

Abstract
The mannose receptor (MR) is an endocytic type I membrane molecule with a broad ligand specificity that is involved in both hemostasis and pathogen recognition. Membrane-anchored MR is cleaved by a metalloproteinase into functional soluble MR (sMR) composed of the extracellular domains of intact MR. Although sMR production was initially considered a constitutive process, enhanced MR shedding has been observed in response to the fungal pathogen Pneumocystis carinii. In this work, we have investigated the mechanism mediating enhanced MR shedding in response to fungi. We show that other fungal species, including Candida albicans and Aspergillus fumigatus, together with zymosan, a preparation of the cell wall of Saccharomyces cerevisiae, mimic the effect of P. carinii on sMR production and that this effect takes place mainly through β-glucan recognition. Additionally, we demonstrate that MR cleavage in response to C. albicans and bioactive particulate β-glucan requires expression of dectin-1. Our data, obtained using specific inhibitors, are consistent with the canonical Syk-mediated pathway triggered by dectin-1 being mainly responsible for inducing MR shedding, with Raf-1 being partially involved. As in the case of steady-state conditions, MR shedding in response to C. albicans and β-glucan particles requires metalloprotease activity. The induction of MR shedding by dectin-1 has clear implications for the role of MR in fungal recognition, as sMR was previously shown to retain the ability to bind fungal pathogens and can interact with numerous host molecules, including lysosomal hydrolases. Thus, MR cleavage could also impact on the magnitude of inflammation during fungal infection.
Keywords: ADAM, ADAMTS, Fungi, Inflammation, Lectin, Macrophage, Metalloprotease, Mouse, Shedding
Introduction
The mannose receptor (MR)3 is an endocytic receptor with three distinct types of domains at its extracellular region, a single transmembrane region, and a cytoplasmic tail. The extracellular region mediates binding of a wide range of ligands; the N-terminal cysteine-rich domain is involved in sulfated sugar recognition in a Ca2+-independent manner; the fibronectin type II domain binds collagen types I–IV; and the eight C-type lectin-like domains are responsible for Ca2+-dependent binding to sugars terminating in mannose, fucose, or N-acetylglucosamine (1). This broad range of ligand binding, including endogenous antigens (such as lysosomal enzymes released into the extracellular milieu during inflammation, as well as thyroglobulin) and microbial antigens (Candida albicans, Leishmania, Mycobacterium tuberculosis, HIV, and Pneumocystis carinii), enables MR to participate in a wide range of processes, including hemostatic clearance, pathogen recognition, and cell migration (1, 2).
In addition to the cell-associated form (cMR), a soluble form of MR (sMR) has been detected previously in the supernatants from human dendritic cells and mouse macrophages (Mφ) and in mouse and human sera (3–6). sMR comprises the extracellular region of the receptor and is produced as a result of proteolytic cleavage of cMR by a metalloprotease (4). sMR has been suggested to play an important role in transferring mannosylated antigens to a subset of macrophages present in secondary lymphoid organs (7) and also in assisting pathogens to escape the immune response (8). Although initial studies indicated that MR shedding was a constitutive process, as the levels of sMR correlated with the amount of cMR present in the cells (5), Fraser et al. (8) observed that P. carinii enhanced sMR production.
β-Glucan is considered to be a major fungus-associated molecular pattern composing nearly 50% of the cell wall. β-Glucan consists of long polymers of β(1,3)-linked glucose with β(1,6)-linked branches. These polysaccharides are highly immunogenic, and in the fungal cell wall, they are mostly masked underneath a layer of mannosylated proteins. In certain fungal species such as C. albicans conidia and Aspergillus fumigatus maturing conidia, β-glucan surface expression can be detected also on restricted regions such as bud scars (9, 10). Dectin-1 does not bind β(1,3/1,4)-glucans to any appreciable extent (11).
Dectin-1 is a non-classical C-type lectin-like receptor with a type II transmembrane protein topology. The extracellular region of dectin-1 is composed of a single C-type lectin-like domain responsible for Ca2+-independent β-glucan binding and a short stalk, which is followed by a transmembrane domain and a cytoplasmic tail that contains an immunoreceptor tyrosine-based activation motif (ITAM)-like motif that mediates intracellular signaling (10). In mice, there are two isoforms as a result of alternative splicing: full-length dectin-1A and “stalkless” dectin-1B (12).
Dectin-1 was shown to recognize several fungal species, including P. carinii, C. albicans, Coccidioides posadasii, and A. fumigatus (10). Upon engagement, dectin-1 triggers intracellular signaling mediating a variety of cell responses, including phagocytosis, endocytosis, oxidative burst, activation and regulation of phospholipase A2 and cyclooxygenase-2, and production of various cytokines and chemokines (e.g. TNF, IL-2, IL-10, IL-6, and IL-23) (10, 13).
Dectin-1 can signal through both Syk-dependent and Syk-independent pathways. The Syk-dependent pathway involves phosphorylation of the cytoplasmic ITAM motif by Src kinases, allowing the recruitment and activation of Syk kinase (14, 15). The Syk-independent pathway is poorly characterized, and the only identified kinase involved is Raf-1 (16).
Dectin-1-mediated signaling can act independently (e.g. IL-2, IL-10, and reactive oxygen species) as well as in cooperation with the Toll-like receptor (TLR) pathway (e.g. TNF-α and IL-12) (10, 13). MyD88 and Syk (17), Raf-1 kinase (16), and CARD9 (18) were found to be required for this collaborative response.
In this study, we investigated if MR shedding could be up-regulated by other fungi in addition to P. carinii. Our results demonstrate that non-opsonic recognition of zymosan (a preparation of the cell wall of Saccharomyces cerevisiae), C. albicans, and A. fumigatus by Mφ leads to enhanced sMR production. Using purified soluble and particulate β-glucan and knock-out (KO) macrophages, we demonstrate that C. albicans-induced shedding is caused by the recognition of β-glucan on the fungal cell wall, that β-glucan binding is sufficient to induce shedding, and that this process requires dectin-1 expression, with inhibition studies supporting the involvement of Syk- and PI3K-dependent pathways and the need for metalloproteinase activity. Additionally, we observed a major effect of actin polymerization inhibitors on MR expression. Our results unveil a novel outcome for dectin-1 engagement, the rapid activation of metalloproteases leading to MR cleavage, which could potentially modulate the contribution of MR to fungal recognition.
EXPERIMENTAL PROCEDURES
Animals
WT C57BL/6 and BALB/c mice were supplied by Charles River, and MR-KO mice were breed at the Biomedical Services Unit of the University of Nottingham. Animals were handled according to institutional and UK Home Office guidelines and were kept under specific pathogen-free conditions. They were used at 7–10 weeks of age. Dectin-1-KO mice and their controls were on a 129S6/SvEv genetic background and were maintained in accordance with institutional guidelines at Cardiff University School of Medicine.
Cells
Thioglycolate-elicited Mφ (thio-Mφ) were obtained by intraperitoneal injection of 4% (w/v) brewers' thioglycolate broth (Sigma) 4 days before harvest. Mice were killed, and the peritoneal cavities were rinsed with PBS (Sigma) containing 5 mm EDTA. Mφ were cultured overnight on 6-well tissue culture plates (1.25 × 106 cells/well; BD Biosciences) in RPMI 1640 medium (Sigma) supplemented with R-10 medium (10% heat-inactivated (56 °C at 30 min) FBS (Sigma), 2 mm glutamine (Sigma), 100 units/ml penicillin, 100 μg/ml streptomycin (Sigma), and 10 mm Hepes (Invitrogen)) and washed three times with PBS prior use.
Experimental Conditions
Thio-Mφ were treated with zymosan (Invitrogen), particulate β-glucan, fixed A. fumigatus, heat-killed (HK) and fixed C. albicans (ATCC 18804), curdlan (Wako), and fixed Staphylococcus aureus (RN6390B strain, kindly contributed by Alan Cockayne, University of Nottingham) at concentrations of 50 particles/cell and/or Pam3-Cys-Ser-Lys4 (Alexis Biochemicals) in serum-free Opti-MEM medium (Invitrogen) supplemented with 2 mm glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin for 3 h at 37 °C and 5% CO2. Before treatment with formaldehyde-fixed stimuli, free aldehyde groups were quenched by incubating with 0.1 m glycine in PBS, followed by three washes with PBS.
For inhibition assays, thio-Mφ were preincubated with β-glucan phosphate, mannan (Sigma), Syk kinase inhibitor IV, wortmannin, Akt inhibitor VI, Raf-1 kinase inhibitor I, GM6001 (N-[(2R)-2(hydroxamidocarbonylmethyl)-4-methylpentanoyl]-l-tryptophan methylamide, a potent broad-spectrum hydroxamic acid inhibitor of matrix metalloproteases), a control for GM6001 (N-t-butoxycarbonyl-l-leucyl-l-tryptophan methylamide), cytochalasin D, latrunculin A (all from Calbiochem) or bafilomycin or chloroquine (Sigma) at the indicated concentrations for 1 h before treatment and were present during the incubation with the stimuli.
Collection of Culture Media and Preparation of Cell Lysates
Cell culture supernatants were collected after the addition of 1× protease inhibitors (Roche Applied Science), 10 mm EDTA, and 1 mg/ml BSA (Sigma). Following PBS washes, Mφ were lysed using lysis buffer (2% Triton X-100, 10 mm Tris-HCl (pH 8), 150 mm NaCl, 2 mm NaN3, and 10 mm EDTA) plus protease inhibitors for 45 min at 4 °C. The cell lysates and the culture media were centrifuged at 2000 rpm for 5 min at 4 °C and at 13,000 rpm for 30 min at 4 °C using a benchtop centrifuge. The cleared cell lysates and supernatants were stored at −20 °C. Protein concentration was determined using the BCA protein assay kit (Perbio) following the manufacturer's protocol.
Western Blotting
Cell lysate (3 μg of protein) and the corresponding fraction of non-concentrated culture supernatant were electrophoresed under nonreducing conditions on a 6% SDS-polyacrylamide gel, and proteins were transferred overnight to Hybond-C extra nitrocellulose membrane (GE Healthcare). After a 1-h incubation in blocking buffer (5% nonfat milk in PBS and 0.1% Tween 20), MR and CD44 were visualized using rat anti-MR mAb (clone 5D3, prepared in house) and rat anti-CD44 mAb (clone KM201, Abcam) respectively, in combination with HRP-conjugated anti-rat IgG (Chemicon) and enhanced chemiluminescence reagent (Amersham Biosciences).
Flow Cytometry Analysis
Thio-Mφ were plated for 2 h at 37 °C and 5% CO2 on bacteriological plastic in Opti-MEM with GlutaMAX containing 100 units/ml penicillin and 100 μg/ml streptomycin. After 2 h, Opti-MEM was replaced with R-10 medium; cells were maintained at 37 °C and 5% CO2 overnight; and Mφ were selected by washing three times with cold PBS. Mφ were collected by scraping in the presence of non-enzymatic cell dissociation buffer (Sigma), washed with FACS block buffer (5% heat-inactivated rabbit serum, 0.5% BSA, 2 mm NaN3, and 5 mm EDTA in PBS), and incubated in FACS block buffer containing 2.4G2 (10 μg/ml) for 30 min at 4 °C. After blocking, Mφ were incubated with Alexa 488-labeled anti-MR Ab (clone 5D3, BioLegend) or FITC-labeled anti-dectin-1 Ab (clone 2A11, AbD Serotec) for 60 min at 4 °C. After staining, cells were washed three times with FACS wash buffer (0.5% BSA, 2 mm NaN3, and 5 mm EDTA in PBS) and fixed in 1% paraformaldehyde in PBS. Isotype-matched Abs were used as controls. Labeling was analyzed using a Beckman Coulter Epics Altra system.
Real-time Quantitative PCR
RNA was extracted and purified using an RNeasy mini kit (Qiagen). DNase digestion was performed using an RNase-free DNase set (Qiagen), and 500–1000 ng of total RNA was used in the reverse transcription reactions. Quantitative PCR was performed using Brilliant SYBR Green QPCR Master Mix (Stratagene) according to the manufacturer's instructions in an Mx3005P instrument (Stratagene). MxPro-Mx3005P software was used to analyze results. The primers used were as follows: HPRT, 5′-GTAATGATCAGTCAACGGGGGAC-3′ (forward) and CCAGCAAGCTTGCAACCTTAACCA-3′ (reverse); MMP-9, 5′-CAGAGGTAACCCACGTCAGC-3′ (forward) and 5′-GGGATCCACCTTCTGAGACTT-3′ (reverse); and MMP-8, 5′-CTTTCAACCAGGCCAAGG-3′ (forward) and 5′-GAGCAGCCACGAGAAATAGG-3′ (reverse).
Cytokine Assays
The levels of TNF-α and KC (CXCL1) were determined using the mouse TNF-α DuoSet and mouse CXCL1/KC DuoSet (R&D Systems), respectively.
RESULTS
MR Shedding Is Promoted by Non-opsonic Recognition of Fungal Particles
To investigate if stimulation of Mφ with other fungal particles in addition to P. carinii (8) could promote MR shedding, thio-Mφ were treated with zymosan, a S. cerevisiae-derived bioparticle, under serum-free conditions for 1.5 and 3 h. After treatment, cell culture supernatants and cell lysates were analyzed for sMR and cMR levels, respectively, by Western blotting. As shown in Fig. 1A, sMR could be detected in the supernatants of zymosan-treated cells at 1.5 h post-treatment with levels increasing at 3 h. No sMR could be detected in the supernatants of untreated cells under these conditions. In support of a more general effect of fungi in enhancing the production of sMR, supernatants collected from Mφ treated with fixed A. fumigatus and fixed and HK C. albicans under serum-free conditions for 3 h also contained increased levels of sMR (Fig. 1B). This effect appears to be pathogen-specific, as increased MR shedding was not evident when fixed S. aureus was used (data not shown).
FIGURE 1.

MR shedding is induced by zymosan, C. albicans, and A. fumigatus and requires β-glucan recognition. Thio-Mφ were incubated with zymosan (50 particles/cell; Z-50) for 1.5 and 3 h at 37 °C (A) or fixed A. fumigatus (A. f.) and fixed or HK C. albicans (50 particles/cell) for 3 h at 37 °C in serum-free medium (B). cMR and sMR levels were assessed in cell lysates and supernatants, respectively, by Western blot analysis as described under “Experimental Procedures.” All fungal particles induced MR shedding (A and B). MR shedding in response to HK C. albicans could be inhibited by soluble β-glucan phosphate (β-Gluc; 1 mg/ml) but not by mannan (Man; 1 mg/ml) (C). Data are representative of three independent experiments. Unt, untreated.
The method of fungal processing appeared to have an effect on the level of sMR production, as HK C. albicans consistently induced more MR shedding than the fixed fungal particles. As the β-glucan component of the cell wall is thought to become more exposed during heat inactivation, we hypothesized that enhanced β-glucan exposure could be responsible for the differences observed between HK and fixed fungi and that the recognition of β-glucan in the fungal cell wall could be responsible for their effect on MR cleavage. We investigated this possibility by performing the treatment with HK C. albicans in the presence of soluble glucan phosphate. Fig. 1C shows that glucan phosphate (but not mannan) treatment considerably blocked sMR production, which suggests that fungus-induced MR shedding is mediated mainly through the recognition of the β-glucan component of the cell wall. These results also demonstrate that the recognition of soluble β-glucan or mannan per se is insufficient for inducing sMR production.
β-Glucan Particles Induce MR Shedding through Dectin-1 Engagement
To investigate if the recognition of particulate β-glucan is sufficient to induce MR cleavage, we analyzed sMR production by thio-Mφ in response to purified β-glucan particles. The β-glucan particles used in these experiments were isolated from S. cerevisiae and consist of β(1–3)-linked glucose with a limited number of β(1–6)-linkages. Particulate β-glucan enhanced MR shedding when used at a particle/cell ratio similar to that used in the case of zymosan, C. albicans, and A. fumigatus, and pretreatment with soluble glucan phosphate demonstrated that this effect was not mediated by any contaminant present in the preparation of particulate β-glucan (Fig. 2A). Using dectin-1-KO thio-Mφ, we determined that dectin-1, the major β-glucan receptor, is essential for the induction of MR shedding in response to particulate β-glucan, as no enhanced sMR production was observed in response to particulate β-glucan when these cells were tested (Fig. 2A). HK C. albicans-induced sMR production was also reduced in dectin-1-KO cells, although to a lesser extent (Fig. 2B).
FIGURE 2.

β-Glucan treatment induces sMR production through dectin-1 engagement independently of the mouse strain and does not promote CD44 shedding. Sex- and age-matched WT and dectin-1-KO thio-Mφ were treated with β-glucan particles (50 particles/cell) in the presence and absence of 1 mg/ml glucan phosphate (β-Gluc) or 1 mg/ml mannan (Man) (A). Sex- and age-matched WT and dectin-1-KO thio-Mφ were treated with HK C. albicans (50 particles/cell) (B). Analysis of cell lysates and culture supernatants by Western blotting demonstrated dectin-1-mediated enhanced sMR production in response to particulate β-glucan that could be inhibited by soluble β-glucan but not by mannan (A). Similarly, sMR production in response to HK C. albicans was reduced in dectin-1-deficient cells (B). Both C57BL/6 and BALB/c Mφ were capable of shedding MR in response to particulate β-glucan (C), despite the different levels of surface dectin-1 expression in each strain (D). CD44 could be detected in cell lysates but not in supernatants from β-glucan-treated WT Mφ, whereas cMR and sMR were detected in cell lysates and supernatants, respectively (E). No signal could be detected in supernatants from MR-KO cells treated with β-glucan (F). Data are representative of two or three independent experiments (A–D and F). Unt, untreated.
When thio-Mφ from C57BL/6 and BALB/c mice were compared, similar levels of sMR production in response to β-glucan were observed (Fig. 2C), indicating that the presence of different alternatively spliced isoforms of dectin-1 in C57BL/6 and BALB/c Mφ and the increased surface expression of dectin-1 in C57BL/6 cells (Fig. 2D) (12) did not affect the shedding process. Confirming the specificity of the MR-shedding process, particulate β-glucan recognition did not lead to a general release of receptors from the cell surface, as it failed to induce the shedding of CD44 (Fig. 2E). CD44 can be shed from the cell surface as a result of multiple signaling pathways by several metalloproteases (19). No band cross-reactive with the anti-MR antibody was detected in supernatants from MR-KO thio-Mφ treated with β-glucan (Fig. 2F).
β-Glucan-mediated MR Shedding Requires the Canonical Signaling Pathway Triggered by Dectin-1 and Is Independent of TLR
Syk kinase, the kinase responsible for most of dectin-1-mediated signaling events, appears to be indispensible for β-glucan-induced sMR production because MR shedding was completely blocked by Syk kinase inhibition (Fig. 3A). Raf-1 kinase seems to be partially responsible for this effect, as reduced sMR production was observed only at high concentrations of Raf-1 kinase inhibitor (Fig. 3B).
FIGURE 3.

Dectin-1-mediated MR shedding is dependent on Syk and partially on Raf-1, utilizes the Akt-independent PI3K pathway, and is not affected by the presence of an agonist for TLR2. Thio-Mφ preincubated for 1 h with Syk kinase inhibitor IV (A), Raf-1 inhibitor I (B), wortmannin (C), or Akt inhibitor VI (D) were treated with particulate β-glucan (β-Gluc; 50 particles/cell) for 3 h at 37 °C in the presence of inhibitors. cMR and sMR levels were assessed in cell lysates and supernatants, respectively, by Western blot analysis as described under “Experimental Procedures.” Thio-Mφ were treated with Pam3-Cys-Ser-Lys4 (10 μg/ml) in the presence or absence of particulate β-glucan (50 particles/cell) for 3 h at 37 °C (E). cMR and sMR levels were assessed in cell lysates and supernatants. Data are representative of three independent experiments. Unt, untreated.
The potential role of the PI3K/Akt (protein kinase B) pathway in β-glucan-induced MR shedding was investigated using PI3K and Akt inhibitors. Phosphoinositides are crucial second messengers for intracellular signaling pathways initiated by dectin-1-mediated Syk kinase activation (20–25). Among several other downstream elements that PI3K activates, Akt was shown to be required for the production of cytokines such as IL-12 and IL-10 as well as for controlling cell proliferation and survival (26–29). Enhanced MR shedding was completely blocked upon PI3K inhibition with wortmannin, a potent, cell-permeable, and irreversible inhibitor of PI3K enzymes (Fig. 3C), but Akt inhibition had no effect (Fig. 3D). Therefore, these results suggest that β-glucan induces MR shedding through an Akt-independent, PI3K-mediated signaling pathway.
We also investigated if collaboration between dectin-1- and TLR-mediated signaling could have an effect on the shedding process by analyzing sMR production in the presence of the TLR2 agonist Pam3-Cys-Ser-Lys4. TLR2 engagement did not affect MR shedding in isolation or in combination with β-glucan (Fig. 3E), indicating that this process is mediated exclusively by a dectin-1-dependent pathway.
β-Glucan-induced MR Shedding Is Phagocytosis-independent but Requires Actin Polymerization
Even though dectin-1 is a phagocytic receptor, it does not require phagocytosis to induce inflammatory responses (30). In fact, dectin-1-mediated intracellular signaling is enhanced by frustrated phagocytosis (31, 32). We investigated if phagocytosis is required for dectin-1-mediated MR shedding using three different approaches: (i) using the non-phagocytosable β-glucan curdlan, (ii) inhibiting actin polymerization with latrunculin A or cytochalasin D, and (iii) inhibiting phagosome acidification.
As shown in Fig. 4A, incubation of thio-Mφ with curdlan, a β(1–3)-glucan preparation from Alcaligenes faecalis that is known to be able to hydrate and form gels, enhanced MR shedding, which suggests that dectin-1-mediated MR shedding is independent of particle internalization. To eliminate the possibility of small curdlan fragments being responsible for the effects of curdlan effect on MR shedding, only curdlan particles repeatedly (three times) retained in a 100-μm cutoff filter were used for these experiments. Microscopic examination of these preparations demonstrated that all small fragments had been eliminated.
FIGURE 4.
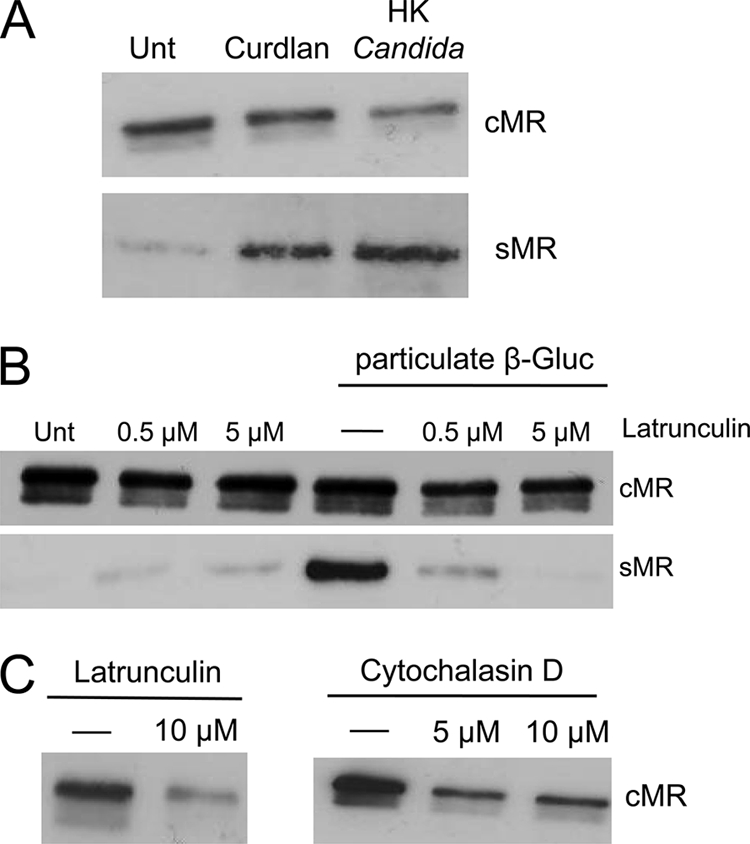
Dectin-1-induced sMR production is phagocytosis-independent but requires actin polymerization. Thio-Mφ were treated with curdlan and HK C. albicans (A) under serum-free conditions at concentrations of four particles/cell (10 μg of curdlan would contain ∼550 particles (32)) and 50 particles/cell, respectively. cMR and sMR levels were determined as described under “Experimental Procedures.” Both particles were found to enhance MR shedding (A). Latrunculin A inhibited the production of sMR in response to β-glucan (β-Gluc) treatment (B). Higher doses of latrunculin A and cytochalasin D reduced the levels of cMR (C). Data are representative of three independent experiments. Unt, untreated.
Despite the ability of curdlan to promote MR shedding, we observed that two inhibitors of actin polymerization that work through different mechanisms, cytochalasin D (used at 0.5–1 μm) and latrunculin A (used at 0.5–5 μm), blocked sMR and that latrunculin A blocked sMR production in response to β-glucan (Fig. 4B). This could be due to an inherent effect of these inhibitors on MR biology, as a drastic reduction is MR expression was observed when higher amounts were used: 10 μm instead of 5 μm for latrunculin and 5 μm instead of 1 μm for cytochalasin D (Fig. 4C). In support of phagocytosis not being required for MR shedding in response to β-glucan particles, inhibition of phagosome acidification using bafilomycin or chloroquine did not alter sMR production in response to β-glucan (data not shown).
Fungus-induced MR Shedding Is Mediated by a Metalloprotease
Comparison of the effects of different protease activity inhibitors on sMR production revealed that MR shedding under steady-state conditions depends on a metalloproteinase activity (4). Treatment of thio-Mφ with GM6001, a broad-spectrum matrix metalloproteinase (MMP)/ADAM (a disintegrin and metalloprotease) inhibitor, blocked HK C. albicans- and β-glucan-induced sMR production (Fig. 5, A and C), which suggests that dectin-1-mediated MR shedding, as in the case of steady-state sMR production, is MMP- or ADAM-dependent. As expected, GM6001 treatment had a drastic effect on TNF-α production (33) but did not affect KC (chemokine CXCL1) production in response to HK C. albicans (Fig. 5B), confirming its specificity. In this study, we also investigated production of IL-1β and IL-6, which could not be detected (data not shown).
FIGURE 5.

Dectin-1-induced MR shedding by a non-secreted metalloprotease. GM6001 treatment inhibited MR shedding (A and C). Mφ were treated with the inhibitor GM6001 (Inh) at 1 h prior and during the 3-h treatment with HK C. albicans (A) or β-glucan (β-Gluc) particles (C), or a GM6001 control (Cont) was used as a negative control. GM6001 treatment reduced TNF-α production but did not affect KC (CXCL1) production in response to HK C. albicans (B). MMP-8- and MMP-9-specific mRNAs in thio-Mφ treated with particulate β-glucan were quantitated (D). Data are representative of three independent experiments. Unt and U, untreated.
We did not obtain any data supporting a role for MMP-2 or MMP-9 in MR shedding in response to β-glucan using gelatin zymography; only a band corresponding to MMP-9 was visualized in cell lysates and supernatants from untreated and β-glucan- or HK C. albicans-treated thio-Mφ (data not shown). These observations correlated with quantitative PCR results demonstrating a lack of MMP-2- and MMP-3-specific mRNAs (data not shown) and the presence of MMP-8- and MMP-9-specific mRNAs in thio-Mφ. The levels of MMP-8- and MMP-9-specific mRNAs were not affected by the presence of particulate β-glucan (Fig. 5D).
DISCUSSION
In this study, we have shown that (i) induction of MR shedding is a broad feature of fungal pathogens such as C. albicans and A. fumigatus; (ii) β-glucan is the cell wall component responsible for most of the fungus-induced MR shedding; and (iii) MR shedding in response to β-glucan depends completely on dectin-1, Syk kinase, PI3K, and MMP/ADAM activity, partially on Raf-1 kinase, and is independent of phagocytosis and Akt.
Induction of MR Shedding Is a Feature of Fungal Pathogens and Is Mediated by Dectin-1
Enhanced MR shedding was initially described in response to P. carinii (8). Using fixed A. fumigatus and both HK and fixed C. albicans, we have shown that MR shedding can also be induced by other opportunistic pathogens (Fig. 1). In correlation with previous studies demonstrating that dectin-1 is a major pattern recognition receptor for fungi (34–36), we showed that fungus-mediated sMR production is induced mainly by β-glucan recognition (Figs. 1C and 2) through dectin-1 (Fig. 2). These results were further supported by the significant reduction in MR shedding induced by HK C. albicans in dectin-1-deficient cells (Fig. 2B), although the residual levels observed under these conditions indicate a potential contribution of other receptors. In agreement with previous studies demonstrating high dependence on ligand valence for dectin-1 signaling, soluble β-glucan (glucan phosphate), in contrast to particulate β-glucan and curdlan, acted as an antagonist. Further supporting the wide relevance of these observations, thio-Mφ from C57BL/6 and BALB/c mice shed equivalent amounts of MR despite the fact that they express different dectin-1 isoforms (12).
Among the two signaling pathways induced by dectin-1, the Syk-dependent pathway is the most characterized because of the common elements shared with other ITAM-mediated pathways. The Syk-independent signaling pathway is still poorly characterized but has been shown to involve Raf-1 kinase activity (16). In our study, we showed that the effect of the Syk-independent pathway on sMR production was significantly weaker than that of the Syk-dependent pathway, whose inhibition completely stopped the enhanced MR shedding (Fig. 3).
The partial dependence of the phenomenon on Raf-1 kinase suggests the requirement of a possible cross-talk between Syk kinase-dependent and Syk kinase-independent pathways. This might occur via PI3K because PI3K and Raf-1 kinase were previously shown to induce activation of each other in response to various growth factors (37–40). Apart from PI3K, Raf-1 also stimulates the downstream kinase MEK, which in turn activates ERK1/2 (37, 41, 42). Future work will need to address the question as to whether this pathway is required for dectin-1-mediated sMR production. Collaboration between TLR and dectin-1 (17) contributing to MR shedding appears to be unlikely, as treatment with the TLR2 agonist Pam3-Cys-Ser-Lys4 on its own or in combination with particulate β-glucan (Fig. 3E), even after prolonged incubation periods (e.g. overnight) (data not shown), did not influence sMR production.
Role of Actin Polymerization in MR Shedding
The requirement for actin polymerization in this model is intriguing because this does not appear to be caused by a requirement for phagosome formation (Fig. 4). A possibility that could explain these observations is the requirement of actin polymerization for MR recycling, as suggested by Deslée et al. (43). In this study, MR-mediated endocytosis of dextran and Der p 1 by human dendritic cells was shown to be reduced significantly in the presence of the actin polymerization inhibitor cytochalasin D (43). This would also explain the drastic reduction in cMR levels observed when actin polymerization inhibitors were used at higher concentrations (Fig. 4C), as non-recycled MR could be targeted for degradation. A detailed analysis of MR function in the absence of actin polymerization would be required to address this issue.
Enhanced MR Shedding in Response to C. albicans and β-Glucan Particles Requires Metalloproteinase Activity
Our results suggest the requirement for a protease sensitive to the broad-spectrum metalloproteinase inhibitor GM6001, known to inhibit both MMPs and ADAM proteins. The lack of soluble CD44 detection in the β-glucan-treated samples (Fig. 2D) suggests the exclusion of membrane-associated MMP-14, ADAM-10, and ADAM-17 from the list of candidate enzymes responsible for the enhanced sMR production, as these metalloproteases have previously been implicated in CD44 ectodomain shedding (19). We attempted to examine further the possible ADAM participation in our model by treating thio-Mφ with ionomycin, as calcium influx has been shown to activate selected metalloproteases (33). Treatment of thio-Mφ with ionomycin had no effect on MR cleavage in the presence of serum. On the other hand, when ionomycin was added in the absence of serum, a major increase in sMR production was detected (data not shown). This could be a consequence of the increased cell death we observed under these conditions. This enhanced level of MR ectodomain shedding under conditions of increased cell death suggested the possibility of the involvement in MR shedding of other ITAM-coupled receptors such as Mincle, known to recognize dead cells (44). Preliminary results indicate that uptake of dead cells by thio-Mφ does not affect MR cleavage, making this possibility unlikely (data not shown). In an attempt to discern whether the metalloprotease responsible for MR shedding was secreted into the medium, WT thio-Mφ were incubated with culture supernatants collected from β-glucan-treated MR-deficient Mφ. We reasoned that this would be a suitable approach because β-glucan recognition is not affected by MR deficiency (19). MR shedding was not altered upon treatment with culture supernatants collected from MR-KO Mφ treated with β-glucan (data not shown), indicating that the protease responsible for MR shedding is probably membrane-anchored.
MR and Dectin-1, the Yin and Yang of Fungal Recognition
MR and dectin-1 bind distinct structures in the fungal cell wall; MR recognizes the external layer of mannoproteins and mannan, whereas dectin-1 recognizes β-glucans. β-Glucans are thought to be largely masked by the mannose-rich outer layer. This arrangement is considered to facilitate fungal immunoevasion, as β-glucans are highly immunogenic and, in isolation or in combination with other microbial compounds, promote inflammation. Despite this arrangement of the fungal cell wall, lack of dectin-1 expression causes defective recognition and uptake of fungal pathogens by Mφ (45). In vitro studies using thio-Mφ suggest that MR involvement during C. albicans recognition is restricted to the phagosomal compartment and leads to enhanced TNF-α and MCP-1 production, indicating that MR can indeed act as a sensor of fungi but, at least in this experimental model, downstream of dectin-1 (45).
In the mouse model, dectin-1 deficiency leads to increased susceptibility to fungal infection in the case of P. carinii and A. fumigatus (34), with conflicting evidence available regarding its involvement in C. albicans infection (46) and no involvement reported in the case of Cryptococcus neoformans (47). In the case of MR, one in vivo study suggested that MR deficiency does not increase susceptibility to C. albicans, and it has been argued that this could be caused by the use of an intraperitoneal model of infection (46). Additionally, even though the survival was similar between WT and MR-KO animals, fungal outgrowth did show a difference. Similarly, in the case of P. carinii, there was no major effect on survival, but increased inflammatory cells in the bronchoalveolar lavage of MR-KO animals were observed. Additionally and consistent with a role for MR in glycoprotein clearance, there was significantly greater glycoprotein accumulation in the lung lavage of MR-KO mice (48). MR-KO mice were more susceptible to infection by C. neoformans, which appears to correlate with reduced development of CD4 T cell responses to cryptococcal mannoproteins (2). An important role of MR in fungal recognition has been suggested by its ability to bind N-bound mannans from C. albicans, its involvement in the induction of Th17 responses to C. albicans (46), and its key role in the recognition of P. carinii by human alveolar macrophages (49, 50).
MR shedding promoted by dectin-1 could represent an additional mechanism for modulating MR involvement in antifungal responses. sMR is capable of binding fungal particles (5), so the generation of sMR-coated fungi is a real possibility as indicated by the presence of MR-coated P. carinii in the lungs of infected animals (8). Additionally, the presence in mice of ligands for the cysteine-rich domain of MR in cells lacking MR expression in lymphoid organs (7, 51, 52) and kidneys (53) suggests the possibility of the delivery of MR ligands (both soluble and particulate) to these organs. Our opinion is that only the generation of mouse and cellular models in which MR cleavage does not take place will clarify the contribution of MR shedding to fungal pathogenesis, particularly in view of the broad effects that metalloproteinase deficiencies and metalloproteinase inhibitors have on biological processes at the cellular and organism levels.
In conclusion, we have identified a novel event triggered by dectin-1 engagement, the enhanced shedding of MR. This intriguing interplay between two major pattern recognition receptors involved in fungal recognition has the potential to bias the innate recognition process and the inflammatory response to fungi.
Acknowledgment
We thank the support of all staff at the Biomedical Services Unit, University of Nottingham.
This work was supported in part by the Mizutani Foundation for Glycoscience (Japan) and Cancer Research UK.
- MR
- mannose receptor
- cMR
- cell-associated MR
- sMR
- soluble MR
- Mφ
- macrophage(s)
- ITAM
- immunoreceptor tyrosine-based activation motif
- TLR
- Toll-like receptor
- KO
- knock-out
- thio-Mφ
- thioglycolate-elicited Mφ
- HK
- heat-killed
- MMP
- matrix metalloproteinase
- Pam3
- (S)-[2,3-Bis(palmitoyloxy)-(2-RS)-propyl]-N-palmitoyl-(R)-Cys-(S)-Ser-(S)-Lys4-OH·3HCl.
REFERENCES
- 1. Gazi U., Martinez-Pomares L. (2009) Immunobiology 214, 554–561 [DOI] [PubMed] [Google Scholar]
- 2. Dan J. M., Kelly R. M., Lee C. K., Levitz S. M. (2008) Infect. Immun. 76, 2362–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taylor P. R., Gordon S., Martinez-Pomares L. (2005) Trends Immunol. 26, 104–110 [DOI] [PubMed] [Google Scholar]
- 4. Martinez-Pomares L., Wienke D., Stillion R., McKenzie E. J., Arnold J. N., Harris J., McGreal E., Sim R. B., Isacke C. M., Gordon S. (2006) Eur. J. Immunol. 36, 1074–1082 [DOI] [PubMed] [Google Scholar]
- 5. Martínez-Pomares L., Mahoney J. A., Káposzta R., Linehan S. A., Stahl P. D., Gordon S. (1998) J. Biol. Chem. 273, 23376–23380 [DOI] [PubMed] [Google Scholar]
- 6. Jordens R., Thompson A., Amons R., Koning F. (1999) Int. Immunol. 11, 1775–1780 [DOI] [PubMed] [Google Scholar]
- 7. Martínez-Pomares L., Crocker P. R., Da Silva R., Holmes N., Colominas C., Rudd P., Dwek R., Gordon S. (1999) J. Biol. Chem. 274, 35211–35218 [DOI] [PubMed] [Google Scholar]
- 8. Fraser I. P., Takahashi K., Koziel H., Fardin B., Harmsen A., Ezekowitz R. A. (2000) Microbes Infect. 2, 1305–1310 [DOI] [PubMed] [Google Scholar]
- 9. Ruiz-Herrera J., Elorza M. V., Valentín E., Sentandreu R. (2006) FEMS Yeast Res. 6, 14–29 [DOI] [PubMed] [Google Scholar]
- 10. Brown G. D. (2006) Nat. Rev. Immunol. 6, 33–43 [DOI] [PubMed] [Google Scholar]
- 11. Adams E. L., Rice P. J., Graves B., Ensley H. E., Yu H., Brown G. D., Gordon S., Monteiro M. A., Papp-Szabo E., Lowman D. W., Power T. D., Wempe M. F., Williams D. L. (2008) J. Pharmacol. Exp. Ther. 325, 115–123 [DOI] [PubMed] [Google Scholar]
- 12. Heinsbroek S. E., Taylor P. R., Rosas M., Willment J. A., Williams D. L., Gordon S., Brown G. D. (2006) J. Immunol. 176, 5513–5518 [DOI] [PubMed] [Google Scholar]
- 13. Willment J. A., Brown G. D. (2008) Trends Microbiol. 16, 27–32 [DOI] [PubMed] [Google Scholar]
- 14. Rogers N. C., Slack E. C., Edwards A. D., Nolte M. A., Schulz O., Schweighoffer E., Williams D. L., Gordon S., Tybulewicz V. L., Brown G. D., Reis e Sousa C. (2005) Immunity 22, 507–517 [DOI] [PubMed] [Google Scholar]
- 15. Underhill D. M., Rossnagle E., Lowell C. A., Simmons R. M. (2005) Blood 106, 2543–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gringhuis S. I., den Dunnen J., Litjens M., van der Vlist M., Wevers B., Bruijns S. C., Geijtenbeek T. B. (2009) Nat. Immunol. 10, 203–213 [DOI] [PubMed] [Google Scholar]
- 17. Dennehy K. M., Ferwerda G., Faro-Trindade I., Pyz E., Willment J. A., Taylor P. R., Kerrigan A., Tsoni S. V., Gordon S., Meyer-Wentrup F., Adema G. J., Kullberg B. J., Schweighoffer E., Tybulewicz V., Mora-Montes H. M., Gow N. A., Williams D. L., Netea M. G., Brown G. D. (2008) Eur. J. Immunol. 38, 500–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gross O., Gewies A., Finger K., Schäfer M., Sparwasser T., Peschel C., Förster I., Ruland J. (2006) Nature 442, 651–656 [DOI] [PubMed] [Google Scholar]
- 19. Stamenkovic I., Yu Q. (2009) J. Invest. Dermatol. 129, 1321–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hiller G., Sternby M., Sundler R., Wijkander J. (2000) Biochim. Biophys. Acta 1485, 163–172 [DOI] [PubMed] [Google Scholar]
- 21. Olsson S., Sundler R. (2007) Mol. Immunol. 44, 1509–1515 [DOI] [PubMed] [Google Scholar]
- 22. Olsson S., Sundler R. (2006) J. Inflamm. 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shah V. B., Ozment-Skelton T. R., Williams D. L., Keshvara L. (2009) Mol. Immunol. 46, 1845–1853 [DOI] [PubMed] [Google Scholar]
- 24. Lee J. Y., Kim J. Y., Lee Y. G., Rhee M. H., Hong E. K., Cho J. Y. (2008) J. Microbiol. Biotechnol. 18, 355–364 [PubMed] [Google Scholar]
- 25. Crowley M. T., Costello P. S., Fitzer-Attas C. J., Turner M., Meng F., Lowell C., Tybulewicz V. L., DeFranco A. L. (1997) J. Exp. Med. 186, 1027–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weichhart T., Säemann M. D. (2008) Ann. Rheum. Dis. 67, iii70–4 [DOI] [PubMed] [Google Scholar]
- 27. Martin M., Schifferle R. E., Cuesta N., Vogel S. N., Katz J., Michalek S. M. (2003) J. Immunol. 171, 717–725 [DOI] [PubMed] [Google Scholar]
- 28. Polumuri S. K., Toshchakov V. Y., Vogel S. N. (2007) J. Immunol. 179, 236–246 [DOI] [PubMed] [Google Scholar]
- 29. Koyasu S. (2003) Nat. Immunol. 4, 313–319 [DOI] [PubMed] [Google Scholar]
- 30. McCann F., Carmona E., Puri V., Pagano R. E., Limper A. H. (2005) Infect. Immun. 73, 6340–6349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hernanz-Falcón P., Joffre O., Williams D. L., Reis e Sousa C. (2009) Eur. J. Immunol. 39, 507–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rosas M., Liddiard K., Kimberg M., Faro-Trindade I., McDonald J. U., Williams D. L., Brown G. D., Taylor P. R. (2008) J. Immunol. 181, 3549–3557 [DOI] [PubMed] [Google Scholar]
- 33. Huovila A. P., Turner A. J., Pelto-Huikko M., Kärkkäinen I., Ortiz R. M. (2005) Trends Biochem. Sci. 30, 413–422 [DOI] [PubMed] [Google Scholar]
- 34. Taylor P. R., Tsoni S. V., Willment J. A., Dennehy K. M., Rosas M., Findon H., Haynes K., Steele C., Botto M., Gordon S., Brown G. D. (2007) Nat. Immunol. 8, 31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Werner J. L., Metz A. E., Horn D., Schoeb T. R., Hewitt M. M., Schwiebert L. M., Faro-Trindade I., Brown G. D., Steele C. (2009) J. Immunol. 182, 4938–4946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brown G. D., Herre J., Williams D. L., Willment J. A., Marshall A. S., Gordon S. (2003) J. Exp. Med. 197, 1119–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang C. C., Cirit M., Haugh J. M. (2009) Mol. Syst. Biol. 5, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sutor S. L., Vroman B. T., Armstrong E. A., Abraham R. T., Karnitz L. M. (1999) J. Biol. Chem. 274, 7002–7010 [DOI] [PubMed] [Google Scholar]
- 39. Wennström S., Downward J. (1999) Mol. Cell. Biol. 19, 4279–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scheid M. P., Woodgett J. R. (2000) Curr. Biol. 10, R191–R194 [DOI] [PubMed] [Google Scholar]
- 41. Yart A., Chap H., Raynal P. (2002) Biochim. Biophys. Acta 1582, 107–111 [DOI] [PubMed] [Google Scholar]
- 42. Hagemann C., Rapp U. R. (1999) Exp. Cell Res. 253, 34–46 [DOI] [PubMed] [Google Scholar]
- 43. Deslée G., Charbonnier A. S., Hammad H., Angyalosi G., Tillie-Leblond I., Mantovani A., Tonnel A. B., Pestel J. (2002) J. Allergy Clin. Immunol. 110, 763–770 [DOI] [PubMed] [Google Scholar]
- 44. Yamasaki S., Ishikawa E., Sakuma M., Hara H., Ogata K., Saito T. (2008) Nat. Immunol. 9, 1179–1188 [DOI] [PubMed] [Google Scholar]
- 45. Heinsbroek S. E., Taylor P. R., Martinez F. O., Martinez-Pomares L., Brown G. D., Gordon S. (2008) PLoS Pathog. 4, e1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Netea M. G., Maródi L. (2010) Trends Immunol. 31, 346–353 [DOI] [PubMed] [Google Scholar]
- 47. Nakamura K., Kinjo T., Saijo S., Miyazato A., Adachi Y., Ohno N., Fujita J., Kaku M., Iwakura Y., Kawakami K. (2007) Microbiol. Immunol. 51, 1115–1119 [DOI] [PubMed] [Google Scholar]
- 48. Swain S. D., Lee S. J., Nussenzweig M. C., Harmsen A. G. (2003) Infect. Immun. 71, 6213–6221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tachado S. D., Zhang J., Zhu J., Patel N., Cushion M., Koziel H. (2007) J. Leukocyte Biol. 81, 205–211 [DOI] [PubMed] [Google Scholar]
- 50. Zhang J., Zhu J., Imrich A., Cushion M., Kinane T. B., Koziel H. (2004) Infect. Immun. 72, 3147–3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Linehan S. A., Martínez-Pomares L., Stahl P. D., Gordon S. (1999) J. Exp. Med. 189, 1961–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Martínez-Pomares L., Kosco-Vilbois M., Darley E., Tree P., Herren S., Bonnefoy J. Y., Gordon S. (1996) J. Exp. Med. 184, 1927–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fiete D., Mi Y., Oats E. L., Beranek M. C., Baenziger J. U. (2007) J. Biol. Chem. 282, 1873–1881 [DOI] [PubMed] [Google Scholar]