes [47]. The authors suggest this may be a consequence of the caveolae-mediated endocytosis of surface BSA by the albumin receptor gp60 [49].
5.4 Shape
Nonspherical particle shapes are only recently being evaluated for altered tissue distribution and cellular uptake. This may be due to a previous lack of readily available techniques for the synthesis of well-defined nonspherical particles [50]. A lithographic method called PRINT (Particle Replication In Non-wetting Templates) has been developed to produce particles with various shapes and surface charges [51–52]. These cationic poly(ethylene glycol)-based particles with different shapes but similar zeta potentials have dramatically different uptake kinetics in HeLa cells. Comparing cylinders with similar particle volume, 150 nm cylinders with aspect ratio of 3 were taken up much more rapidly than 200 nm cylinders with aspect ratio of 1. The uptake was abolished if the particles were rendered anionic by conversion of protonated surface amines to amides. The authors also interrogated the cells with a series of endocytic inhibitors, revealing important roles for clathrin- and caveolae-dependent endocytosis in the uptake of small particles. The high aspect ratio 150 nm cylinders may have been taken up efficiently due to their utilization of all of the endocytic pathways probed. Highly elongated lipoplexes have also demonstrated improved efficiency in vivo [53], but the elongated structure has not been clearly correlated with improved transfection efficiency in vitro [54].
Alexander et al. have shown that polymeric micro-doughnuts are internalized much less efficiently than similarly-sized microspheres in a variety of non-phagocytic cell types [55]. This could be explained by the necessitation of a greater membrane curvature to engulf nonspherical particles of equal volumes [56]. Also, oblong nanoparticles with a minor axis that is smaller than nuclear membrane pores have been shown to transfect post-mitotic cells following cytoplasmic injection [57]. Taken together, these effects suggest particle shape could be engineered to investigate, avoid, or exploit various endocytic pathways and barriers to transfection by presentation of well-defined lengths and curvatures.
5.5 Ligand modification
Nanoparticles can also be targeted to specific arms of endocytosis through modification with species known to ligate endocytosable receptors, though this enormous body of work will not be the focus of this review. These strategies are particularly useful for targeting cell types that uniquely or over-express a particular receptor. Many of the classic endocytic ligands have been added to the surface of DNA complexes to increase transfection efficiency, including EGF-modified PEI polyplexes [58–60] and transferrin-modified lipoplexes [61–63] targeted to clathrin-mediated endocytosis. As previously mentioned, heparan sulfate proteoglycans act as “nonspecific receptors” for the binding of particles modified with arginine-rich peptides [64–65]. Arginine decoration increases uptake by macropinocytosis [26] and clathrin-mediated endocytosis [66], depending on the orientation and concentration of the peptide on the particle surface. Interestingly, the uptake of octaarginine-modified particles can be switched from macropinocytosis to a caveolae-mediated pathway by substitution of only two of the peptide’s residues [67].
6. Relationship between substrate surface nanotechnology and uptake and activity of adsorbed gene carriers
As discussed above, cell-particle interactions are critically important for efficient uptake and transfection. Through these interactions, particle morphology and chemistry can either enable or prohibit entry into and expression from target cells. However, nonviral vectors not only interact with target cells, but also with the substrates on which the cells are cultured. While these particle-substrate interactions may quietly impact the success of many “forward” transfection systems, their effects are most apparent during reverse transfection (also referred to as substrate-mediated transfection). Reverse transfection differs from forward transfection in that cells are seeded on top of particles that have been previously immobilized onto a surface, rather than adding particles to previously seeded cells. Complex immobilization is thought to increase transfection efficiency by increasing the local concentration of DNA at the cell surface, and can out-perform bolus delivery at times [68–70]. In one such example, Okazaki et al. found that reverse transfection of DNA-spermine-pullulan complexes maintained better hMSC viability, and produced a more intense and sustained expression of reporter transgene compared to forward transfection in the presence of serum - an important consideration for in vivo translation [71]. Substrate-mediated gene delivery also provides a simple method to locally deliver genetic material from the surface of porous implants. However, if the association of complexes with a substrate is too tight, endocytic uptake and transfection can suffer.
The context wherein adsorbed complexes are presented to cells can be modified with surface chemistry and the deposition of serum or ECM proteins. These modifications can alter transfection efficiency in a manner uncoupled from the extent of uptake, suggesting alternate processing for complexes presented in different substrative contexts. The consequences of altering the density and tightness of particle adsorption, as well as the benefit of matrix proteins, are presented in Figure 4. Furthermore, substrate-mediated transfection may change the rate-limiting barrier to expression. For instance, Bengali et al. report that internalization of lipoplexes is impaired but nuclear trafficking is improved in reverse compared to forward transfection. On the other hand, internalization of polyplexes was unaffected, but nuclear trafficking was weakened upon substrate-mediated rather than bolus delivery [72]. Reverse transfection may therefore be well-indicated for switching a bottleneck barrier to one that a certain particle type is more able to overcome.
Figure 4. Effect of density and presentation of surface-bound complexes on the efficiency of reverse (substrate-mediated) transfection.

Low densities of adsorbed complexes lead to low levels of particle uptake and expression upon cell seeding (A). Increasing the density of adsorbed complexes may lead to proportionally increased expression (B), but over-tight immobilization of complexes renders cells unable to internalize bound complexes, and diminishes transgene expression (C). Complexes co-immobilized with extracellular matrix components often support superior internalization and transfection (D) by incompletely-understood mechanisms.
6.1 Concentration of DNA at the cell surface
The concentration of DNA at the cell surface has been suggested as a limiting factor in nonviral gene delivery [73]. For forward transfection, the delivery of complexes to the cell surface is typically a diffusion-limited process, whereas reverse transfection can pre-load complexes at high levels onto the cell-substrate interface through drying, electrostatic and hydrophobic interactions, or ligand-receptor binding. Similar to forward transfection, increasing the amount of DNA adsorbed to a surface during reverse transfection increases expression levels, up to a limit. PEI polyplexes with increasingly positive zeta potentials adhere in greater numbers to acellular intestinal submucosa (rich in negatively charged glycosaminoglycans), leading to increased transgene expression in fibroblasts seeded in direct contact with the adsorbed complexes [74]. The size of printed lipoplex spots depends on the hydrophobicity of the substrate; more hydrophobic substrates produce smaller spot sizes, higher local DNA concentrations, and elevated expression levels [75].
6.2 Strength and nature of complex adsorption
Aggregation may translate to weaker binding between complexes and their substrate [76]. Polyplexes formed from a block copolymer of cationic poly N,N-dimethylaminopropyl acrylamide and thermoresponsive N-isopropylacrylamide do not produce strong expression in forward transfection or when dried to the culture surface. However, heat-induced hydrophobic transition, aggregation, and deposition of the complexes onto the substrate produced expression levels rivaling that of PEI in conventional transfection; the authors suggested that dried complexes were too tightly attached to the surface for uptake to proceed [77]. The inclusion of an increasing amount of cationic peptide to lipoplex preparations can also induce aggregation onto cell culture substrate, thereby increasing vector release, shifting to a non-lysosomal caveolar pathway, and enhancing transfection efficiency [76].
The balancing act between concentrating vector at the surface and facilitating cellular internalization is well-illustrated by the tethering of biotinylated polylysine complexes to a neutravidin-coated surface [78]. While the immobilization of particles increased with increasing biotinylation, the transfection efficiency was maximal for only a small amount of biotin functionalization; a low level of biotinylation encouraged complex deposition but was simultaneously permissive to internalization, whereas highly-biotinylated complexes bound too tightly to the surface to be internalized. In another study, stamping PEI complexes onto a layer of cells did not result in transfection unless the complexes were first released by an underlying pH-sensitive polymer layer [79].
Substrate surface chemistry can also have a marked effect on the immobilization and expression of nonviral vectors. Immobilization of Lipofectamine 2000 complexes onto self-assembled monolayers of alkanethiols with varying endgroups allows the comparison of substrate-mediated gene delivery from surfaces with controlled ionization and hydrophilicity [80]. Surfaces with a high ratio of anionic (carboxylic) to neutral (hydroxyl) groups supported the highest levels of complex immobilization and transfection of NIH/3T3 fibroblasts. Hydrophobic decane surface chemistry also bound high levels of lipoplex, but did not transfect cells, putatively from over-tight complex-surface interactions. Inclusion of PEG-like moieties can also increase the transfection efficiency of PEI polyplexes adsorbed to monolayers of carboxylic endgroups [81]. This increase cannot be attributed to an increase in complex binding or release, but the size and shape of adsorbed complexes is markedly affected. The ionic association of complexes with a substrate may be preferable to hydrophobic association, because ionic interactions can be displaced upon introduction of serum proteins, freeing complexes for internalization during reverse transfection.
6.3 Co-presentation of adsorbed protein with nonviral vectors
Protein pre-adsorbed to surfaces used in reverse transfection can aid in the subsequent deposition of complexes. Adsorbed protein can also improve transgene expression in a manner that is not fully explainable by increased immobilization or uptake. Complexes delivered with protein may maintain conformations favorable for cellular uptake, or may be differentially trafficked. Fibronectin deposited onto surfaces dramatically increased the reverse transfection efficiency of polyplexes in hMSCs, though cell adhesion and spreading were not apparently affected [69]. This suggests an active role of fibronectin in complex internalization. A similar effect was demonstrated with co-deposition of antibodies against various integrin subunits. Engagement of integrin subunits through adsorbed antibodies, particularly anti-CD29 (engages the ubiquitously-expressed integrin β1 [82]), resulted in an increase in transfection efficiency compared to control IgG for all of the cell types analyzed [83]. Intriguingly, placing RGD (an integrin-binding peptide sequence) directly on the surface of adsorbed PEI polyplexes leads to a decrease in transfection efficiency [84].
Fibronectin has been implicated elsewhere in the enhancement of substrate-mediated gene delivery [85]; drying a layer of fibronectin, collagen, laminin, FBS, BSA, and collagen was demonstrated to control the extent of PEI complex deposition, cellular association, endocytic internalization, and transfection efficiency following reverse transfection, whereas transgene expression was identical for all coating types following forward transfection. All coatings other than laminin mediated high levels of complex deposition. With the exception of FBS, complexes were found to be highly associated with the remaining coated surfaces (collagen, fibronectin, and BSA). Collagen and fibronectin had identically high levels of complex internalization compared to other coatings, but fibronectin supported a significantly higher level of transgene expression, suggesting differences in the trafficking of complexes internalized from collagen- and fibronectin-coated surfaces. Uptake in this system depended more heavily on caveolae-than clathrin-mediated endocytosis, and may be a functional manifestation of the co-localization of fibronectin with integrin β1 and caveolin in adhesion complexes [86] (discussed further in the next section). Similarly, substrates with adsorbed rather than dried FBS immobilized similar quantities of PEI and Lipofectamine 2000 complexes [70]. This FBS coating boosted the transgene expression of PEI 1500-fold compared to delivery from an uncoated surface. Expression of Lipofectamine 2000 from FBS-coated surfaces was unchanged, but the number of transfected cells was increased compared to uncoated substrates. Adsorption of fibronectin for improvement of substrate-mediated transfection has also been applied to 3D PLGA scaffolds for spinal cord regeneration [87–88].
7. Modulation of endocytic phenotype by substrate surface nanotechnology
The use of surface nanotechnology to modify particulate parameters has gained well-deserved attention in non-viral gene delivery, as these parameters are becoming increasingly well-understood modulators of uptake and transfection efficiency. Another approach worth considering is the engineering of desirable endocytic cellular phenotypes through substrate surface nanotechnology. A suite of parameters such as cell morphology, adhesion, proliferation, differentiation, and protein expression are tunable with substrate parameters including stiffness, chemistry, and physical topography (Figure 5). Cell biology offers hints towards mechanisms that may mediate useful interactions between these substrate-mediated effects on cell phenotype, the endocytic machinery, and subsequent transfection. Further, perhaps the simplest method whereby substrate nanotopography could enhance the transgene expression of interacting cells would be by increasing the loading of nonviral carriers onto patterned surfaces through an increase in effective surface area afforded by adsorption to feature sidewalls. Cells stretched over substrate topography may also expose an increased proportion of their basal surface, creating a larger area for complexes to attach and to be internalized. In general these interactions have yet to be directly investigated and exploited for gene delivery. Advances in patterning technology have made micro- and nano-patterned substrates more widely available; the following is an exploratory discussion of how these substrates may already be affecting endocytosis and transfectability, despite a lack of explicit, functional studies.
Figure 5. Known micro- and nano-topographical effects on cell phenotype, and their possible impact on nonviral gene delivery.
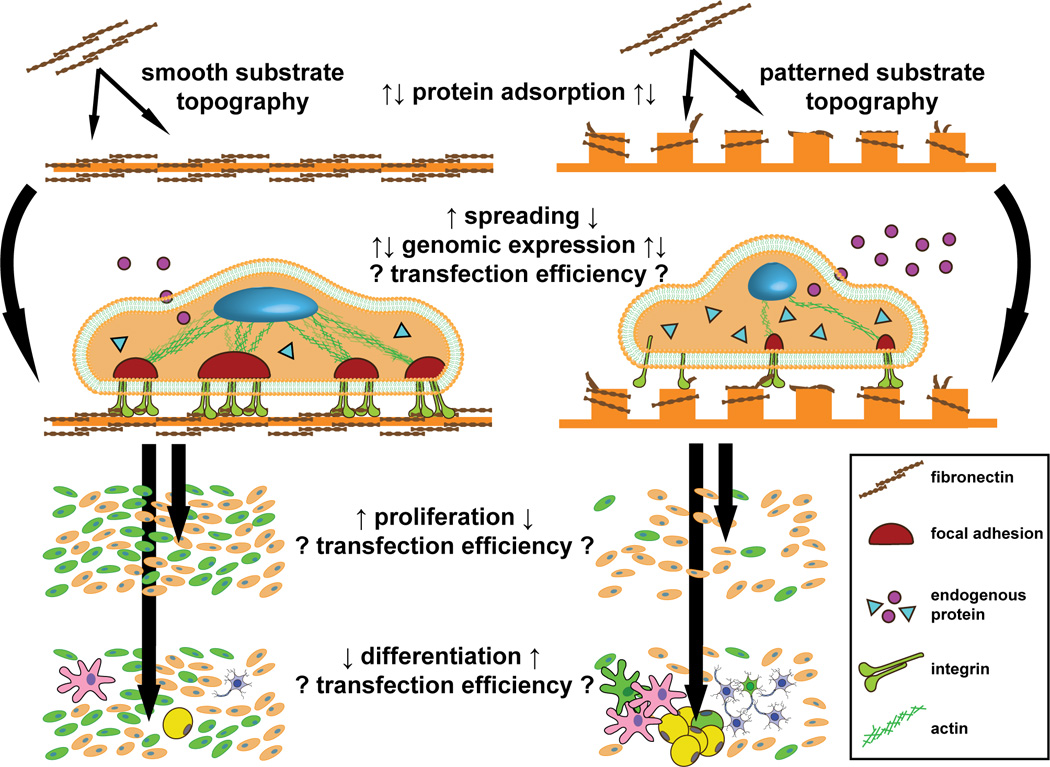
The quantity and denaturation of matrix proteins adsorbed to patterns can be increased or decreased, depending on the specifics of the substrate topography and chemistry. These differences in the adsorbed protein layer mediate alterations in a number of cell phenotypes. In general, cells cultured on patterned topographies have decreased spreading and proliferation, reduced integrin clustering, and smaller focal adhesion complexes compared to smooth controls. Proliferative cells are often more susceptible to nonviral gene delivery. Further, actin-mediated transduction of tension from integrin-nucleated focal adhesions to the nucleus alters the expression of a multitude of secreted and intracellular proteins; many of these proteins play a role in endocytosis, and therefore likely in the endocytic uptake of nonviral carriers. Finally, patterned topography can control the differentiation state of many cell types, plausibly leading to altered transfectability.
7.1 Protein adsorption on nanofeatures
Cells interact with the substrates they are cultured on through integrins. Integrins are a family of transmembrane receptors that bind to an assortment of extracellular matrix proteins through shared RGD domains, including: fibronectin, laminin, collagen, and fibrinogen. Integrin binding and clustering initiates the assembly of adaptor proteins vinculin, talin, paxillin, and phosphorylative activation of focal adhesion kinase (FAK), comprising the focal adhesion complex [89]. FAK goes on to provoke downstream effects such as cell movement and cytoskeletal contractility [90]. Therefore, cell-substrate interactions are mediated by the proteins that are deposited from solution onto to a given substrate, as actuated through integrin signaling. Substrate topography and chemistry alter the amount and conformation of integrin ligands, and may form the general basis for many if not all subsequent topographical effects on cell behavior.
Nano- and micro-scale topographies affect the amount and conformation of protein binding by presenting heterogeneous surface energies, altering exposed surface area, or restricting wettable area [91–92]. Nanoislands prepared by polymer demixing with heights of 14 nm supported 50% more fibronectin binding than 45 nm islands, despite a negligible change in exposed surface area [93]. Furthermore, the fibronectin preferentially adsorbed to the valleys between 14 nm islands. Similarly, 4 µm wide, 1 µm tall PDMS posts encouraged the adsorption of 50% more fibrinogen compared to smooth control despite only an 8% increase in surface area [94]. In contrast, higher aspect ratio PLGA support decreased fibrinogen adsorption [95].
The conformation of proteins adsorbed to surfaces also changes with physical patterning, which has implications for integrin binding. Adsorption of fibronectin to a nano-rough tantalum surface was increased compared to smooth tantalum, and the adsorbed layer’s stiffness increased, accompanied by a decreased susceptibility to antibody interrogation, indicative of a more extended fibronectin conformation [96]. This result is echoed for fibronectin adsorbed to colloidally-roughened silica substrates [97]. The rearrangement of collagen into fibers on smooth substrates can also be hindered by nanocolumn patterning [98], and F-actin aligns on 2 nm- but not 4 nm- tall ridges [99]. Globular proteins have been shown to retain their structure and biological activity on small, highly curved particles, whereas large proteins are denatured when forced to adsorb to such geometries [100–101]. These effects may be responsible for the differences observed for cells interacting with ECM adsorbed or secreted onto various nanopatterned substrates.
Altered protein conformation has also been directly implicated in the control of adherent cell behavior. Osteoblastic differentiation of MC3TC cells can be controlled by substrates that influence the conformation of adsorbed fibronectin [102]. Antibodies against specific fibronectin epitopes alter fibronectin binding when adsorbed to different substrate chemistries, subsequently affecting myoblast proliferation and differentiation [103]. Fibroblasts can sense the conformational change of fibronectin elicited by the subtle replacement of a substrate polymer’s methylene groups with oxygen [104]. Cells are able to detect changes in substrate topography and chemistry through the high sensitivity of protein adsorption to these physical and chemical features.
7.2 Proliferation and endocytosis
Rapidly dividing cells are generally more susceptible to nonviral transfection, an effect attributed to greater access to the nucleus during cell division [4]. For example, low rates of proliferation elicited by contact-inhibition of epithelial cells leads to decreased lipoplex uptake and expression [105]. The decrease in transfectability with cellular confluence and age could also be due in part to decreased endocytosis in these cells. Indeed, late passage fibroblasts down-regulate the expression of amphiphysin-1, a linker between the clathrin coat and dynamin, causing a decrease in receptor-mediated endocytosis that can be resurrected with reexpression of amphiphysin-1 [106–107]. Therefore, substrates that control cell proliferation are also expected to control cellular susceptibility to nonviral gene delivery.
Researchers have repeatedly noted changes in the metabolic and proliferative rates of cells cultured on patterned topographies. A decrease in proliferation for cells on micro- and nano-structures is most common, but increases are also observed [108]. For example, smooth muscle cells cultured on 350 nm wide gratings proliferated significantly slower, as measured by BrdU incorporation [109]. Fibroblasts cultured on quartz micropits with 7–25 µm diameters and 20–40 µm spacing proliferated most slowly for the smallest, most closely spaced pits [110]. There are some exceptions which report increased proliferation on patterns; connective tissue progenitor cells grown on 10 µm diameter, 6 µm high posts exited post-seeding lag phase days before cohorts cultured on smooth control PDMS [111]. Substrates eliciting elevated proliferation rates may therefore be beneficial for gene delivery.
7.3 Differentiation and endocytosis
The differentiation of certain cell types can be controlled with substrate topography and stiffness. Naïve hMSC are sensitive to nanotopography, encouraged by nanogratings to more readily commit to a neuronal lineage than by a chemical differentiation factor (retinoic acid) delivered to cells on a smooth substrate [112]. hMSCs are also sensitive to the stiffness of their substrate (a property that could be modified locally with substrate topography), preferring osteogenic, myogenic, or neurogenic differentiation when cultured on gels with stiffnesses mimicking that of bone, muscle, or brain, respectively [113]. Mouse embryonic stem cells and osteoprogenitors can be maintained in undifferentiated phenotypes with culture on 2.4 µm tall, 1 µm diameter posts discovered by high-throughput screening of hundreds of microtopographies [114–115].
The differentiation state of a cell population can determine the transfectability and endocytic phenotype of those cells. Nonviral transfection is notoriously difficult for neurons [116], macrophages [117], dendritic cells [118], and differentiated chondrocytes [119] and adipocytes [120]. Further, the expression of caveolae is up-regulated in terminally differentiated fat, endothelial, muscle, and transdifferentiated lens epithelial cells [121–122], and the uptake of LDL by clathrin-mediated endocytosis decreases with adipogenic differentiation of hMSCs [123]. hMSCs directed to the neuronal lineage by nanopatterns express less caveolin-1, and alter the expression profile of a number of integrin subunits [112]. In addition, the production of heparan sulfate proteoglycans (the ubiquitous receptors for cationic particles) is down-regulated with differentiation of myogenic satellite cells [124]. Given these differences in transfectability and endocytosis between various cell types, substrate nanotechnology-mediated differentiation is another tool that could be leveraged to engineer the nonviral expression of transgenes.
7.4 Expression of endocytic proteins
Irrespective of differentiation, culturing cells on patterned topography undergo genome-level changes in the expression of proteins that are implicated in endocytosis. Changes in gene expression of cells cultured on patterned topography are believed to be mediated, at least in part, by nuclear deformation. Many different cell types have been observed to align and elongate with nano- and micro-grooved topography. The lower size limit for this alignment may be cell type-specific, but has been demonstrated to be around 100 nm wide, 70 nm deep gratings for the early alignment of rat fibroblasts [125]. Smooth muscle cells [109] and hMSCs [112] also align and elongate on 350 nm PDMS gratings. Alignment of the cell membrane and cytoskeleton can translate force to the nucleus via intermediate filaments, and can result in nuclear alignment and deformation [126–129]. Stress applied to the nucleus alters histone deacetylation, chromatin condensation [130], and centromere arrangement [131], leading to global changes in gene expression.
The changes in protein expression on patterned topography include those explicitly implicated in endocytosis such as clathrin and caveolin, growth factors and cytokines that have established effects on endocytosis, matrix proteins that may alter cell-substrate interactions, and signaling proteins that are needed for endocytosis but are not necessarily as well understood in their mode of action. The latter category has recently been elucidated in an impressive manuscript where genomic libraries of siRNA were assessed for effects on the endocytosis of EGF and transferrin as directly observed with automated confocal microscopy [132]. The 4,609 genes affecting endocytosis can be cross-referenced with the handful of gene array studies that have been performed on cells interacting with patterned topography. For instance, human fibroblasts cultured on micro-grooved quartz [126] and 13 nm polymer demixed islands [133–134] upregulated their expression of Grk6, integrin α6, integrin β5, the growth factor receptor tyrosine kinase Ryk, REP-2 (targets Rab5 to the plasma membrane for early endosome trafficking [135]), Jnk2, and many more; RNA interference with any of these proteins leads to changes in the uptake of EGF and transferrin or in the intracellular distribution of endosomes [132].
Dalby et al. have suggested that endocytosis is altered (qualitatively) for cells cultured on patterned topography. Human fibroblasts attempt to endocytose nanocolumns with 100 nm diameter and 160 nm height, as supported by increased dynamin and peripheral clathrin staining, and TEM visualization of nascent endocytic vesicles near columns [136]. Fibroblasts cultured on nanopits with similar dimensions formed clathrin tracks indicative of “high rates of endocytosis”, and upregulated the expression of Epsin 2 [137], a stimulator of endocytic vesicle fissure from the plasma membrane [138].
Cytokines and growth factors are potent stimulators of cell function, and endocytosis is no exception. hMSCs cultured on patterned PMMA surfaces upregulate the expression of EGF and FGFb [112]. EGF can stimulate uptake of its own receptor as well as fluid phase endocytosis [139], and FGFb has been noted to downregulate the expression of surface-bound heparan sulfate proteoglycans [140]. Macrophages cultured on increasing nanograting widths increase their secretion of TNF-α [141], an inflammatory cytokine that accelerates clathrin-mediated endocytosis in Sertoli [142] and endothelial cells [143]. The expression of signals which increase endocytosis may be up-regulated in cells interacting with patterns and act in an autocrine fashion, thereby altering the uptake of nonviral vectors.
7.5 Cell spreading and endocytosis
Cell spreading may be the best-described effect of culture on nano- and microtopographic surfaces. The heterogeneous presentation of extracellular matrix proteins adsorbed to topography supports varying degrees of integrin engagement. Adhesive substrates are predicted to sustain increased spreading with patterning (increased adhesive surface area), whereas non-adhesive substrates are expected to maintain better spreading if smooth (maintaining a minimal degree of cell deformation) [144]. Indeed, NIH/3T3 cells cultured on rough, super-hydrophobic silicon nanospikes were rounded, while those on rough, hydrophilic spikes were well-spread [145]. Fibroblasts [146] and endothelial cells [147] cultured on 13 nm tall polystyrene islands were more spread than those on flat control, and those cultured on 120 nm diameter, 100 nm depth pits in PMMA were much less spread than control [137]. This dependence of spreading on topography may be due to the altered ability of integrin to cluster for various substrates; gold nanospheres with RGD functionalization were deposited onto a passivated surface with well-defined spacing to study this possibility [148]. The nanospheres were small enough that only a single integrin could be expected to be able to bind to each. Cells grown on RGD spaced 108 nm apart had delayed spreading and a smaller projected area, compared to those on RGD spaced 58 nm apart.
The act of integrin engagement and spreading itself may have an immediate effect on endocytosis, before changes in protein synthesis could be expected (Figure 6, right). Caveolin-1 is the lynchpin of caveolar endocytosis, and convincing evidence has appeared implicating it in integrin signaling [149]. Integrin β1 colocalizes with fibronectin and caveolin in focal adhesions [86], and its binding results in phosphorylation of caveolin-1 [150]. Caveolin-1 acts as a membrane adaptor protein to couple integrins to downstream signaling partners, and to facilitate integrin clustering by oligomerization [149]. Caveolin-1 knockouts are unable to internalize lipid rafts after detachment, and this behavior can be restored with subsequent expression of caveolin-1 [151]. Also, a loss of integrin-mediated adhesion results in the dramatic internalization of caveolae [152]. These studies demonstrate a strong link between integrin function and caveolar trafficking, but it is unclear what the functional implications are for the uptake of cargo molecules that are nearby or directly involved in this process.
Figure 6. Two possible routes for the direct modulation of nonviral carrier uptake by integrin signaling.

Actin filaments localized to the cell surface by integrin-containing focal adhesions bind to the HIP1/HIP1R complex, which recruits AP-2 to assemble the clathrin coat on nascent vesicles. Cortactin is thought to induce and localize the polymerization of actin at internalizing vesicles by linking dynamin and the actin-nucleating complex Arp2/3. Integrin disassembly and internalization is a clathrin-mediated process, and high rates of this activity on patterned topography could compete with or augment complex uptake. Integrin engagement also results in local sequestration of caveolin-1 and stabilization of caveolae at the cell surface. Subsequent integrin release induces caveolae internalization, but it is unknown what effect this may have on the uptake of nearby gene carriers. Nanotopographical control of integrin engagement and turnover (depicted) may be a useful tool in the study of these effects.
7.6 Endocytic turnover of focal adhesions
The formation, strengthening, contraction, and disassembly of focal adhesions allow cells to explore and move across their substrates. This cycle is controlled by substrate topography, as mediated through the availability of adsorbed integrin ligands. When integrins are not allowed to cluster, the small focal adhesions that form are unable to hold on upon contraction of the actin cytoskeleton [127], leading to a more rapid turnover of adhesions as the cell searches for stable contact [148]. Integrin clustering is likely affected by the adsorption of ECM to nano- and microtopography, producing differences in the size, strength, and number of adhesions formed on different structures. Tall features restrict binding to the tops of patterns, small feature size (< 70 nm) prevents adhesions from forming at all, and large interfeature spacing reduces integrin clustering [89]. Indeed, fibroblasts cultured on 50 nm diameter pits have much smaller focal adhesions, and normal actin stress fibers are not formed [153]. On the other hand, shallow 30 µm diameter PMMA pits encourage a higher number of adhesions in hMSCs [154]. Furthermore, 350 nm PDMS gratings decrease the expression of vinculin in hMSCs compared to smooth control, and lower the cells’ elastic moduli as measured by AFM, indicative of structural changes in the organization of the actin cytoskeleton [155].
These effects of topography on focal adhesion formation and turnover may be instructive of the situation in vivo; natural 3D extracellular matrices encourage the formation of small focal adhesions, while flattening the same matrix into two dimensions recovers large adhesions [156]. 3D fibrillar matrix also increases the rate of fibronectin remodeling [157]; this remodeling proceeds by caveolar endocytosis, as demonstrated with inhibition by low temperature, and siRNA against caveolin-1, genistein, β -cyclodextrin, and staursporin [158]. Substrate topography introduces a degree of three-dimensionality to the presentation of adsorbed proteins, and may therefore modulate this remodeling process.
A number of recently described molecular interactions have linked components of the focal adhesion complex and the endocytic and cytoskeletal machineries (Figure 6, left). Disassembly of focal adhesions is a clathrin-dependent endocytic process; clathrin and AP-2 colocalize with focal adhesions, and knockdown of clathrin, AP-2, and dynamin activity results in lowered integrin internalization [90]. As well, recently described proteins including cortactin, Abp1p, Hip1R, and intersectin-1 link actin-nucleating enzymes such as Arp2/3 to their substrates [159–160], and recruit clathrin to the plasma membrane to stimulate its assembly and association with actin [161–162]. Actin contractility has indeed been suggested to drive the internalization of cholesterol-rich lipid rafts containing cationic complexes bound to proteoglycans [163]. Interestingly, inhibition of PKC with staurosporine inhibits uptake and expression of PEI complexes, but not their binding. These interactions suggest differences in the strength of integrin binding and the speed of integrin turnover induced by substrate topography can be expected to alter endocytic activity. This could manifest as a coincident uptake of complexes or as a downregulation of particle uptake through increased competition for the endocytic machinery by the process of integrin internalization.
8. Summary
Optimization of nonviral gene delivery has so far mostly focused on design of particulate carriers that are endowed with desirable membrane targeting, internalization, and endosomal escape properties. Comparatively little attention has been paid to understand and exploit the factors driving nonviral vectors into one of the variably attractive endocytic pathways. Surface nanotechnology at the particulate level has been an established approach adopted by researchers to manipulate the endocytic process. Surface nanotechnology at the substrate level, however, remains a largely unexplored but potentially attractive strategy. Emerging literature has highlighted the influence of cell-topography interactions on modulation of many cell phenotypes, including endocytosis. Improvement in transfection efficiency has been noted for nonviral vectors delivered with a substrate-mediated approach, but the reasons behind this enhancement remain unclear.
Practically, the use of substrate topography to improve nonviral gene delivery will require a fundamental understanding of the dominant mechanisms of the modulation, so surfaces could then be intelligently selected. Therefore first and foremost, this understanding must be developed with pilot studies that first directly demonstrate the functional differences in endocytic phenotypes and transfectability as a result of interaction with a range of controlled substrate topographies, and to subsequently elucidate whether the modulation is a consequence of altered uptake, trafficking, proliferation, cytoskeletal tension, focal adhesion turnover, etc. This principal aim of this review is to stimulate interest in the performance of these experiments, which are expected to reveal exciting new insights into the interaction of the cytoskeleton, endocytosis, and transgene expression, culminating in applications which employ surface nanopatterning to enhance transfection in vitro and at the surface of gene-delivering bioerodible scaffolds.
Research Highlights.
Endocytic processes can be manipulated with both particulate and substrate nanotechnology to enhance nonviral gene transfer
There are emerging molecular links between the endocytic and cytoskeletal machineries
Substrate topography-mediated control of endocytosis is a new paradigm to be elucidated and leveraged towards the design of efficient nonviral gene delivery systems
Acknowledgements
The authors gratefully acknowledge support by the NIH (HL89764 and DK068399) and the Center for Biomolecular and Tissue Engineering (CBTE, Duke University) NIH Training Program.
Biographies

Andrew F. Adler received his B.S. in Biomedical Engineering in 2008, summa cum laude, from Northwestern University (Chicago, IL), where he worked with Prof. Lonnie D. Shea on implantable gene-releasing scaffolds for the regeneration of injured spinal cords. Currently, Andrew is pursuing a PhD in Biomedical Engineering at Duke University (Durham, NC), and is a NIH Training Grant Fellow in the Center for Biomolecular and Tissue Engineering. His interests include the elucidation of substrate-mediated modulation of endocytosis and transfectability, the development of novel Surface Enhanced Raman Spectroscopy (SERS) techniques, and surfing.

Kam W. Leong is the James B. Duke Professor of Biomedical Engineering in the Pratt School of Engineering of Duke University, at which he also holds a joint appointment in the Department of Surgery of the School of Medicine. His research concentrates on understanding and exploiting the interactions of cells with nanostructures for therapeutic applications. Discrete nanostructures in the form of multi-functional nanoparticles are applied to deliver drug, antigen, protein, siRNA, mRNA and DNA to cells for drug, gene, and immunotherapy. Continuous nanostructures in the form of electrospun nanofibers and imprinted nanopatterns are applied to biomaterials design and regenerative medicine applications.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ratko TA, Cummings JP, Blebea J, Matuszewski KA. The American Journal of Medicine. 2003;115:560–569. doi: 10.1016/s0002-9343(03)00447-9. [DOI] [PubMed] [Google Scholar]
- 2.van Gaal EVB, Oosting RS, Hennink WE, Crommelin DJA, Mastrobattista E. International Journal of Pharmaceutics. In Press, Corrected Proof. [Google Scholar]
- 3.Belting M, Sandgren S, Wittrup A. Adv Drug Deliver Rev. 2005;57:505–527. doi: 10.1016/j.addr.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Medina-Kauwe LK, Xie J, Hamm-Alvarez S. Gene Ther. 2005;12:1734–1751. doi: 10.1038/sj.gt.3302592. [DOI] [PubMed] [Google Scholar]
- 5.Grigsby CL, Leong KW. Journal of the Royal Society Interface. 7:S67–S82. doi: 10.1098/rsif.2009.0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hama S, Akita H, Iida S, Mizuguchi H, Harashima H. Nucleic Acids Res. 2007;35:1533–1543. doi: 10.1093/nar/gkl1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kogure K, Akita H, Yamada Y, Harashima H. Adv Drug Deliver Rev. 2008;60:559–571. doi: 10.1016/j.addr.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Pollard Hln, Remy J-S, Loussouarn G, Demolombe S, Behr J-P, Escande D. Journal of Biological Chemistry. 1998;273:7507–7511. doi: 10.1074/jbc.273.13.7507. [DOI] [PubMed] [Google Scholar]
- 9.Payne CK, Jones SA, Chen C, Zhuang X. Traffic. 2007;8:389–401. doi: 10.1111/j.1600-0854.2007.00540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lechardeur D, Verkman AS, Lukacs GL. Adv Drug Deliver Rev. 2005;57:755–767. doi: 10.1016/j.addr.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Conner SD, Schmid SL. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 12.Sonawane ND, Szoka FC, Jr, Verkman AS. J Biol Chem. 2003;278:44826–44831. doi: 10.1074/jbc.M308643200. [DOI] [PubMed] [Google Scholar]
- 13.Wagner E. Journal of Controlled Release. 1998;53:155–158. doi: 10.1016/s0168-3659(97)00249-6. [DOI] [PubMed] [Google Scholar]
- 14.Rejman J, Bragonzi A, Conese M. Mol Ther. 2005;12:468–474. doi: 10.1016/j.ymthe.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 15.Pelkmans L, Bürli T, Zerial M, Helenius A. Cell. 2004;118:767–780. doi: 10.1016/j.cell.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Pelkmans L. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2005;1746:295–304. doi: 10.1016/j.bbamcr.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 17.Hoekstra D, Rejman J, Wasungu L, Shi F, Zuhorn I. Biochem Soc Trans. 2007;35:68–71. doi: 10.1042/BST0350068. [DOI] [PubMed] [Google Scholar]
- 18.Zuhorn IS, Kalicharan R, Hoekstra D. Journal of Biological Chemistry. 2002;277:18021–18028. doi: 10.1074/jbc.M111257200. [DOI] [PubMed] [Google Scholar]
- 19.Zuhorn IS, Oberle V, Visser WH, Engberts JB, Bakowsky U, Polushkin E, Hoekstra D. Biophys J. 2002;83:2096–2108. doi: 10.1016/S0006-3495(02)73970-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindberg J, Fernandez MA, Ropp JD, Hamm-Alvarez SF. Pharm Res. 2001;18:246–249. doi: 10.1023/a:1011001022570. [DOI] [PubMed] [Google Scholar]
- 21.HammAlvarez SF, Sonee M, LoranGoss K, Shen WC. Pharmaceut Res. 1996;13:1647–1656. doi: 10.1023/a:1016432505275. [DOI] [PubMed] [Google Scholar]
- 22.Akinc A, Thomas M, Klibanov AM, Langer R. The Journal of Gene Medicine. 2005;7:657–663. doi: 10.1002/jgm.696. [DOI] [PubMed] [Google Scholar]
- 23.Wrobel I, Collins D. Bba-Biomembranes. 1995;1235:296–304. doi: 10.1016/0005-2736(95)80017-a. [DOI] [PubMed] [Google Scholar]
- 24.Rémy-Kristensen A, Clamme J-P, Vuilleumier C, Kuhry J-G, Mély Y. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2001;1514:21–32. doi: 10.1016/s0005-2736(01)00359-5. [DOI] [PubMed] [Google Scholar]
- 25.Douglas KL, Piccirillo CA, Tabrizian M. European Journal of Pharmaceutics and Biopharmaceutics. 2008;68:676–687. doi: 10.1016/j.ejpb.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 26.Khalil IA, Kogure K, Futaki S, Harashima H. J Biol Chem. 2006;281:3544–3551. doi: 10.1074/jbc.M503202200. [DOI] [PubMed] [Google Scholar]
- 27.Hess GT, Humphries Iv WH, Fay NC, Payne CK. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2007;1773:1583–1588. doi: 10.1016/j.bbamcr.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujimoto T, Kogo H, Nomura R, Une T. J Cell Sci. 2000;113:3509–3517. doi: 10.1242/jcs.113.19.3509. [DOI] [PubMed] [Google Scholar]
- 29.Tenchov BG, Wang L, Koynova R, MacDonald RC. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2008;1778:2405–2412. doi: 10.1016/j.bbamem.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 30.Ma B, Zhang S, Jiang H, Zhao B, Lv H. Journal of Controlled Release. 2007;123:184–194. doi: 10.1016/j.jconrel.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 31.Masotti A, Mossa G, Cametti C, Ortaggi G, Bianco A, Grosso ND, Malizia D, Esposito C. Colloids and Surfaces B: Biointerfaces. 2009;68:136–144. doi: 10.1016/j.colsurfb.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 32.Mishra S, Webster P, Davis ME. European Journal of Cell Biology. 2004;83:97–111. doi: 10.1078/0171-9335-00363. [DOI] [PubMed] [Google Scholar]
- 33.Kunath K, von Harpe A, Fischer D, Petersen H, Bickel U, Voigt K, Kissel T. Journal of Controlled Release. 2003;89:113–125. doi: 10.1016/s0168-3659(03)00076-2. [DOI] [PubMed] [Google Scholar]
- 34.Sahoo SK, Panyam J, Prabha S, Labhasetwar V. Journal of Controlled Release. 2002;82:105–114. doi: 10.1016/s0168-3659(02)00127-x. [DOI] [PubMed] [Google Scholar]
- 35.Almofti MR, Harashima H, Shinohara Y, Almofti A, Li WH, Kiwada H. Molecular Membrane Biology. 2003;20:35–43. doi: 10.1080/09687680210035104. [DOI] [PubMed] [Google Scholar]
- 36.Ross PC, Hui SW. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1999;1421:273–283. doi: 10.1016/s0005-2736(99)00132-7. [DOI] [PubMed] [Google Scholar]
- 37.Ramezani M, Khoshhamdam M, Dehshahri A, Malaekeh-Nikouei B. Colloids and Surfaces B: Biointerfaces. 2009;72:1–5. doi: 10.1016/j.colsurfb.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 38.Li D, Li G, Li P, Zhang L, Liu Z, Wang J, Wang E. Biomaterials. 2010;31:1850–1857. doi: 10.1016/j.biomaterials.2009.11.027. [DOI] [PubMed] [Google Scholar]
- 39.Prabha S, Zhou W-Z, Panyam J, Labhasetwar V. International Journal of Pharmaceutics. 2002;244:105–115. doi: 10.1016/s0378-5173(02)00315-0. [DOI] [PubMed] [Google Scholar]
- 40.Zauner W, Farrow NA, Haines AMR. Journal of Controlled Release. 2001;71:39–51. doi: 10.1016/s0168-3659(00)00358-8. [DOI] [PubMed] [Google Scholar]
- 41.Lai SK, Hida K, Man ST, Chen C, Machamer C, Schroer TA, Hanes J. Biomaterials. 2007;28:2876–2884. doi: 10.1016/j.biomaterials.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 42.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Biochem J. 2004;377:159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiethoff CM, Koe JG, Koe GS, Middaugh CR. Journal of Pharmaceutical Sciences. 2004;93:108–123. doi: 10.1002/jps.10519. [DOI] [PubMed] [Google Scholar]
- 44.He C, Hu Y, Yin L, Tang C, Yin C. Biomaterials. In Press, Corrected Proof. [Google Scholar]
- 45.Radwan Almofti M, Harashima H, Shinohara Y, Almofti A, Baba Y, Kiwada H. Archives of Biochemistry and Biophysics. 2003;410:246–253. doi: 10.1016/s0003-9861(02)00725-7. [DOI] [PubMed] [Google Scholar]
- 46.Liu DX. Adv Drug Deliver Rev. 1997;24:201–213. [Google Scholar]
- 47.Jung SH, Kim SK, Jung SH, Kim EH, Cho SH, Jeong K-S, Seong H, Shin BC. Colloids and Surfaces B: Biointerfaces. 2010;76:434–440. doi: 10.1016/j.colsurfb.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 48.Caracciolo G, Callipo L, De Sanctis SC, Cavaliere C, Pozzi D, Laganà A. Biochimica et Biophysica Acta (BBA) - Biomembranes. doi: 10.1016/j.bbamem.2009.11.007. In Press, Corrected Proof. [DOI] [PubMed] [Google Scholar]
- 49.Schnitzer JE. Adv Drug Deliver Rev. 2001;49:265–280. doi: 10.1016/s0169-409x(01)00141-7. [DOI] [PubMed] [Google Scholar]
- 50.Champion JA, Katare YK, Mitragotri S. Journal of Controlled Release. 2007;121:3–9. doi: 10.1016/j.jconrel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM. P Natl Acad Sci USA. 2008;105:11613–11618. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gratton SEA, Pohlhaus PD, Lee J, Guo J, Cho MJ, DeSimone JM. Journal of Controlled Release. 2007;121:10–18. doi: 10.1016/j.jconrel.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sternberg B, Hong K, Zheng W, Papahadjopoulos D. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1998;1375:23–35. doi: 10.1016/s0005-2736(98)00129-1. [DOI] [PubMed] [Google Scholar]
- 54.Xu Y, Hui S-W, Frederik P, Szoka FC., Jr Biophysical Journal. 1999;77:341–353. doi: 10.1016/S0006-3495(99)76894-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alexander L, Dhaliwal K, Simpson J, Bradley M. Chem Commun (Camb) 2008:3507–3509. doi: 10.1039/b805323e. [DOI] [PubMed] [Google Scholar]
- 56.Decuzzi P, Ferrari M. Biophysical Journal. 2008;94:3790–3797. doi: 10.1529/biophysj.107.120238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu G, Li D, Pasumarthy MK, Kowalczyk TH, Gedeon CR, Hyatt SL, Payne JM, Miller TJ, Brunovskis P, Fink TL, Muhammad O, Moen RC, Hanson RW, Cooper MJ. J Biol Chem. 2003;278:32578–32586. doi: 10.1074/jbc.M305776200. [DOI] [PubMed] [Google Scholar]
- 58.Blessing T, Kursa M, Holzhauser R, Kircheis R, Wagner E. Bioconjug Chem. 2001;12:529–537. doi: 10.1021/bc0001488. [DOI] [PubMed] [Google Scholar]
- 59.Kloeckner J, Boeckle S, Persson D, Roedl W, Ogris M, Berg K, Wagner E. J Control Release. 2006;116:115–122. doi: 10.1016/j.jconrel.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 60.Wolschek MF, Thallinger C, Kursa M, Rossler V, Allen M, Lichtenberger C, Kircheis R, Lucas T, Willheim M, Reinisch W, Gangl A, Wagner E, Jansen B. Hepatology. 2002;36:1106–1114. doi: 10.1053/jhep.2002.36372. [DOI] [PubMed] [Google Scholar]
- 61.Girão da Cruz MT, Simões S, Pedroso de Lima MC. Experimental Neurology. 2004;187:65–75. doi: 10.1016/j.expneurol.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 62.Singh M, Hawtrey A, Ariatti M. International Journal of Pharmaceutics. 2006;321:124–137. doi: 10.1016/j.ijpharm.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 63.Tros de Ilarduya C, Düzgünes N. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2000;1463:333–342. doi: 10.1016/s0005-2736(99)00225-4. [DOI] [PubMed] [Google Scholar]
- 64.Khalil IA, Kogure K, Futaki S, Harashima H. International Journal of Pharmaceutics. 2008;354:39–48. doi: 10.1016/j.ijpharm.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 65.Fretz MM, Koning GA, Mastrobattista E, Jiskoot W, Storm G. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2004;1665:48–56. doi: 10.1016/j.bbamem.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 66.Vandenbroucke RE, De Smedt SC, Demeester J, Sanders NN. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2007;1768:571–579. doi: 10.1016/j.bbamem.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 67.Mudhakir D, Akita H, Tan E, Harashima H. Journal of Controlled Release. 2008;125:164–173. doi: 10.1016/j.jconrel.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 68.Fujita S, Ota E, Sasaki C, Takano K, Miyake M, Miyake J. Journal of Bioscience and Bioengineering. 2007;104:329–333. doi: 10.1263/jbb.104.329. [DOI] [PubMed] [Google Scholar]
- 69.Yoshikawa T, Uchimura E, Kishi M, Funeriu DP, Miyake M, Miyake J. Journal of Controlled Release. 2004;96:227–232. doi: 10.1016/j.jconrel.2004.01.024. [DOI] [PubMed] [Google Scholar]
- 70.Bengali Z, Pannier AK, Segura T, Anderson BC, Jang JH, Mustoe TA, Shea LD. Biotechnol Bioeng. 2005;90:290–302. doi: 10.1002/bit.20393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Okazaki A, Jo J-I, Tabata Y. Tissue Engineering. 2007;13:245–251. doi: 10.1089/ten.2006.0185. [DOI] [PubMed] [Google Scholar]
- 72.Bengali Z, Rea JC, Gibly RF, Shea LD. Biotechnology and Bioengineering. 2009;102:1679–1691. doi: 10.1002/bit.22212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Luo D, Saltzman WM. Nature Biotechnology. 2000;18:893–895. doi: 10.1038/78523. [DOI] [PubMed] [Google Scholar]
- 74.Tseng Sj, Chuang C-J, Tang S-C. Acta Biomaterialia. 2008;4:799–807. doi: 10.1016/j.actbio.2008.01.018. [DOI] [PubMed] [Google Scholar]
- 75.Delehanty JB, Shaffer KM, Lin B. Biosensors and Bioelectronics. 2004;20:773–779. doi: 10.1016/j.bios.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 76.Rea JC, Gibly RF, Barron AE, Shea LD. Acta Biomaterialia. 2009;5:903–912. doi: 10.1016/j.actbio.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhou Y-M, Ishikawa A, Okahashi R, Uchida K, Nemoto Y, Nakayama M, Nakayama Y. Journal of Controlled Release. 2007;123:239–246. doi: 10.1016/j.jconrel.2007.08.026. [DOI] [PubMed] [Google Scholar]
- 78.Segura T, Shea LD. Bioconjug Chem. 2002;13:621–629. doi: 10.1021/bc015575f. [DOI] [PubMed] [Google Scholar]
- 79.Mehrotra S, Lee I, Chan C. Acta Biomaterialia. 2009;5:1474–1488. doi: 10.1016/j.actbio.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pannier AK, Anderson BC, Shea LD. Acta Biomaterialia. 2005;1:511–522. doi: 10.1016/j.actbio.2005.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pannier AK, Wieland JA, Shea LD. Acta Biomaterialia. 2008;4:26–39. doi: 10.1016/j.actbio.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.ffrench-Constant C, Colognato H. Trends in Cell Biology. 2004;14:678–686. doi: 10.1016/j.tcb.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 83.Uchimura E, Yamada S, Nomura T, Matsumoto K, Fujita S, Miyake M, Miyake J. Journal of Bioscience and Bioengineering. 2007;104:152–155. doi: 10.1263/jbb.104.152. [DOI] [PubMed] [Google Scholar]
- 84.Segura T, Chung PH, Shea LD. Biomaterials. 2005;26:1575–1584. doi: 10.1016/j.biomaterials.2004.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bengali Z, Rea JC, Shea LD. Journal of Gene Medicine. 2007;9:668–678. doi: 10.1002/jgm.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boyd ND, Chan BMC, Petersen NO. Biochemistry and Cell Biology-Biochimie Et Biologie Cellulaire. 2003;81:335–348. doi: 10.1139/o03-063. [DOI] [PubMed] [Google Scholar]
- 87.De Laporte L, Lei Yan A, Shea LD. Biomaterials. 2009;30:2361–2368. doi: 10.1016/j.biomaterials.2008.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.De Laporte L, Huang A, Ducommun MM, Zelivyanska ML, Aviles MO, Adler AF, Shea LD. Acta Biomaterialia. doi: 10.1016/j.actbio.2010.02.018. In Press, Corrected Proof. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Biggs MJP, Richards RG, Dalby MJ. Nanomedicine: Nanotechnology, Biology and Medicine. doi: 10.1016/j.nano.2010.01.009. In Press, Uncorrected Proof. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chao WT, Kunz J. FEBS Lett. 2009;583:1337–1343. doi: 10.1016/j.febslet.2009.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lord MS, Foss M, Besenbacher F. Nano Today. 5:66–78. [Google Scholar]
- 92.Carpenter J, Khang D, Webster TJ. Nanotechnology. 2008;19 doi: 10.1088/0957-4484/19/50/505103. [DOI] [PubMed] [Google Scholar]
- 93.Gonzalez-Garcia C, Sousa SR, Moratal D, Rico P, Salmeron-Sanchez M. Colloids Surf B Biointerfaces. 77:181–190. doi: 10.1016/j.colsurfb.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 94.Chen H, Song W, Zhou F, Wu Z, Huang H, Zhang J, Lin Q, Yang B. Colloids and Surfaces B: Biointerfaces. 2009;71:275–281. doi: 10.1016/j.colsurfb.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 95.Koh LB, Rodriguez I, Venkatraman SS. Biomaterials. 2010;31:1533–1545. doi: 10.1016/j.biomaterials.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 96.Hovgaard MB, Rechendorff K, Chevallier J, Foss M, Besenbacher F. The Journal of Physical Chemistry B. 2008;112:8241–8249. doi: 10.1021/jp801103n. [DOI] [PubMed] [Google Scholar]
- 97.Lord MS, Cousins BG, Doherty PJ, Whitelock JM, Simmons A, Williams RL, Milthorpe BK. Biomaterials. 2006;27:4856–4862. doi: 10.1016/j.biomaterials.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 98.Denis FA, Hanarp P, Sutherland DS, Gold J, Mustin C, Rouxhet PG, Dufrêne YF. Langmuir. 2002;18:819–828. [Google Scholar]
- 99.Galli C, Coen MC, Hauert R, Katanaev VL, Wymann MP, Groning P, Schlapbach L. Surface Science. 2001;474:L180–L184. [Google Scholar]
- 100.Vertegel AA, Siegel RW, Dordick JS. Langmuir. 2004;20:6800–6807. doi: 10.1021/la0497200. [DOI] [PubMed] [Google Scholar]
- 101.Roach P, Farrar D, Perry CC. Journal of the American Chemical Society. 2006;128:3939–3945. doi: 10.1021/ja056278e. [DOI] [PubMed] [Google Scholar]
- 102.Stephansson SN, Byers BA, García AJ. Biomaterials. 2002;23:2527–2534. doi: 10.1016/s0142-9612(01)00387-8. [DOI] [PubMed] [Google Scholar]
- 103.Garcia AJ, Vega MD, Boettiger D. Mol Biol Cell. 1999;10:785–798. doi: 10.1091/mbc.10.3.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bae YH, Johnson PA, Florek CA, Kohn J, Moghe PV. Acta Biomaterialia. 2006;2:473–482. doi: 10.1016/j.actbio.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 105.Fasbender A, Zabner J, Zeiher BG, Welsh MJ. Gene Ther. 1997;4:1173–1180. doi: 10.1038/sj.gt.3300524. [DOI] [PubMed] [Google Scholar]
- 106.Park SC. Mechanisms of Ageing and Development. 2002;123:917–926. doi: 10.1016/s0047-6374(02)00029-5. [DOI] [PubMed] [Google Scholar]
- 107.Park JS, Park WY, Cho KA, Kim DI, Jhun BH, Kim SR, Park SC. Faseb J. 2001;15 doi: 10.1096/fj.00-0723fje. 1625-+ [DOI] [PubMed] [Google Scholar]
- 108.Martinez E, Engel E, Planell JA, Samitier J. Annals of Anatomy-Anatomischer Anzeiger. 2009;191:126–135. doi: 10.1016/j.aanat.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 109.Yim EK, Reano RM, Pang SW, Yee AF, Chen CS, Leong KW. Biomaterials. 2005;26:5405–5413. doi: 10.1016/j.biomaterials.2005.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Berry CC, Campbell G, Spadiccino A, Robertson M, Curtis AS. Biomaterials. 2004;25:5781–5788. doi: 10.1016/j.biomaterials.2004.01.029. [DOI] [PubMed] [Google Scholar]
- 111.Kim EJ, Boehm CA, Mata A, Fleischman AJ, Muschler GF, Roy S. Acta Biomater. 6:160–169. doi: 10.1016/j.actbio.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yim EK, Pang SW, Leong KW. Exp Cell Res. 2007;313:1820–1829. doi: 10.1016/j.yexcr.2007.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 114.Markert LD, Lovmand J, Foss M, Lauridsen RH, Lovmand M, Fuchtbauer EM, Fuchtbauer A, Wertz K, Besenbacher F, Pedersen FS, Duch M. Stem Cells and Development. 2009;18:1331–1342. doi: 10.1089/scd.2009.0114. [DOI] [PubMed] [Google Scholar]
- 115.Lovmand J, Justesen E, Foss M, Lauridsen RH, Lovmand M, Modin C, Besenbacher F, Pedersen FS, Duch M. Biomaterials. 2009;30:2015–2022. doi: 10.1016/j.biomaterials.2008.12.081. [DOI] [PubMed] [Google Scholar]
- 116.Washbourne P, McAllister AK. Current Opinion in Neurobiology. 2002;12:566–573. doi: 10.1016/s0959-4388(02)00365-3. [DOI] [PubMed] [Google Scholar]
- 117.Van De Parre TJL, Martinet W, Schrijvers DM, Herman AG, De Meyer GRY. Biochemical and Biophysical Research Communications. 2005;327:356–360. doi: 10.1016/j.bbrc.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 118.Lenz P, Bacot SM, Frazier-Jessen MR, Feldman GM. FEBS Letters. 2003;538:149–154. doi: 10.1016/s0014-5793(03)00169-8. [DOI] [PubMed] [Google Scholar]
- 119.Qureshi HY, Ahmad R, Zafarullah M. Analytical Biochemistry. 2008;382:138–140. doi: 10.1016/j.ab.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 120.Jiang ZY, Zhou QL, Coleman KA, Chouinard M, Boese Q, Czech MP. P Natl Acad Sci USA. 2003;100:7569–7574. doi: 10.1073/pnas.1332633100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cho KA, Park SC. Mechanisms of Ageing and Development. 2005;126:105–110. doi: 10.1016/j.mad.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 122.Perdue N, Yan Q. Experimental Eye Research. 2006;83:1154–1161. doi: 10.1016/j.exer.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 123.Prawitt J, Niemeier A, Kassem M, Beisiegel U, Heeren J. Experimental Cell Research. 2008;314:814–824. doi: 10.1016/j.yexcr.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 124.McFarland DC, Liu XS, Velleman SG, Zeng CY, Coy CS, Pesall JE. Comparative Biochemistry and Physiology C-Toxicology & Pharmacology. 2003;134:341–351. doi: 10.1016/s1532-0456(02)00272-7. [DOI] [PubMed] [Google Scholar]
- 125.Loesberg WA, te Riet J, van Delft FCMJM, Schon P, Figdor CG, Speller S, van Loon JJWA, Walboomers XF, Jansen JA. Biomaterials. 2007;28:3944–3951. doi: 10.1016/j.biomaterials.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 126.Dalby MJ, Riehle MO, Yarwood SJ, Wilkinson CD, Curtis AS. Exp Cell Res. 2003;284:274–282. doi: 10.1016/s0014-4827(02)00053-8. [DOI] [PubMed] [Google Scholar]
- 127.Dalby MJ, Riehle MO, Sutherland DS, Agheli H, Curtis ASG. European Journal of Cell Biology. 2004;83:159–169. doi: 10.1078/0171-9335-00369. [DOI] [PubMed] [Google Scholar]
- 128.Chalut KJ, Chen S, Finan JD, Giacomelli MG, Guilak F, Leong KW, Wax A. Biophysical Journal. 2008;94:4948–4956. doi: 10.1529/biophysj.107.124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chalut KJ, Kulangara K, Giacomelli MG, Wax A, Leong KW. Soft Matter. 2010;6:1675–1681. doi: 10.1039/B921206J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Le Beyec J, Xu R, Lee S-Y, Nelson CM, Rizki A, Alcaraz J, Bissell MJ. Experimental Cell Research. 2007;313:3066–3075. doi: 10.1016/j.yexcr.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dalby MJ, Riehle MO, Yarwood SJ, Wilkinson CDW, Curtis ASG. Experimental Cell Research. 2003;284:272–280. doi: 10.1016/s0014-4827(02)00053-8. [DOI] [PubMed] [Google Scholar]
- 132.Collinet C, Stoter M, Bradshaw CR, Samusik N, Rink JC, Kenski D, Habermann B, Buchholz F, Henschel R, Mueller MS, Nagel WE, Fava E, Kalaidzidis Y, Zerial M. Nature. 464:243–249. doi: 10.1038/nature08779. [DOI] [PubMed] [Google Scholar]
- 133.Dalby MJ, Yarwood SJ, Johnstone HJ, Affrossman S, Riehle MO. IEEE Trans Nanobioscience. 2002;1:12–17. doi: 10.1109/tnb.2002.806930. [DOI] [PubMed] [Google Scholar]
- 134.Dalby MJ, Yarwood SJ, Riehle MO, Johnstone HJH, Affrossman S, Curtsi ASG. Experimental Cell Research. 2002;276:1–9. doi: 10.1006/excr.2002.5498. [DOI] [PubMed] [Google Scholar]
- 135.Seabra MC. Journal of Biological Chemistry. 1996;271:14398–14404. doi: 10.1074/jbc.271.24.14398. [DOI] [PubMed] [Google Scholar]
- 136.Dalby MJ, Berry CC, Riehle MO, Sutherland DS, Agheli H, Curtis ASG. Experimental Cell Research. 2004;295:387–394. doi: 10.1016/j.yexcr.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 137.Dalby MJ, Gadegaard N, Wilkinson CDW. Journal of Biomedical Materials Research Part A. 2008;84A:973–979. doi: 10.1002/jbm.a.31409. [DOI] [PubMed] [Google Scholar]
- 138.Chen H, Slepnev VI, Di Fiore PP, De Camilli P. Journal of Biological Chemistry. 1999;274:3257–3260. doi: 10.1074/jbc.274.6.3257. [DOI] [PubMed] [Google Scholar]
- 139.Barbieri MA, Roberts RL, Gumusboga A, Highfield H, Alvarez-Dominguez C, Wells A, Stahl PD. The Journal of Cell Biology. 2000;151:539–550. doi: 10.1083/jcb.151.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tzanakakis GN, Karamanos NK, Hjerpe A. Exp Cell Res. 1995;220:130–137. doi: 10.1006/excr.1995.1299. [DOI] [PubMed] [Google Scholar]
- 141.Chen SL, Jones JA, Xu YG, Low HY, Anderson JM, Leong KW. Biomaterials. 2010;31:3479–3491. doi: 10.1016/j.biomaterials.2010.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Xia W, Wong EWP, Mruk DD, Cheng CY. Developmental Biology. 2009;327:48–61. doi: 10.1016/j.ydbio.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Martinez I, Sveinbjornsson B, Vidalvanaclocha F, Asumendi A, Smedsrod B. Biochemical and Biophysical Research Communications. 1995;212:235–241. doi: 10.1006/bbrc.1995.1961. [DOI] [PubMed] [Google Scholar]
- 144.Decuzzi P, Ferrari M. Biomaterials. 2010;31:173–179. doi: 10.1016/j.biomaterials.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 145.Ranella A, Barberoglou M, Bakogianni S, Fotakis C, Stratakis E. Acta Biomater. doi: 10.1016/j.actbio.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 146.Dalby MJ, Riehle MO, Johnstone HJ, Affrossman S, Curtis AS. Tissue Eng. 2002;8:1099–1108. doi: 10.1089/107632702320934191. [DOI] [PubMed] [Google Scholar]
- 147.Dalby MJ, Riehle MO, Johnstone H, Affrossman S, Curtis ASG. Biomaterials. 2002;23:2945–2954. doi: 10.1016/s0142-9612(01)00424-0. [DOI] [PubMed] [Google Scholar]
- 148.Cavalcanti-Adam EA, Volberg T, Micoulet A, Kessler H, Geiger B, Spatz JP. Biophysical Journal. 2007;92:2964–2974. doi: 10.1529/biophysj.106.089730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wary KK, Mariotti A, Zurzolo C, Giancotti FG. Cell. 1998;94:625–634. doi: 10.1016/s0092-8674(00)81604-9. [DOI] [PubMed] [Google Scholar]
- 150.Echarri A, Muriel O, Del Pozo MA. Semin Cell Dev Biol. 2007;18:627–637. doi: 10.1016/j.semcdb.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 151.Echarri A, Del Pozo MA. Cell Cycle. 2006;5:2179–2182. doi: 10.4161/cc.5.19.3264. [DOI] [PubMed] [Google Scholar]
- 152.del Pozo MA, Alderson NB, Kiosses WB, Chiang HH, Anderson RGW, Schwartz MA. Science. 2004;303:839–842. doi: 10.1126/science.1092571. [DOI] [PubMed] [Google Scholar]
- 153.Gallagher JO, McGhee KF, Wilkinson CD, Riehle MO. IEEE Trans Nanobioscience. 2002;1:24–28. doi: 10.1109/tnb.2002.806918. [DOI] [PubMed] [Google Scholar]
- 154.Dalby MJ, McCloy D, Robertson M, Wilkinson CD, Oreffo RO. Biomaterials. 2006;27:1306–1315. doi: 10.1016/j.biomaterials.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 155.Yim EK, Darling EM, Kulangara K, Guilak F, Leong KW. Biomaterials. 31:1299–1306. doi: 10.1016/j.biomaterials.2009.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cukierman E, Pankov R, Stevens DR, Yamada KM. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 157.Mao Y, Schwarzbauer JE. J Cell Sci. 2005;118:4427–4436. doi: 10.1242/jcs.02566. [DOI] [PubMed] [Google Scholar]
- 158.Sottile J, Chandler J. Mol Biol Cell. 2005;16:757–768. doi: 10.1091/mbc.E04-08-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wiesner S, Lange A, Fassler R. Trends Cell Biol. 2006;16:327–329. doi: 10.1016/j.tcb.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 160.Jeng RL, Welch MD. Current Biology. 2001;11:R691–R694. doi: 10.1016/s0960-9822(01)00410-9. [DOI] [PubMed] [Google Scholar]
- 161.Bader MF, Doussau F, Chasserot-Golaz S, Vitale N, Gasman S. Biochimica Et Biophysica Acta-Molecular Cell Research. 2004;1742:37–49. doi: 10.1016/j.bbamcr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 162.McPherson PS. Trends in Cell Biology. 2002;12:312–315. doi: 10.1016/s0962-8924(02)02309-7. [DOI] [PubMed] [Google Scholar]
- 163.Kopatz I, Remy J-S, Behr J-P. The Journal of Gene Medicine. 2004;6:769–776. doi: 10.1002/jgm.558. [DOI] [PubMed] [Google Scholar]